

Abstract
Macrophages mediate kidney disease and are prominent in a mouse model (MRL-Faslpr) of lupus nephritis. Colony stimulating factor-1 (CSF-1) is the primary growth factor for macrophages, and CSF-1 deficiency protects MRL-Faslpr mice from kidney disease and systemic illness. Whether this renoprotection derives from a reduction of macrophages and whether systemic CSF-1, as opposed to intrarenal CSF-1, promotes macrophage-dependent lupus nephritis remain unclear. Here, we found that increasing systemic CSF-1 hastened the onset of lupus nephritis in MRL-Faslpr mice. Using mutant MRL-Faslpr strains that express high, moderate, or no systemic CSF-1, we detected a much higher tempo of kidney disease in mice with the highest level of CSF-1. Furthermore, we uncovered a multistep CSF-1-dependent systemic mechanism central to lupus nephritis. CSF-1 heightened monocyte proliferation in the bone marrow (SSClowCD11b+), and these monocytes subsequently seeded the circulation. Systemic CSF-1 skewed the frequency of monocytes toward “inflammatory” (SSClowCD11b+Ly6Chigh) and activated populations that homed to sites of inflammation, resulting in a more rapid accumulation of intrarenal macrophages (CD11b+CSF-1R+ or CD68+) that induced apoptosis of tubular epithelial cells, damaging the kidney. In humans, we found increased levels of CSF-1 in the serum, urine, and kidneys of patients with lupus compared with healthy controls. Furthermore, serum and urine CSF-1 levels correlated with lupus activity, and intrarenal CSF-1 expression correlated with the histopathology activity index of lupus nephritis. Taken together, circulating CSF-1 is a potential therapeutic target for lupus nephritis.
Identifying molecules that mediate experimental lupus nephritis may uncover therapeutic targets and biomarkers. MRL-Faslpr mice develop a systemic autoimmune disease akin to human lupus nephritis and thus are a powerful tool to probe for molecules that regulate kidney disease in these patients.1,2 Kidney disease in MRL-Faslpr mice is rapid, progressive, and predictable.3 Moreover, the time frame is sufficiently slow to tease apart the pathogenesis, and sufficiently fast to be efficient. Thus, these mice are a powerful tool to probe for therapeutic targets and biomarkers in human lupus nephritis.
Macrophages (Mø) regulate kidney disease.4 Mø originate from pluripotent stem cells in the bone marrow that differentiate into mature monocytes (Mo), which enter the blood stream5,6 and traffic to the kidney. Growing evidence implicates Mø as mediators of lupus nephritis because intrarenal Mø (CD68+, F4/80+) increase with advancing disease in MRL-Faslpr mice.7 Mø require the colony stimulating factor-1 (CSF-1), their principle growth factor, to differentiate, survive, and multiply.8 Our prior studies indicate that CSF-1 is central to lupus nephritis. Implanting cells generating CSF-1 into the kidney of MRL-Faslpr mice incites local Mø-rich inflammation.9,10 Moreover, CSF-1-deficient mice (Csf1op/op;MRL-Faslpr) are protected from kidney disease and systemic illness.11,12 However, the Csf1op/op;MRL-Faslpr mice are frail and have skeletal abnormalities and numerous other defects.13–16 Thus, it is possible that the effect of deleting CSF-1 on lupus in MRL-Faslpr mice is, at least in part, not directly related to the reduction of Mø. Moreover, CSF-1-generating cells implanted into the kidney induce inflammation that is restricted to the area adjacent to the implant site.9 Thus, the systemic effect of CSF-1 during the initiation and progression of Mø-dependent lupus nephritis remains unclear.
Understanding the effect of circulating and tissue CSF-1 expression is key to designing a therapeutic treatment. CSF-1 is expressed in the circulation and is upregulated in the kidney in MRL-Faslpr mice with lupus nephritis.17–19 Intrarenal CSF-1 expression occurs during inflammation and expression is largely limited to tubular epithelial cells (TECs).20 Moreover, the rise in circulating CSF-1 precedes intrarenal CSF-1 expression and is bimodal in MRL-Faslpr mice. CSF-1 is upregulated in neonates, declines to normal levels, and then progressively rises with advancing kidney disease in MRL-Faslpr mice.19 Moreover, an increase in CSF-1 in the circulation precedes overt kidney pathology in MRL-Faslpr mice.18,19 However, it is not clear whether CSF-1 in the circulation, apart from intrarenal CSF-1, is central to the progression of lupus nephritis in MRL-Faslpr mice. Therefore, we propose to test the hypothesis that systemic CSF-1 hastens the progression of Mø-rich lupus nephritis. Furthermore, we hypothesize that circulating CSF-1 increases the frequency of circulating Mo (SSClowCD11b+), which are more readily recruited to the kidney and, in turn, induce injury.
Finally, preclinical studies are a first step in identifying therapeutic targets and biomarkers for lupus nephritis and require validation in humans. Therefore, we propose to test the hypothesis that CSF-1 is upregulated in the circulation, urine, and kidneys of patients with active lupus nephritis.
Results
The CSF-1 Transgene Drives Disease-Related Tissue Expression of CSF-1 in TgC/+;MRL-Faslpr Mice
To verify that the CSF-1 promoter/first intron-driven full-length CSF-1 transgene21 used to overexpress CSF-1 restored disease-related tissue expression of CSF-1, we created TgC/+;Csf1op/op;MRL-Faslpr mice, in which the only source of CSF-1 was encoded by the TgC transgene. We compared CSF-1 in the serum and tissues and the restoration of the phenotypic features characteristic of MRL-Faslpr mice (kidney, skin disease, lymphadenopathy, and splenomegaly) in this transgenic mouse during the disease progression (1.5, 3.0, and 5.0 mo of age) with MRL-Faslpr mice. Csf1op/op;MRL-Faslpr mice deficient in CSF-1 were used as negative controls. In the TgC/+;Csf1op/op;MRL-Faslpr mouse, CSF-1 expression in the serum, kidney, and other tissues (skin, salivary gland, lung, skin, spleen, and lymph nodes), as well as kidney pathology, lymphadenopathy, and splenomegaly were restored to the values of age-matched MRL-Faslpr mice [Supplement Figure 1 (3.0 mo data) and 1.5 and 5.0 mo data (not shown)]. Similarly, skin, salivary gland, and lung pathology in TgC/+;Csf1op/op;MRL-Faslpr mice were restored to MRL-Faslpr levels (data not shown). Thus, the TgC transgene, previously shown to direct normal tissue-specific and temporal expression of CSF-1,21 drives the disease-related tissue expression of CSF-1 in TgC/+;Csf1op/op;MRL-Faslpr mice in a manner mimicking the expression of CSF-1 in nontransgenic MRL-Faslpr mice.
The CSF-1 Transgene Increases CSF-1 in the Serum, Kidney, and Other Tissues in MRL-Faslpr Mice
To overexpress CSF-1 in MRL-Faslpr mice, we constructed TgC/+;MRL-Faslpr mice in which CSF-1 is expressed from the wild-type CSF-1 gene and the TgC transgene. To determine whether this strategy amplified CSF-1 expression, we compared CSF-1 levels in the serum, kidney, and other tissues in TgC/+;MRL-Faslpr and MRL-Faslpr mice using Csf1op/op;MRL-Faslpr as a negative control. CSF-1 expression in MRL-Faslpr mice progressively rose in the serum (Figure 1A) and tissues (Figure 1B) with advancing age and disease (1.5, 3.0, and 5.0 mo of age). The increase in CSF-1 in the TgC/+;MRL-Faslpr was, as anticipated, higher than in MRL-Faslpr mice. By comparison, serum CSF-1 levels in age-matched MRL-++ and normal B6 mice remained unchanged (dotted line, Figure 1A). Thus, we have constructed mutant MRL-Faslpr strains with varying levels of CSF-1 expression in the serum and kidneys: TgC/+;MRL-Faslpr (high), MRL-Faslpr (intermediate), and Csf1op/op;MRL-Faslpr (none).
Figure 1.
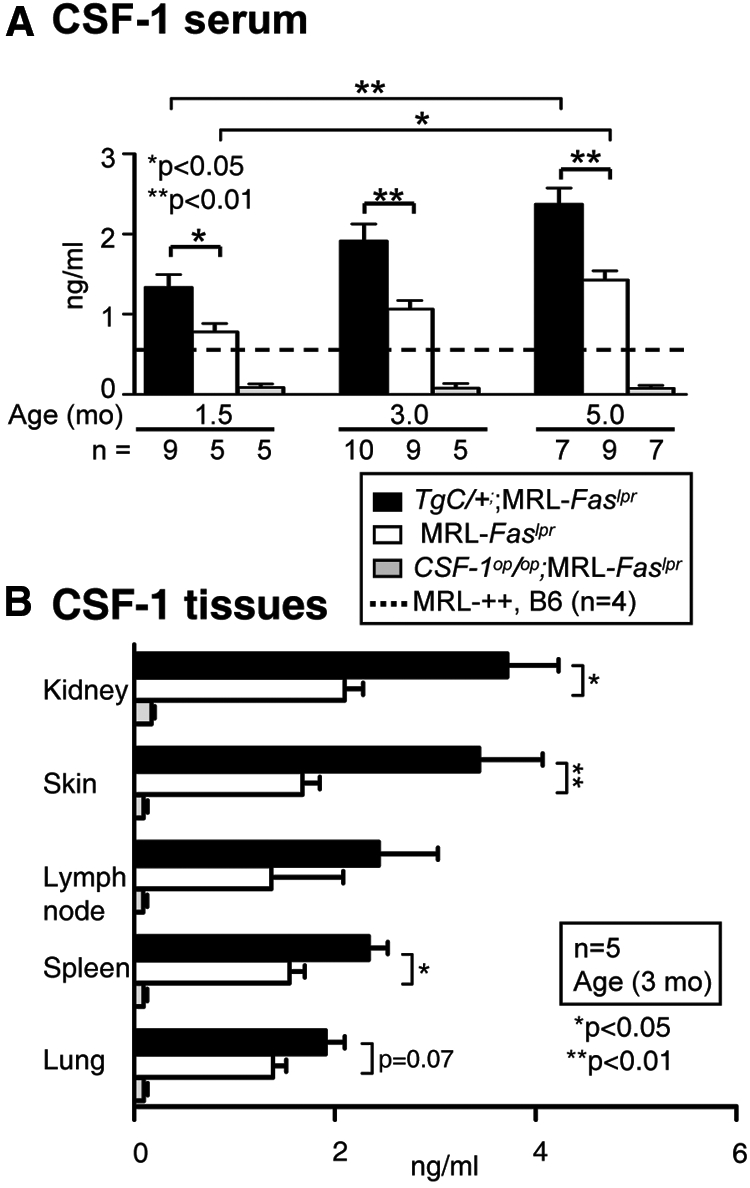
CSF-1 expression increases with age in serum and tissues of TgC/+;MRL-Faslpr mice. (A) Serum CSF-1 levels in TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf1op/op;MRL-Faslpr mice at 1.5, 3.0, and 5.0 mo of age; MRL-++ and B6 (dotted line) mice served as controls. B. CSF-1 levels in tissues (kidney, skin, lymph node, spleen, lung; 3 mo of age) determined from the supernatant fraction of the homogenates by ELISA. Values are the mean ± SEM.
CSF-1 Exacerbates Kidney Disease and Shortens Survival in MRL-Faslpr Mice
To determine whether an increase in CSF-1 promotes kidney disease, we compared the time-related renal pathology and function in TgC/+;MRL-Faslpr, MRL-Faslpr (WT), and Csf1op/op;MRL-Faslpr mice. The severity of renal pathology (glomerular, interstitial, perivascular) in TgC/+;MRL-Faslpr mice was increased at 1.5 and 3.0 mo of age as compared with WT mice (Figure 2A1). Renal disease in the WT and TgC/+;MRL-Faslpr mice was equivalent by 5 mo of age. The progression in renal pathology is consistent with an increased loss of renal function (BUN, albuminuria/creatinine ratio) in the TgC/+;MRL-Faslpr mice (Figure 2A2). The Csf1op/op;MRL-Faslpr mice remain protected from renal disease11 and MRL-++ and B6 kidneys did not have any renal pathology between 1.5 and 5.0 mo of age. Because CSF-1 is increased with advancing age and is higher in TgC/+;MRL-Faslpr than WT mice, we conclude that the tempo of kidney disease is accelerated by amplifying CSF-1 expression in MRL-Faslpr mice.
Figure 2.

Increasing CSF-1 expression in MRL-Faslpr mice accelerates kidney disease and shortens survival. (A1) Kidney histopathology comparing TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf1op/op;MRL-Faslpr mice at 1.5, 3.0, and 5.0 mo of age. MRL-++ and B6 mice without kidney disease (dotted line) are controls. Representative photomicrographs of the kidney (periodic acid-Schiff reagent). Magnification 40×. (A2) Loss of renal function is accelerated in TgC/+;MRL-Faslpr mice in comparison to MRL-Faslpr mice. In contrast, Csf1op/op;MRL-Faslpr mice remain protected from loss of renal function. Serum BUN and albuminuria/creatinine ratio in TgC/+; MRL-Faslpr, MRL-Faslpr, and Csf1op/op mice; MRL-++ and B6 mice (dotted line) served as controls. (B) Survival is decreased in TgC/+;MRL-Faslpr compared with MRL-Faslpr mice. Values are the mean ± SEM. (C) CSF-1 reporter expression in the kidney of MRL-Faslpr mice (1.5 and 3 mo of age).
Because increasing CSF-1 accelerates the tempo of kidney disease in MRL-Faslpr mice, we hypothesized that the rate of mortality is accelerated in TgC/+;MRL-Faslpr compared with WT mice. As hypothesized, TgC/+;MRL-Faslpr mice had an accelerated rate of mortality compared with WT mice (Figure 2B). Taken together, increasing CSF-1 in MRL-Faslpr mice in the circulation (Figure 1A) and kidney (primarily in TECs and to a lesser extent within glomeruli) (Figure 2C) hastens the tempo of kidney disease and shortens survival.
CSF-1 Increases Intrarenal Mø in MRL-Faslpr Mice
To determine whether the intrarenal Mø in MRL-Faslpr mice are CSF-1-dependent, we examined the time-related intrarenal accumulation of CD68+ cells in MRL-Faslpr mice with varying levels of CSF-1. The number of CD68+ cells progressively increased in the TgC/+;MRL-Faslpr and WT mice from 1.5 to 3.0 to 5.0 mo of age. However, TgC/+;MRL-Faslpr mice accumulated more CD68+ in the kidney than WT mice (Figure 3A). In contrast, CD68+ cells were rare in Csf1op/op;MRL-Faslpr kidneys (Figure 3A). Furthermore, we detected an increase in the leukocyte populations of B220+ (largely expressed by double-negative T cells22), CD4+, and CD8+ known to accompany Mø that was proportional to the level of CSF-1 in nephritic MRL-Faslpr mice (Supplement Figure 2). Of note, the magnitude of double-stranded DNA antibodies (dsDNA Abs) did not rise along with increasing CSF-1 levels and the CSF-1-dependent increase in renal disease (Supplement Figure 3). Thus, increasing CSF-1 hastens the accumulation of intrarenal Mø (CD68+) and the spectrum of leukocytes characteristic of MRL-Faslpr kidney disease.
Figure 3.

CSF-1 increases circulating Mo (SSClowCD11bhigh) as a result of heightened BM Mo proliferation and shifts circulating Mo to the inflammatory type that are recruited to the kidney. (A) Number of intrarenal CD68+ cells (Mø, DC) in TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf1op/op;MRL-Faslpr mice at 1.5, 3.0, and 5.0 mo of age. Representative photomicrographs from mice at 3 mo of age. Data are the mean ± SEM. (B) Flow cytometric analysis of the frequency of circulating SSClowCD11b+ leukocytes and extent of blood leukocyte proliferation evaluated by Brdu uptake. For Brdu uptake, freshly isolated TgC/+;MRL-Faslpr and MRL-Faslpr blood leukocytes were cultured for 6 h in Brdu. (C) Proliferation of BM CD11b+ leukocytes by flow cytometry after 24 and 48 h after administration of Brdu (intraperitoneally) into TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf-1op/op;MRL-Faslpr mice. (D) Comparison of Ly6Chigh CX3CR1low Mo in blood and Ly6Chigh cells in the kidney of TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf-1op/op;MRL-Faslpr mice by flow cytometry. (B through D) Data use mice at 3 mo of age and are the mean ± SEM. Representative FACS plots are shown.
CSF-1 Elevates the Proliferation of SSClowCD11bhigh Cells in the Bone Marrow Resulting in a Rise in Circulating Mo in MRL-Faslpr Mice
To determine whether the CSF-1-dependent accumulation of intrarenal Mø (CD68+) in MRL-Faslpr mice resulted from a rise of circulating Mo (SSClowCD11bhigh), we evaluated the frequency and total number of circulating SSClowCD11bhigh leukocytes in TgC/+;MRL-Faslpr and WT mice. We detected an enhanced frequency of SSClowCD11bhigh leukocytes in the circulation of TgC/+:MRL-Faslpr mice compared with WT mice (Figure 3B1, Supplement Figure 4A). To determine whether the heightened SSClowCD11bhigh leukocytes in the blood resulted from enhanced SSClowCD11bhigh leukocyte proliferation in the blood or bone marrow (BM), we evaluated the frequency and total number of proliferating (Brdu+) SSClowCD11bhigh leukocytes in both locations at 24 and 48 h. We detected a rise in SSClowCD11bhigh cell proliferation in the BM (Figure 3C), but not in the blood (Figure 3B2, Supplement Figure 4B), proportional to the level of CSF-1 (TgC/+;MRL-Faslpr > WT > Csf1op/op;MRL-Faslpr mice). We wish to point out that adult Csf1op/op;MRL-Faslpr mice are not monocytopenic (data not shown). Taken together, this suggests that increasing CSF-1 elevates circulating Mo (SSClowCD11bhigh) as a result of heightened BM Mo proliferation.
CSF-1 Shifts the Circulating Mo toward the “Inflammatory,” Activated Phenotype in MRL-Faslpr Mice
Our findings thus far indicate that CSF-1 increases circulating Mo and thereby provides a larger pool of cells to be recruited into the kidney. However, there are two functional subsets among blood Mo (SSClowCD11bhigh): a short-lived Ly6ChighCX3CR1lowCCR2+ subset that is actively recruited to inflamed tissues and a Ly6ClowCX3CR1highCCR2− “resident” subset characterized by CCR1-dependent recruitment to noninflamed tissues.6,23 To determine if an increased CSF-1 expression shifted the circulating Mo (SSClowCD11bhigh) to the actively recruited “inflammatory” Mo phenotype, we probed for the expression of these Mo subsets in the circulation of TgC/+;MRL-Faslpr, WT, and Csf1op/op;MRL-Faslpr mice. We detected a higher frequency and total number of Ly6ChighCX3CR1low cells in the circulation and kidney of TgC/+;MRL-Faslpr mice compared with WT mice (Figure 3D, Supplement Figure 4C). Moreover, we detected a decrease in Ly6ChighCX3CR1low subsets in the circulation and kidneys of Csf1op/op;MRL-Faslpr compared with TgC/+;MRL-Faslpr, but not WT mice (Figure 3D, Supplement Figure 4C), and an increase in the Ly6ChighCCR2+ Mo subset in the circulation in Csf1op/op;MRL-Faslpr compared with TgC/+;MRL-Faslpr but not WT mice (Supplement Figure 5). Of note, the resident Mo subset has recently been shown to patrol blood vessels and rapidly (1 to 2 h) invade tissues after an infection.24 However, our studies focus on CSF-1-dependent events at later time points (24 to 48 h) after adoptive transfer of enhanced green fluorescent protein (EGFP+) cells. Taken together, our findings suggest that CSF-1 shifts the Mo phenotype toward the Ly6ChighCX3CR1lowCCR2+ subset that are actively recruited into inflamed tissues at 24 to 48 h.
CSF-1 Fosters the Recruitment of Activated Mo (CD45.1+CSF-1R+) into the Kidney that Induces Apoptosis in MRL-Faslpr Mice
To determine whether increasing CSF-1 in the kidney more actively recruits Mo (CD11b+CSF-1R+), we injected EGFP+ BM cells into MRL-Faslpr mice expressing varying levels of CSF-1 (TgC/+;MRL-Faslpr, WT, and Csf1op/op;MRL-Faslpr). Most of these BM Mo (SSClowCD11bhigh) derived from MRL-Faslpr mice are Ly6Chigh (78% ± 4,3), a finding that is similar to nonlupus susceptible mice.6 Dependent on their milieu in the circulation, the Ly6C expression on Mo will be downregulated, resulting in an decreased recruitment to inflamed tissue.6 We detected an increase in hematopoetic cells (CD45.1+) expressing the CSF-1R promoter EGFP in the kidneys of MRL-Faslpr with the highest level of CSF-1 (TgC/+;MRL-Faslpr > WT > Csf1op/op;MRL-Faslpr), but not in the blood at 24 h posttransplant (Figure 4A, Supplement Figure 6A). This increase in EGFP+ leukocytes in the kidney was 50% higher in TgC/+;MRL-Faslpr compared with WT mice, 80% higher in WT compared with Csf1op/op;MRL-Faslpr mice, and 90% higher in TgC/+;MRL-Faslpr compared with Csf1op/op;MRL-Faslpr mice. Because cultured BM Mo derived from MRL-Faslpr mice do not double until 48 h (Supplement Figure 7), it is likely that most EGFP+ BM in the kidney reflects recruitment rather than proliferation.6
Figure 4.

CSF-1 recruits Mo (CD45.1+CSF-1R+) into the kidney, increases activated Mo (SSClowCD11bhigh) in the circulation, activated Mø (CD68+) in the kidney and, in turn, apoptosis in MRL-Faslpr kidneys. (A) We transferred BM cells from MacGreen (EGFP+);MRL-Faslpr mice into TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf1op/op;MRL-Faslpr mice and evaluated the recruitment of EGFP+ cells into the kidney at 24 h. Data are representative of two separate experiments. (B) Mo (CD11b+) activation (CD86+) in the blood and Mø (CD68+) activation (CD86+,CD69+) in the kidney analyzed by flow cytometry in TgC/+;MRL-Faslpr, MRL-Faslpr and Csf1op/op;MRL-Faslpr mice at 3 mo of age. (C) CSF-1-dependent rise in apoptosis (caspase 3) in TgC/+;MRL-Faslpr, MRL-Faslpr, and Csf1op/op;MRL-Faslpr mice. Representative photomicrographs are shown. (A through C) Data use mice at 3 mo of age and are the mean ± SEM. (A and B) Representative FACS plots are displayed.
CSF-1 directly activates Mø (CD68+),25 and these Mø, in turn, release mediators that induce TEC apoptosis in vitro.26 Therefore, we hypothesized that the number of circulating Mo (SSClowCD11bhigh) and intrarenal activated Mø (CD68+) and apoptotic TECs in TgC/+;MRL-Faslpr mice is greater than WT mice. We detected a higher frequency and total number of activated (CD69+ and CD86+) cells in TgC/+;MRL-Faslpr mice with increased levels of CSF-1 in the circulation (Figure 4B1, Supplement Figure 6B1) and the kidney (Figure 4B2, Supplement Figure 6B2). Moreover, in the circulation we detected a CSF-1-dependent shift to an increased population of activated Ly6ChighCD69+ and Ly6ChighCD86+ Mo (SSClowCD11bhigh) (Supplement Figure 8), MRL-Faslpr mice at 3 and 5 mo of age that are primarily recruited to inflamed tissue.6 Similarly, the number of intrarenal apoptotic TECs rose in proportion to the level of CSF-1 in MRL-Faslpr mice (Figure 4C). Taken together, enhancing CSF-1 expression in MRL-Faslpr mice increases the number of Mo seeded from the BM into the circulation; shifts these Mo toward an inflammatory and activated phenotype; and, in turn, hastens the rise of activated Mø in the kidney that induces TEC apoptosis.
Circulating CSF-1, apart from Intrarenal CSF-1, Increases Mø in the Kidney
To determine whether a rise in circulating CSF-1 alone, apart from upregulated intrarenal CSF-1, contributes to the CSF-1-dependent increase in intrarenal Mø (CD45.1+CSF-1R+) in the kidney, we compared Csf1op/op;MRL-Faslpr mice injected with CSF-1 versus PBS for 2 d (Supplement Figure 9). Hence, the sole source of CSF-1 in the Csf1op/op;MRL-Faslpr mice was in the circulation. After adoptive transfer of EGFP+ BM cells (derived from MacGreen;MRL-Faslpr mice) into Csf1op/op;MRL-Faslpr mice, the frequency and total number of EGFP+ cells in the kidney was substantially greater in CSF-1 versus PBS-treated mice (Figure 5A). This suggests that increasing circulating CSF-1 enhances Mø accumulation in the kidney.
Figure 5.

Circulating CSF-1 alone increases BM Mo proliferation; increases circulating, activated Mo (SSClowCD11bhigh) and intrarenal inflammatory activated Mø (F4/80+); and recruits Mo (CD45.1+CSF-1R+) into MRL-Faslpr kidneys. (A) More adoptively transferred EGFP+ cells were recruited into the kidney in CSF-1 versus PBS-injected Csf1op/op;MRL-Faslpr mice. (B) Increase in circulating CD11b+ cells in the blood in CSF-1 versus PBS-injected Csf1op/op;MRL-Faslpr mice. (C) Rise in BM Mo proliferation in CSF-1- versus PBS-injected Csf1op/op;MRL-Faslpr mice. (D) Increase in CD11b+ expressing Ly6Chigh Mo in the blood and kidney in CSF-1 versus PBS-injected Csf1op/op;MRL-Faslpr mice. (A through D) Data used mice at 3 mo of age and are mean ± SEM (flow cytometry). Representative plots are shown.
Circulating CSF-1 Enhances BM Mo Proliferation, the Frequency of Inflammatory Mo in the Circulation, and, in Turn, Mø Accumulation in MRL-Faslpr Kidneys
Consistent with the action of CSF-1 in increasing blood Mo (SSClowCD11bhigh), we detected an elevation in the frequency and total number of SSClowCD11b+ cells in the blood in Csf1op/op;MRL-Faslpr mice injected with CSF-1 compared with PBS (Figure 5B). Moreover, there was a robust increase in the frequency and total number of proliferating (Brdu+) SSClowCD11b+ cells in the BM in CSF-1 versus PBS-treated Csf1op/op;MRL-Faslpr mice (Figure 5C). Furthermore, we detected a considerable rise in the frequency and total number of Ly6ChighCD11b+ inflammatory Mo in the circulation and Ly6ChighF4/80+ activated Mø (F4/80+) in the kidney of CSF-1- versus PBS-treated mice (Figure 5D). Taken together, this suggests that elevation of circulating CSF-1 alone is responsible for increasing the number of Mo released from the BM into the circulation and for shifting the circulating Mo populations toward a Ly6Chigh activated Mo phenotype that is more readily recruited into the kidney.
CSF-1 is Expressed by TECs in Patients with Lupus Nephritis
To determine whether our findings in lupus nephritis in mice may be applicable to human lupus, we probed for CSF-1 expression in kidney sections in patients with lupus nephritis, in proteinuric patients with other kidney diseases (minimal-change and membranous GN), and in control patients (patient's characteristics; Table 1). Similar to the MRL-Faslpr mice, CSF-1 is highly (nearly 50%) expressed by TECs in patients with lupus nephritis as compared with other kidney diseases and controls (Figure 6A). By comparison, CSF-1 is more robustly expressed in lupus nephritis as compared with other kidney diseases (minimal-change and membranous GN) with far fewer intrarenal leukocytes (CD68: 36 ± 8 versus 10 ± 3; CD3: 18 ± 5 versus 4 ± 1, respectively, n = 10/group). Note CSF-1 expression is specific because we did not detect staining using a goat IgG control, and CSF-1 staining was blocked by preabsorption of the anti-CSF-1 antibody (Figure 6A). To determine whether elevated CSF-1 expression by TECs correlates with the activity and chronicity of the lupus nephritis, we evaluated the histopathological activity index and chronicity index.27,28 We determined that the CSF-1 expression in TECs correlates with the activity index but not with the chronicity index (Figure 6B). Thus, TECs are the principle source of CSF-1 in human lupus nephritis and the level of CSF-1 expression is an index of disease activity.
Table 1.
Characteristics of patients analyzed for CSF-1 expression in serum and urine
| Group | n | Gender (Male/Female) | Age (yr)a | C3/C4b (g/L) | dsDNAc >200 (IU/ml) | Serum Creatinine (mg/dl)a |
|---|---|---|---|---|---|---|
| SLE with lupus nephritis | 20 | 0/20 | 19.88 ± 4.68 | >0.9/>0.1d | 6 of 20 | 1.01 ± 0.27 |
| WHO Class IV | 14 | 2/12 | 37.67 ± 5.07 | <0.9/<0.1e | 12 of 14 | 1.13 ± 0.17 |
| SLE with lupus nephritis | 7 | 0/7 | 37.51 ± 4.25 | >0.9/>0.1d | 2 of 7 | 1.38 ± 0.28 |
| WHO Class III or V | 2 | 0/2 | 39.29 ± 6.12 | <0.9/<0.1e | None | 1.24 ± 0.17 |
| SLE without nephritis | 10 | 0/10 | 45.34 ± 7.3 | >0.9/>0.1d | 4 of 10 | 0.81 ± 0.24 |
| 10 | 0/10 | 40.14 ± 6.9 | <0.9/<0.1e | 8 of 10 | 0.84 ± 0.15 | |
| Other kidney diseases | 8 | 4/4 | 40.78 ± 8.32 | >0.9/>0.1d | None | 1.67 ± 0.78 |
| Healthy controls | 17 | 6/11 | 32.71 ± 2.86 | >0.9/>0.1d | 1 of 17 | 0.97 ± 0.17 |
aData are mean ± SD.
bC3, complement 3 in the serum; C4 complement 4 in the serum.
cdsDNA, dsDNA Ab in the serum.
dC3/C4 → normal.
eC3/C4 → reduced.
SLE, systemic lupus erythematosus; WHO, World Health Organization; 190 × 113 mm (300 × 300 DPI).
Figure 6.

CSF-1 expression is abundant in TECs from patients with lupus nephritis and correlates with the histopathologic activity. (A) We detected far more robust CSF-1 expression in the kidney of patients with lupus nephritis compared with minimal expression in other kidney diseases (minimal-change and membranous GN) with far fewer inflammatory infiltrates and with controls. Boxed area is enlargement. (B) CSF-1 expression in TECs correlates with the histopathology activity but not chronicity index in patients with lupus nephritis. Values are the mean ± SEM.
CSF-1 Is Increased in the Serum and Urine in Patients with Serologically Active Lupus Nephritis
To determine whether the pathologic effect of elevated systemic CSF-1 on lupus nephritis may be relevant in humans, we measured the concentrations of CSF-1 in serum and urine of patients with lupus nephritis, proteinuric patients with other kidney diseases (minimal-change and membranous GN), lupus patients without nephritis, and healthy controls. We detected a marked rise in CSF-1 in the serum and urine in patients with lupus nephritis, as compared with other kidney diseases and healthy controls (Figure 7A, Table 1). Moreover, CSF-1 was elevated in the serum, but not in the urine, in patients with lupus nephritis compared with patients with lupus without nephritis (Figure 7A, Table 1). To determine whether CSF-1 expression correlated with disease activity in lupus, we evaluated serologic parameters and renal function. Lupus activity parameters included low levels of serum C3 and C4 and high levels of serum anti-dsDNA Ab. We determined that circulating CSF-1 concentrations correlated with the low levels of C3 and C4 and elevated dsDNA Ab levels in the sera (Figure 7B). By comparison, we did not detect CSF-1 in the serum of healthy individuals. Furthermore, CSF-1 levels in the urine correlated with low, but not high, proteinuria/creatinine ratios in patients with serologically active lupus nephritis. This finding most likely reflects an increased loss of TECs that generate CSF-1 with advancing loss of renal function (Figure 7C1). Of note, CSF-1 was higher in patients with active lupus nephritis compared with those in remission (Figure 7C2). Taken together, a rise of CSF-1 expression in TECs and elevated CSF-1 in the serum and urine are indicators of active lupus nephritis.
Figure 7.

CSF-1 is elevated in urine and serum of patients with lupus nephritis and correlates with disease activity. (A) CSF-1 levels in serum and urine are elevated in patients with lupus nephritis compared with other kidney disease and healthy controls (ELISA). CSF-1 is elevated in the serum, but not urine, of lupus patients with nephritis compared with those without nephritis. (B) Serum CSF-1 levels of patients with lupus nephritis correlate with serologically active lupus (low C3/C4, high anti-dsDNA Ab). (C) Urine CSF-1 levels in patients with lupus nephritis correlates with low, but not high, proteinuria/creatinine ratios. The criteria for proteinuria/creatinine ratios is high >1000 mg/24 h; and low <1000 mg/24 h). CSF-1 in the urine is elevated in patients with active lupus compared with those in remission (proteinuria <300 mg/24 h, creatinine <1.3 mg/dl). The clinical data and histopathology type of GN of the patient collective are summarized in Table 1. Values are the mean ± SEM.
Discussion
CSF-1 may be a “master switch” regulating the multiorgan disease characteristic of MRL-Faslpr mice. The concept that CSF-1 expression in the circulation and tissues promotes lupus is in keeping with our prior studies.17,29,30 Using the CSF-1 transgenic MRL-Faslpr line that promotes lupus nephritis we have used in the study presented here, we previously detected a more rapid onset of cutaneous disease (discoid lupus) compared with WT mice.25 Moreover, sunlight exposure induces CSF-1 in keratinocytes in the epidermis that spills over into the circulation and, in turn, leads to the recruitment and activation of Mø (F4/80+CD68+) in the skin. Thus, CSF-1 sets in motion Mø-mediated events leading to the destruction of keratinocytes and discoid lupus.25 Because disease in MRL-Faslpr is a multiorgan disease involving kidney, skin, and other tissues,31 many tissues may contribute and respond to increasing circulating CSF-1 levels. Because our study indicates that circulating CSF-1 is instrumental in mediating renal injury, we speculate that CSF-1 generated within the kidney that is released into the circulation has an effect on cutaneous disease and other tissues, and vice versa. Thus, systemic CSF-1 expression may regulate multitissue injury in lupus.
We now report that CSF-1 regulates the generation of inflammatory Ly6Chigh Mo (SSClowCD11bhigh).23 Evidence suggests that inflamed tissues increase circulating CSF-1 and thereby stimulate the production of Mo (SSClowCD11bhigh) in the BM and their subsequent release into the circulation.6 When this is substantial, there is a skewing of the Mo population toward a higher frequency of Ly6Chigh cells. Our data suggest that this skewing toward circulating Ly6Chigh cells is CSF-1-dependent. This is based on the CSF-1-dependent increase in the immature Ly6Chigh Mo proliferation and the rise in circulating Ly6Chigh Mo that are more readily recruited to the kidney. The MRL-Faslpr kidney may be self-destructive. Because renal TECs are a rich source of CSF-1 during inflammation,20 CSF-1 originating from the kidney may, in part, account for the rise in circulating CSF-1 that drives Mø (CD68+) to become activated and destroy the kidney.25,26 On the other hand, CSF-1 is increased in the circulation in neonates, well in advance of renal injury in MRL-Faslpr mice.19 Thus, it is possible that a circulating CSF-1-dependent mechanism leads to amplifying intrarenal CSF-1. Regardless, our findings indicate that CSF-1 is the conduit between the circulation and tissue that regulates the destiny of Mo and, in turn, inflammation in MRL-Faslpr mice.
CSF-1 levels in the serum and urine may be a biomarker for active lupus nephritis. This is consistent with a prior finding indicating that urinary CSF-1 (also known as M-CSF), regulated upon activation, normal T cell expressed and secreted protein, and monocyte chemoattractant protein-1 are increased during renal flares in patients with lupus.32 Our data agree with a report indicating that elevated CSF-1 in the sera of patients with lupus, including those with nephritis, correlates with disease activity.33 However, our findings extend prior reports because our data indicate that urinary and serum CSF-1 and CSF-1 levels in the kidney correlate with active lupus. Although our findings indicate that low serum C3/C4, an index of active lupus, have a high correlation with increasing serum CSF-1 concentrations, the correlation of high serum dsDNA Ab, another indicator of active lupus, is less impressive. This may be related to the extreme variability between dsDNA Ab commercial kits (10 kits tested, unpublished data). It is worth noting that urine CSF-1 correlates with moderate proteinuria (>1000 mg/24h), but not higher urinary protein levels. This is in keeping with the generation of CSF-1 at the onset of inflammation and the destruction of TECs, the major source of CSF-1, during persistent inflammation. Consistent with this concept, CSF-1 levels in TECs do not correlate with increasing histopathologic chronicity. Although urine and serum CSF-1 may be a biomarker for active lupus, it is unlikely that it is unique for lupus nephritis because CSF-1 is upregulated in the kidney after other forms of renal injury. For example, CSF-1 is expressed in TECs and to a lesser extent in glomeruli after acute kidney injury (after ischemia/reperfusion in mice and in patients with loss of renal function and tubular pathology postengraftment34). Future studies will determine whether CSF-1 is a biomarker and therapeutic target for other human Mø-dependent kidney diseases.35
CSF-1 expression during lupus nephritis is primarily within TECs, because expression in glomeruli is far less abundant. This is at odds with a prior report pinpointing the expression of CSF-1 in humans exclusively to glomeruli.36 This may be related to the method of detecting CSF-1. For example, our prior findings in MRL-Faslpr mice indicated that CSF-1 is expressed by TECs and mesangial cells using antibody detection,17 whereas in this study we localize CSF-1 in TECs, mesangial cells, and podocytes by morphologic criteria and using mice expressing a CSF-1 reporter gene. However, pinpointing the precise cell types expressing CSF-1 during each stage of progressive lupus will require a more comprehensive analysis. Nevertheless, our findings indicate that CSF-1 expression in the kidney correlates with the histopathology activity index for lupus nephritis. However, to more fully explore CSF-1 as a biomarker for lupus nephritis, follow-up studies will correlate the pathology findings with the clinical disease activity score (SLEDAI) for each patient and determine whether serum and urine CSF-1 levels longitudinally track with the progression of lupus nephritis.
Understanding the effect of circulating versus tissue CSF-1 has profound therapeutic implications. We have compelling evidence that CSF-1 in the circulation alone enhances Mø-dependent lupus nephritis in MRL-Faslpr mice. Injecting CSF-1 stimulates the proliferation of Mo (SSClowCD11bhigh) in the BM, which subsequently leads to a rise in circulating Mo populations, which in turn are more readily recruited to the kidney and induce injury. This is consistent with a study in which injecting CSF-1 accelerates the intrarenal accumulation of Mø and proteinuria in LPS-treated nonlupus susceptible mice.37 Within this context, our human translational findings indicate that the rise in circulating CSF-1 in these patients with lupus nephritis is not only a biomarker of disease activity but is instrumental in mediating lupus nephritis. This suggests that strategies to diminish circulating CSF-1 will halt escalating lupus nephritis in humans. Thus, purging CSF-1 from the circulation may tilt the balance of Mo back to a more mature population that is not readily recruited to tissues. Taken together, circulating CSF-1 is a potential therapeutic target and may be an attractive biomarker for lupus.
Concise Methods
Mice
Mice heterozygous for the osteopetrotic mutation (Csf1op) on the C57BL/6JxC3Heb/FeJ-a/a background, BALB/c, C57BL/6 (B6), MRL/MpJ-+/+ (MRL-++), and MRL/MpJ-Faslpr/Faslpr (MRL-Faslpr) mice were purchased from Jackson Laboratories (Bar Harbor, ME). The Csf1op mutation was backcrossed onto the MRL-Faslpr background for ten generations. Transgenic Tgfms-EGFP mice that express EGFP under the control of the CSF-1R (c-fms) promoter and first intron,38 were provided by Dr. D.A. Hume (University of Edinburgh, Edinburgh, Scotland). These mice were backcrossed onto the MRL-Faslpr background (N7) and are referred to as MacGreen;MRL-Faslpr mice. The TgN(FLCsf1)9Ers/+ mice expressing the TgN(FLCsf1)Ers (TgC) transgene (full-length CSF-1 gene driven by the CSF-1 promoter/first intron)21 were backcrossed onto the MRL-Faslpr and Csf-1op/op;MRL-Faslpr background (N7) generations and were referred to as TgC/+;MRL-Faslpr mice. Transgenic mice in which lacZ expression is driven by the same CSF-1 promotor first intron sequence used to construct the transgene were backcrossed onto the MRL-Faslpr background (N7) to identify CSF-1 expressing cells in the kidney.20 Mice were bred and housed at Harvard Medical School. Only female mice were used. The use of mice in this study was reviewed and approved by the Standing Committee on Animals at the Harvard Medical School in adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
CSF-1 ELISA
We measured mouse CSF-1 levels in the serum, urine, and tissue homogenates using an ELISA method.25 Undiluted serum and urine samples were analyzed directly. The tissues used for homogenates were from mice perfused with PBS through the heart to flush out CSF-1 in the circulation. Briefly, we homogenized tissue samples using a MixMill 300 (Qiagen, Valencia, CA). Samples were spinned down and the supernatant fraction of the homogenates was used for the ELISA.39 We determined the protein concentration of each sample (supernatant of homogenate) using the BCA Protein Assay Kit (PIERCE, Rockford, IL) and evaluated 200 μg of protein per tissue sample. To detect CSF-1 in human serum and urine, we used a human CSF-1 ELISA kit according to the manufacturer's instructions. The ELISA antibodies and reagents were purchased from BD Bioscience (San Jose, CA).
Renal Histopathology
We fixed kidneys in 10% neutral buffered formalin for 24 h and stained paraffin sections (4 μm) with periodic acid-Schiff reagent. We evaluated kidney pathology as described previously.40 Briefly, we assessed glomerular pathology by scoring each glomerulus on a semiquantitative scale: 0 = normal [35 to 40 cells/glomerular cross section (GCS)]; 1 = mild [glomeruli with few lesions showing slight proliferative changes, mild hypercellularity (41 to 50 cells/GCS)]; 2 = moderate [glomeruli with moderate hypercellularity (51 to 60 cells/GCS, including segmental and/or diffuse proliferative changes, hyalinosis)], 3 = severe [glomeruli with segmental or global sclerosis and/or severe hypercellularity (>60 cells/GCS), necrosis, and crescent formation]. We scored 20 gcs/kidney. Interstitial/tubular pathology was assessed semiquantitatively on a scale of 0 to 3 in 10 randomly selected high-power fields. We determined the largest and average number of infiltrates and damaged tubules and adjusted the grading system accordingly: 0 = normal, 1 = mild, 2 = moderate, 3 = severe. Perivascular cell accumulation was determined semiquantitatively by scoring the number of cell layers surrounding most vessel walls (score: 0 = none, 1 = <5 cell layers, 2 = 5 to 10 cell layers, 3 = >10 cell layers).
Renal Function
We measured BUN, creatinine, and albuminuria as described previously.41
Splenomegaly/Lymphadenopathy
Splenomegaly and lymph node weight were determined at the time of sacrifice by comparing the ratios of the tissue to body weight.
Immunostaining
Mouse Tissue.
Kidney tissue was processed and stained for the presence of CD68, CD4, CD8, B220, and anticleaved caspase-3 antibody (Asp175) (Cell-Signaling, Danvers, MA), as described previously for the kidney.41 To identify β-galactosidase encoded by lacZ (CSF-1 expression), we treated frozen kidney sections as described previously.20
Human Tissue.
We deparaffinized and rehydrated serial sections (4 μm). Antigen retrieval was performed by immersion in citrate buffer followed by blocking endogenous peroxidase activity and nonspecific binding of avidin and biotin as described previously.41 We incubated these sections with a primary antibody, goat anti-human CSF-1 antibody (N-16; Santa Cruz, CA), and we detected the primary antibody by incubation with biotinylated rabbit anti-goat Ab followed by development with 3′3-diaminobenzidine (Vector; Burlingame, CA). We verified staining specificity by replacing the primary antibody with goat IgG (eBioscience, San Diego, CA) and by preabsorbing with peptide for CSF-1 antibody (Santa Cruz). We determined the percentage of positive TECs in ten randomly selected high-power fields.
Flow Cytometry
We prepared and stained single-cell suspensions from kidneys, blood leukocytes, and BM Mo as described previously.42 We collected 0.5 × 106 to 1.0 × 106 total kidney cells and 0.5 × 105 to 1.0 × 105 blood leukocytes/BM Mo using a FACSCalibur (Becton Dickinson, San Jose, CA) and analyzed the data using Flowjo software (Tree Star, Palo Alto, CA). The BM Mo and circulating Mo were characterized by gating on the SSClowCD11bhigh population.6
Antibodies
We used the following antibodies from eBioscience (San Diego, CA) for FACS analysis: FITC-conjugated anti-CD4 (L3T4), anti-CD8 (53 to 6.7), and anti-CD11b (M1/70) band anti-CD45.2 (104); phycoerythrin (PE)-conjugated anti-CD4, anti-CD45.2, anti-CD69 (H1.2F3), and anti-CD86 (GL1); PE-Cy5-conjugated anti-CD8; and allophycocyanin-conjugated anti-CD4, anti-CD45.2, and anti-F4/80 (BM8). We used FITC- and allophycocyanin-conjugated anti-CD68 antibody (FA11) and PE-conjugated Ly6C (Serotec, Oxford, United Kingdom), purified CX3CR1 (2A9-1; MBL, Woburn, MA),and purified rabbit anti-mouse CCR2 (NovusBiologicals). For the secondary PE- or allophyocyanin-conjugated Ab, we used goat-anti-rabbit from Jackson ImmunoResearch Laboratories (West Grove, PA) and biotin-conjugated rabbit anti-goat antibody (Vector Laboratories, Burlingame, CA). To detect biotin-conjugated secondary antibodies, we used strepavidin PE or allophyocyanin (Jackson ImmunoResearch Laboratories, West Grove, PA).
Brdu Treatment
In Vivo.
We injected mice with Brdu (2 mg/mouse intraperitoneally, every 12 h, Sigma, St. Louis, MO) for 24 and 48 h before sacrifice. Brdu+ cells were analyzed by flow cytometry with an anti-Brdu antibody (eBioscience, San Diego, CA).
In Vitro.
We incubated freshly isolated blood leukocytes for 6, 12, and 24 h in media containing Brdu (1 mg/ml) and analyzed the Brdu+ cells by flow cytometry.
Adoptive Transfer
We isolated BM from MacGreen;MRL-Faslpr mice43 and adoptively transferred these EGFP+ cells (2 × 107) by intravenous injection into the tail. We sacrificed these mice after 24 and 48 h and prepared the kidney and blood samples to detect EGFP+ cells using flow cytometry as described previously.41 We adoptively transferred EGFP+ BM cells into Csf-1op/op;MRL-Faslpr mice. These mice were then injected with rCSF-1 (50 μg/kg body wt) every 12 h, and sacrificed after 24 and 48 h.
BM-Derived Mø
We isolated BM Mo as described previously.43,44 When the cultured cells were confluent, we incubated cells for 24 h without CSF-1 to synchronize the cells, before proceeding with specific stimulations.
Proliferation
BM Mo (1 × 104 well) were stimulated with mouse CSF-1 (10 ng/ml, Preprotech, Rocky Hill, NJ) for 24 and 48 h. We analyzed proliferation using the MTT colorimetric assay (Roche, Palo Alto, CA) according to the manufacturer's instructions.
Serum, Urine, and Renal Biopsy Samples
Human kidney sections with a confirmed pathologic diagnosis lupus nephritis type III, IV, and V (according to the International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 classification of lupus nephritis45 and activity and chronicity index27,28 or minimal-change GN and membranous GN were provided by the Department of Pathology, Rush University Medical Center, Chicago, Illinois. Urine and serum samples were collected at the Department of Medicine, Johannes-Gutenberg University Mainz, Germany. Serum and urine samples were stored at −20°C. The use of the samples was reviewed and approved by the Standing Committee for Clinical Studies of the Johannes-Gutenberg University in adherence to the Declaration of Helsinki. Samples were taken from patients attending the outpatient clinic who fulfilled at least four of the American College of Rheumatology criteria for the classification of systemic lupus erythematosus after informed consent. The characteristics of the patients are summarized in Table 1. The clinical parameters (C3/C4; dsDNA, creatinine, and proteinuria) were evaluated at the Johannes-Gutenberg University Mainz, Germany. Note that we define C3/C4 levels according to the following criteria: C3 (high >0.09g/L; low C3<0.09; and C4 (high C4>0.01; low C4<0.01). The control human kidney tissue was taken from uninvolved areas of nephrectomies for malignancy and preimplantation donor kidney biopsies.
Statistical Analysis
The data representing the mean ± SEM were prepared using GraphPad PRISM version 4.0. We used the nonparametric Mann–Whitney U test to evaluate P values. For correlation analysis, we used the Spearman correlation calculation. For statistical evaluation of survival, we used the log rank test.
Disclosures
None.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants DK 36149 (V.R.K.), CA32551 (E.R.S.), and CA26504 (E.R.S.). J. Menke is supported by the Deutsche Forschungsgemeinschaft (ME-3194/1-1). This work was supported by National Institutes of Health grants DK36149 (V.R.K.), CA32551, and CA26504 (E.R.S.) and the Lupus Research Institute (V.R.K.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental information for this article is available online at http://www.jasn.org/.
References
- 1.Theofilopoulos AN, Dixon FJ: Eito-pathogenesis of murine SLE. Immunol Rev 55: 179–216, 1981 [DOI] [PubMed] [Google Scholar]
- 2.Moyer CF, Strandberg JD, Reinisch CL: Systemic mononuclear-cell vasculitis in MRL/Mp-lpr/lpr mice. A histologic and immunocytochemical analysis. Am J Pathol 127: 229–242, 1987 [PMC free article] [PubMed] [Google Scholar]
- 3.Kelley VE, Roths JB: Interaction of mutant lpr gene with background strain influences renal disease. Clin Immunol Immunopathol 37: 220–229, 1985 [DOI] [PubMed] [Google Scholar]
- 4.Ricardo SD, van Goor H, Eddy AA: Macrophage diversity in renal injury and repair. J Clin Invest 118: 3522–3530, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katsiari CG, Liossis SN, Sfikakis PP: The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: A reappraisal. Semin Arthritis Rheum, 2009. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ: Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol 172: 4410–4417, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR: Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med 190: 1813–1824, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pixley FJ, Stanley ER: CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol 14: 628–638, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Naito T, Yokoyama H, Moore KJ, Dranoff G, Mulligan RC, Kelley VR: Macrophage growth factors introduced into the kidney initiate renal injury. Mol Med 2: 297–312, 1996 [PMC free article] [PubMed] [Google Scholar]
- 10.Moore KJ, Naito T, Martin C, Kelley VR: Enhanced response of macrophages to CSF-1 in autoimmune mice: A gene transfer strategy. J Immunol 157: 433–440, 1996 [PubMed] [Google Scholar]
- 11.Lenda DM, Kikawada E, Stanley ER, Kelley VR: Reduced macrophage recruitment, proliferation, and activation in colony-stimulating factor-1-deficient mice results in decreased tubular apoptosis during renal inflammation. J Immunol 170: 3254–3262, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Lenda DM, Stanley ER, Kelley VR: Negative role of colony-stimulating factor-1 in macrophage, T cell, and B cell mediated autoimmune disease in MRL-Fas(lpr) mice. J Immunol 173: 4744–4754, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW, Jr, Ahmed-Ansari A, Sell KW, Pollard JW, Stanley ER: Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A 87: 4828–4832, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Begg SK, Radley JM, Pollard JW, Chisholm OT, Stanley ER, Bertoncello I: Delayed hematopoietic development in osteopetrotic (op/op) mice. J Exp Med 177: 237–242, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cecchini MG, Dominguez MG, Mocci S, Wetterwald A, Felix R, Fleisch H, Chisholm O, Hofstetter W, Pollard JW, Stanley ER: Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development 120: 1357–1372, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Cohen PE, Chisholm O, Arceci RJ, Stanley ER, Pollard JW: Absence of colony-stimulating factor-1 in osteopetrotic (csfmop/csfmop) mice results in male fertility defects. Biol Reprod 55: 310–317, 1996 [DOI] [PubMed] [Google Scholar]
- 17.Wada T, Naito T, Griffiths RC, Coffman TM, Kelley VR: Systemic autoimmune nephritogenic components induce CSF-1 and TNF-alpha in MRL kidneys. Kidney Int 52: 934–941, 1997 [DOI] [PubMed] [Google Scholar]
- 18.Rubin Kelley V, Bloom RD, Yui MA, Martin C, Price D: Pivotal role of colony stimulating factor-1 in lupus nephritis. Kidney Int Suppl 45: S83–S85, 1994 [PubMed] [Google Scholar]
- 19.Yui MA, Brissette WH, Brennan DC, Wuthrich RP, Rubin-Kelley VE: Increased macrophage colony-stimulating factor in neonatal and adult autoimmune MRL-lpr mice. Am J Pathol 139: 255–261, 1991 [PMC free article] [PubMed] [Google Scholar]
- 20.Jang MH, Herber DM, Jiang X, Nandi S, Dai XM, Zeller G, Stanley ER, Kelley VR: Distinct in vivo roles of colony-stimulating factor-1 isoforms in renal inflammation. J Immunol 177: 4055–4063, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Ryan GR, Dai XM, Dominguez MG, Tong W, Chuan F, Chisholm O, Russell RG, Pollard JW, Stanley ER: Rescue of the colony-stimulating factor 1 (CSF-1)-nullizygous mouse (Csf1(op)/Csf1(op)) phenotype with a CSF-1 transgene and identification of sites of local CSF-1 synthesis. Blood 98: 74–84, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Schwarting A, Moore K, Wada T, Tesch G, Yoon HJ, Kelley VR: IFN-gamma limits macrophage expansion in MRL-Fas(lpr) autoimmune interstitial nephritis: A negative regulatory pathway. J Immunol 160: 4074–4081, 1998 [PubMed] [Google Scholar]
- 23.Geissmann F, Jung S, Littman DR: Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19: 71–82, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F: Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317: 666–670, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Menke J, Hsu MY, Byrne KT, Lucas JA, Rabacal WA, Croker BP, Zong XH, Stanley ER, Kelley VR: Sunlight triggers cutaneous lupus through a CSF-1-dependent mechanism in MRL-Fas(lpr) mice. J Immunol 181: 7367–7379, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tesch GH, Schwarting A, Kinoshita K, Lan HY, Rollins BJ, Kelley VR: Monocyte chemoattractant protein-1 promotes macrophage-mediated tubular injury, but not glomerular injury, in nephrotoxic serum nephritis. J Clin Invest 103: 73–80, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Austin HA, III, Muenz LR, Joyce KM, Antonovych TA, Kullick ME, Klippel JH, Decker JL, Balow JE: Prognostic factors in lupus nephritis. Contribution of renal histologic data. Am J Med 75: 382–391, 1983 [DOI] [PubMed] [Google Scholar]
- 28.Austin HA, III, Muenz LR, Joyce KM, Antonovych TT, Balow JE: Diffuse proliferative lupus nephritis: Identification of specific pathologic features affecting renal outcome. Kidney Int 25: 689–695, 1984 [DOI] [PubMed] [Google Scholar]
- 29.Naito T, Griffiths RC, Coffman TM, Kelley VR: Transplant approach establishes that kidneys are responsible for serum CSF-1 but require a stimulus in MRL-lpr mice. Kidney Int 49: 67–74, 1996 [DOI] [PubMed] [Google Scholar]
- 30.Yokoyama H, Naito T, Dranoff G, Mulligan RC, Rubin-Kelley V: Gene transfer of macrophage growth factors into the kidney of lpr mice initiates renal injury. Contrib Nephrol 118: 94–99, 1996 [DOI] [PubMed] [Google Scholar]
- 31.Mountz JD, Gause WC, Jonsson R: Murine models for systemic lupus erythematosus and Sjogren's syndrome. Curr Opin Rheumatol 3: 738–756, 1991 [DOI] [PubMed] [Google Scholar]
- 32.Tian S, Li J, Wang L, Liu T, Liu H, Cheng G, Liu D, Deng Y, Gou R, Wan Y, Jia J, Chen C: Urinary levels of RANTES and M-CSF are predictors of lupus nephritis flare. Inflamm Res 56: 304–310, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Yang PT, Kasai H, Xiao WG, Zhao LJ, He LM, Yamashita A, Deng XW, Ito M: Increased expression of macrophage colony-stimulating factor in ankylosing spondylitis and rheumatoid arthritis. Ann Rheum Dis 65: 1671–1672, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menke J, Iwata Y, Rabacal WA, Basu R, Yeung YG, Humphreys BD, Wada T, Schwarting A, Stanley ER, Kelley VR: CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. J Clin Invest 119:2330–2342, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang N, Isbel NM, Nikolic-Paterson DJ, Li Y, Ye R, Atkins RC, Lan HY: Local macrophage proliferation in human glomerulonephritis. Kidney Int 54: 143–151, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Matsuda M, Shikata K, Makino H, Sugimoto H, Ota Z: Glomerular expression of macrophage colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in patients with various forms of glomerulonephritis. Lab Invest 75: 403–412, 1996 [PubMed] [Google Scholar]
- 37.Utsunomiya Y, Omura K, Yokoo T, Imasawa T, Kawamura T, Abe A, Hirano K, Mitarai T, Maruyama N, Sakai O: Macrophage-colony stimulating factor (M-CSF) enhances proteinuria and recruitment of macrophages into the glomerulus in experimental murine nephritis. Clin Exp Immunol 106: 286–296, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA: A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101: 1155–1163, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Botran R, Gorantla V, Sun X, Ren X, Perez-Abadia G, Crespo FA, Oliver R, Orhun HI, Quan EE, Maldonado C, Ray M, Barker JH: Targeting of glycosaminoglycan-cytokine interactions as a novel therapeutic approach in allotransplantation. Transplantation 74: 623–629, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Kikawada E, Lenda DM, Kelley VR: IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J Immunol 170: 3915–3925, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Menke J, Lucas JA, Zeller GC, Keir ME, Huang XR, Tsuboi N, Mayadas TN, Lan HY, Sharpe AH, Kelley VR: Programmed death 1 ligand (PD-L) 1 and PD-L2 limit autoimmune kidney disease: Distinct roles. J Immunol 179: 7466–7477, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Lenda D, Kikawada E, Stanley ES, Kelley VR: Reduced macrophage recruitment, proliferation, and activation in colony-stimulating factor-1-deficient mice, results in decreased tubular apoptosis during renal inflammation. J Immunol, 170: 3254–3262, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH: Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203: 883–895, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanley ER: The macrophage colony-stimulating factor, CSF-1. Methods Enzymol 116: 564–587, 1985 [DOI] [PubMed] [Google Scholar]
- 45.Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, Fogo AB, Ginzler EM, Hebert L, Hill G, Hill P, Jennette JC, Kong NC, Lesavre P, Lockshin M, Looi LM, Makino H, Moura LA, Nagata M: The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int 65: 521–530, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, Kelley VR: IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRL- Fas(lpr) mice. J Immunol 163: 6884–6891, 1999 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.