

Abstract
Background
Wolbachia (wBm) is an obligate endosymbiotic bacterium of Brugia malayi, a parasitic filarial nematode of humans and one of the causative agents of lymphatic filariasis. There is a pressing need for new drugs against filarial parasites, such as B. malayi. As wBm is required for B. malayi development and fertility, targeting wBm is a promising approach. However, the lifecycle of neither B. malayi nor wBm can be maintained in vitro. To facilitate selection of potential drug targets we computationally ranked the wBm genome based on confidence that a particular gene is essential for the survival of the bacterium.
Results
wBm protein sequences were aligned using BLAST to the Database of Essential Genes (DEG) version 5.2, a collection of 5,260 experimentally identified essential genes in 15 bacterial strains. A confidence score, the Multiple Hit Score (MHS), was developed to predict each wBm gene's essentiality based on the top alignments to essential genes in each bacterial strain. This method was validated using a jackknife methodology to test the ability to recover known essential genes in a control genome. A second estimation of essentiality, the Gene Conservation Score (GCS), was calculated on the basis of phyletic conservation of genes across Wolbachia's parent order Rickettsiales. Clusters of orthologous genes were predicted within the 27 currently available complete genomes. Druggability of wBm proteins was predicted by alignment to a database of protein targets of known compounds.
Conclusion
Ranking wBm genes by either MHS or GCS predicts and prioritizes potentially essential genes. Comparison of the MHS to GCS produces quadrants representing four types of predictions: those with high confidence of essentiality by both methods (245 genes), those highly conserved across Rickettsiales (299 genes), those similar to distant essential genes (8 genes), and those with low confidence of essentiality (253 genes). These data facilitate selection of wBm genes for entry into drug design pipelines.
Background
Exponential growth in the amount of available genomic information has produced unprecedented opportunities to computationally predict functional genomics in biologically intractable organisms. One application of these data is facilitation of the rational drug design process. Most high throughput drug discovery techniques screen compounds for biological activity, only determining target and mechanism post hoc. An alternative approach, rational drug design, seeks to utilize genomic information to specifically identify and inhibit targets. Often these methods utilize in silico sequence analysis to choose a target protein that is important to the survival of the organism and accessible to small molecule drugs. It has been suggested that ideally a target should fulfill four properties: 1--Essentiality to the survival or pathogenesis of the target organism, 2--Druggability, having protein structure characteristics making it amenable to binding small molecule inhibitors, 3--Functional and structural characterization with established assays for screening small molecule inhibition, 4--Distinctness from current drug targets to avoid resistance [1].
These parameters are not strict rules, however. In reality, few if any pathogenic organisms have sufficiently comprehensive functional genomics information to rigorously screen based on these parameters. A large portion of the target discovery process involves weighing compromises in the selection parameters based on the quality of information available. In silico drug target prediction relies on various approximations and comparisons to identify genes which fit these parameters. Arguably, the most important parameter to assess is gene essentiality. For a compound to serve as an effective antimicrobial or anthelmintic, binding of its target gene product should kill, or at least severely attenuate the growth of the targeted organism. Knock-out and knock-down studies have been used to assess gene essentiality in a number of diverse model and disease organisms. Recently, many of these studies have been assembled into collection databases [2,3] allowing analyses that examine patterns of essential genes across multiple organisms [4]. In organisms in which a genome wide essentiality survey has not been completed, additional approaches have been used to predict essential genes. If gene essentiality has been determined in a closely related model organism, orthology between genes can predict shared essentiality [5-10]. Alternatively, systems biology approaches examine the global enzymatic and metabolic requirements of the organism. Among these are studies which define a minimal genome for a generic bacterial organism [11-13], or model the total metabolic interactions of the cell [14,15]. For organisms with no functional genomics information in nearby species, methods based purely on gene sequence are being developed, though these provide lower accuracy than functional comparisons [16,17]. Among the purely sequence based methods, gene conservation across taxa is the strongest indicator of gene essentiality [11,16,18,19]. Genes whose protein sequences have been tightly conserved across lineages are assumed to be more likely to be important to the survival of the organism [20]. Each of the essential gene prediction methods described above requires different levels of a priori information about the target organism or closely related organisms. As the amount of functional genomics information available decreases, predicting essential genes and drug targets becomes a significantly more difficult task. Here we present the results of our analysis of one such organism having no such functional data, the Wolbachia endosymbiont of Brugia malayi, (wBm).
B. malayi is a parasitic filarial nematode of humans which, along with Wuchereria bancrofti and Onchocerca volvulus, are the causative agents of lymphatic filariasis and onchocerciasis, more commonly known as elephantiasis and river blindness, respectively. Together, filarial parasites infect approximately 150 million people worldwide with 1.5 billion at risk of infection [21]. Current treatments utilize diethylcarbamazine, benzimidazoles (e.g., albendazole) and avermectins (e.g., ivermectin), however, these treatments are predominately only effective during the larval stages of the parasite [22]. Because the life-span of the adult worm is up to 15 years, long treatment courses are required to effectively eliminate the infection. Additionally, the emergence of drug resistance is becoming increasingly apparent [23,24]. The α-proteobacterium Wolbachia is an obligate endosymbiont of most filarial nematodes, and in several, including B. malayi, is required for worm viability. Clearance of the Wolbachia by antibiotics results in worm growth retardation, infertility and killing, while antibiotic treatment of non-Wolbachia carrying nematode species has no effect [25,26]. This makes Wolbachia an attractive target for control of filarial parasites.
Neither Wolbachia nor B. malayi have a life-cycle that can be maintained in vitro. Because of this, traditional drug discovery by high throughput compound screening is not feasible, nor are the basic gene essentiality experiments which are informative to rational drug design. The genomes of both B. malayi and wBm have been sequenced [27,28]; however, only B. malayi has a closely related, well characterized model organism, Caenorhabditis elegans. Previous work has used C. elegans functional genomics data to predict drug targets in B. malayi [9]. Wolbachia, however, has no close relatives in which functional genomics data is available.
Functional genomics information from a large number of more distantly related bacteria can be used to infer similar information in an intractable species [29,30]. Here we present such an approach, utilizing bioinformatic techniques to rank the likelihood of gene essentiality across the wBm genome, for the purpose of facilitating the selection of potential new drug targets. A combination of approaches were used to predict genes likely to be important to the survival of wBm. First, we used comparative sequence analysis to identify wBm genes with strong protein sequence similarity to experimentally identified essential genes in more distantly related bacteria. Second, in order to identify genes important to the biological niche inhabited by wBm, gene conservation across its parent order, Rickettsiales was evaluated. The first approach identifies genes broadly important across bacterial life. The second approach reinforces the genes identified by the first, while additionally identifying genes likely to have importance specifically within Rickettsiales. Consideration of these properties during drug target selection can optimize for development of either a more broad spectrum antibiotic, or a more targeted compound, reducing the side effects related to clearing of the natural biotic flora.
Results
Predicting essential genes in wBm by protein sequence comparison to essential genes in distantly related bacteria
While wBm is not amenable to experimental gene essentiality analysis, knockout and knockdown studies in multiple other bacterial species can serve as a proxy. The results of a number of these analyses are compiled in a publicly available resource called the Database of Essential Genes (DEG). This database contains 5,260 genes from 15 different bacterial strains [3] (Table 1). In most cases, the genes within DEG were identified by large scale knock-out or knock-down screens performed under rich media conditions. Rich media conditions are thought to approximate the growth environment of intracellular bacteria [16]. This makes the collection of genes within DEG a useful model for the gene requirements of wBm. DEG contains a binary description of gene essentiality. Genes included in DEG are considered essential to the organism, while genes omitted are considered dispensable, within the specific conditions of the experiments used. In order to computationally predict essential genes, we used BLAST to compare the protein sequences of all protein-coding wBm genes to the genes contained within DEG. The most straightforward method to evaluate the results from the BLAST analysis is to examine the e-value of the best BLAST hit between a wBm gene and DEG. However, because DEG consists of information on essential genes in multiple bacterial organisms, we wished to evaluate the BLAST results in a manner which accounts for the statistical significance of hits to multiple DEG organisms. A wBm gene with a significant BLAST hit to an essential gene in a single DEG organism represents a quite different result than a wBm gene with significant BLAST hits to essential genes in multiple DEG organisms. While a single alignment to a DEG gene implies similar function and likely shared essentiality, alignments to DEG genes within multiple organisms suggests membership in a class of essential genes conserved across species and increases our confidence in predicting that a given wBm gene is essential. A ranking metric, termed the multiple-hit score (MHS), was developed to evaluate the BLAST results in this context. This metric produced a score for each wBm gene. A gene with high-scoring BLAST hits to each organism within DEG received a high MHS score. In its basic form, the MHS for a wBm gene was calculated by averaging the top BLAST alignment against each DEG organism divided by the smallest e-value able to be returned by BLAST, 1 × 10-200 in this case. The scale of e-values generated by BLAST are dependent on the size of the database searched [31]. Preliminary analysis indicated that when searching against the DEG database, e-values less significant than 1 × 10-25 were predominately partial alignments (data not shown). To reduce the effect of these lower significance alignments, which appeared to be domain alignments instead of full length gene alignments, all e-values were scaled by their square before averaging. The resulting score could range between 0 and 1, with 1 being alignments with an e-value of 1 × 10-200 to all organisms within DEG. Figure 1 is a graph of the MHS scores for the full wBm genome, ordered by MHS score [see Additional file 1]. This graph reveals several properties of the wBm MHS distribution. There is a sharp peak containing fewer than 10 genes which have very good alignments to nearly all DEG organisms. This tapers to a shoulder containing, first, genes with high quality alignments to several DEG organisms, then later, mostly genes with lower quality alignments to multiple DEG organisms. The distribution of actual alignments for the top 20 genes is shown in Figure 2. Because the MHS indicates our confidence that a specific gene is essential, the optimal usage of this ranking is to begin manually examining from the highest ranked genes, progressing through genes with a lower confidence of essentiality. Based on the shape of the MHS curve and examination of the individual alignments, a conservative MHS threshold of 7.3 × 10-3 was chosen. At this threshold, we see alignments to 7 of the 15 taxa in DEG with e-values of 1 × 10-25. This threshold predicts that 250 out of 805 genes have reasonable confidence of essentiality. This should not, however, be mistaken as a prediction that two-thirds of the genome is non-essential. As an obligate endosymbiont of the nematode B. malayi, wBm has undergone significant genome shrinkage compared to other bacteria, thus a large percentage of its genome is expected to be essential [28]. Instead, the MHS result predicts that roughly one-quarter of the wBm genes are involved in basic bacterial processes important for growth across a diversity of species. Identification of a supplementary set of genes consisting of genes likely to be important specifically to members of the order Rickettsiales was accomplished in the second phase of our analysis.
Table 1.
DEG Members
| Organism Name | Taxon ID | Ess. Genes | Refseq Gene Count | % Ess. |
|---|---|---|---|---|
| Acinetobacter baylyi ADP1γ | 202950 | 499 | 3325 | 15% |
| Bacillus subtilis 168B | 224308 | 271 | 4105 | 7% |
| Escherichia coli MG1655γ | 511145 | 712 | 4132 | 17% |
| Francisella novicida U112γ | 401614 | 392 | 1719 | 23% |
| Haemophilus influenzae Rd KW20γ | 71421 | 642 | 1657 | 39% |
| Helicobacter pylori 26695ϵ | 85962 | 323 | 1576 | 20% |
| Mycobacterium tuberculosis H37RvA | 83332 | 614 | 3989 | 15% |
| Mycoplasma genitalium G37M | 243273 | 381 | 477 | 80% |
| Mycoplasma pulmonis UAB CTIPM | 272635 | 310 | 782 | 40% |
| Pseudomonas aeruginosa UCBPP-PA14γ | 208963 | 335 | 5892 | 6% |
| Salmonella typhimurium LT2γ | 99287 | 230 | 4527 | 5% |
| Staphylococcus aureus N315B | 158879 | 302 | 2619 | 12% |
| Streptococcus pneumoniae R6B | 171101 | 133 | 2043 | 12% |
| Streptococcus pneumoniae TIGR4B | 170187 | 111 | 2105 | 12% |
| Vibrio choleraeγ | 243277 | 5 | 3835 | 0% |
(γ): γ-proteobacteria, (B): bacilli, (ϵ): ϵ-proteobacteria, (A): actinobacteria, (M): mollicutes.
Figure 1.

Distribution of MHS values by rank in wBm. The X-axis indicates the 805 protein coding genes in the wBm genome, ranked by MHS. The Y-axis shows the value of the MHS for each protein.
Figure 2.

E-values of the BLAST alignments producing the top 20 MHS. The black bars indicate the e-value of the best alignment to each organism within DEG. The y-axis is a linear scale of the negative log10 of the e-value, ranging from 1 to a maximal alignment of 200. The x-axis bins correspond to the 15 organisms contained within DEG.
Evaluation and validation of the MHS ranked wBm gene list
The annotations of the top 20 wBm genes ranked by MHS can be used to qualitatively assess our ranking metric (Table 2). Many of the top-20 genes fall into the classes of genes targeted by current antibiotics and are annotated in categories likely essential for bacterial growth. The gyrase and topoisomerase family, targeted by quinolones [32], is heavily represented. The DNA-directed RNA polymerase RpoB is the target of rifampin [33], and the tRNA synthetases are targets of several recently developed compounds [34-36]. In addition to qualitatively examining our ranking method, we wanted to quantitatively assess its ability to place essential genes at the top of the ranked list. However, quantitatively validating the ranking of the wBm genome is stymied by the lack of an effective positive control set. To address this we developed a jackknifing methodology which is able to utilize the organisms within DEG as a positive control set with which to validate the ranking methods. The Refseq sets of predicted proteins for organisms included in DEG were acquired from NCBI. Each organism's protein sequences were individually analyzed by comparison to a version of DEG filtered to remove sequences from just that organism, then ordered by MHS. Because essential genes in these organisms have already been experimentally identified, it is possible to assess our ranking methods by their ability to prioritize these genes. In order to quantitate the ranking, each genome was ordered by highest to lowest prediction of essentiality and the cumulative sum of the number of positive control DEG genes was plotted. The area under the curve (AUC) for the experimental ranking was compared to that of an ideal ranking which artificially placed all DEG genes at the beginning of the list, and 1000 replicates of a randomized assortment (Figure 3). The shape of the ideal and sorted curves varies with the percentage of DEG genes within each organism. The important component to examine is the shape of the experimental sorting curve compared to the randomized assortment and the ideal ranking. For each organism a p-value was calculated, comparing the experimental sorting with the randomly assorted population. Additionally, the percentage sorting was calculated by scaling the area under the curve for the experimental sorting to between 100% for the area under the curve in the ideal ranking, and 0% for the AUC for the diagonal line representing random assortment. Qualitatively, for most organisms our methods performed relatively well in recovering DEG genes. In nearly all organisms the sorted curve appears well differentiated from the randomized sorting and in some cases begins to approach the ideal case. For all organisms the experimental sorting was statistically different from random assortment. B. subtilis, S. aureus, and M. pulmonis are examples of organisms with large, medium and small genomes which were especially well sorted by MHS, with 74.2%, 73.3% and 67.1% sorting respectively. On the other hand, H. influenzae and H. pylori and to a lesser extent E. coli performed quite poorly in this validation with 13.7% 12.8% and 32.5% sorting respectively. Further consideration of these outliers can be found in the discussion. Overall, the results from the jackknife analysis indicate that the MHS based ranking effectively predicts essential genes and prioritizes them within the top of the ranked genome.
Table 2.
Top 20 wBm genes ranked by MHS. Annotations taken from the Refseq release of the wBm proteome.
| Rank | MHS | GI | Annotation |
|---|---|---|---|
| 1 | 0.772 | 58584904 | DNA-directed RNA polymerase: RpoB/RpoC |
| 2 | 0.733 | 58584602 | Translation elongation factor GT-Pase: FusA |
| 3 | 0.656 | 58585021 | DNA gyrase, topoisomerase II, B sub-unit: GyrB |
| 4 | 0.585 | 58584662 | DNA gyrase subunit A |
| 5 | 0.550 | 58584524 | Translocase |
| 6 | 0.539 | 58584756 | DNA polymerase III alpha subunit |
| 7 | 0.497 | 58584618 | Alanyl-tRNA synthetase |
| 8 | 0.482 | 58584729 | Threonyl-tRNA synthetase |
| 9 | 0.425 | 58584862 | Leucyl-tRNA synthetase |
| 10 | 0.414 | 58584752 | Molecular chaperone: DnaK |
| 11 | 0.361 | 58584429 | CTP synthetase |
| 12 | 0.310 | 58584410 | ATP-dependent Zn protease: HflB |
| 13 | 0.276 | 58584946 | ATP synthase subunit B |
| 14 | 0.269 | 58584379 | Enolase |
| 15 | 0.267 | 58584441 | ATP-binding subunit of Clp protease and DnaK/DnaJ chaperones |
| 16 | 0.267 | 58584652 | 2-oxoglutarate dehydrogenase complex, E1 component |
| 17 | 0.258 | 58584572 | ATP synthase subunit A |
| 18 | 0.249 | 58584805 | NAD-dependent DNA ligase: Lig |
| 19 | 0.246 | 58584298 | Topoisomerase IA: TopA |
| 20 | 0.245 | 58584921 | Transketolase |
Figure 3.

Essential gene prediction by MHS was validated through a jackknife methodology. For each organism within DEG, the ability of the MHS to place experimentally validated essential genes at the top of a ranked genome was evaluated. All graphs correspond to the schematic found in the upper left. The X-axis represents the ranked genome of the organism, ranked from left to right as strongest to weakest prediction of essentiality. The Y-axis is the cumulative count of essential genes encountered moving left to right through the ranked genome. Line A is the ideal sorting, in which all essential genes are placed at the top of the ranking. Line B is the sorting by MHS. Lines C are 10 random assortments of the genome. Percent sorting achieved by MHS and the p-value for the difference between the MHS score ranking B and 1000 random assortments such as in C are shown in the lower right. Graphs are ordered by descending genome size of the organism. E. coli, F. novicida, and M. genitalium show 10, 2 and 2 fewer total essential genes, respectively, than shown in Table 1 because the corresponding DEG genes are not able to be resolved to genomic genes and are omitted from the jackknife analysis.
Prediction of essential genes in wBm by gene conservation across the order Rickettsiales
While we are confident in the predictions of gene essentiality by MHS, those predictions only identify genes common to the reference set of bacteria in DEG. As there are no α-proteobacteria in DEG, genes uniquely essential to wBm might be missed by MHS analysis. We wished to perform a complementary analysis to predict additional genes important specifically to wBm and closely related organisms. wBm is a highly specialized obligate endosymbiont with a reduced genome [28]. While it seems reasonable that roughly 250 out of 805 wBm genes are essential across bacteria in general, it is likely that there is an additional set of genes essential specifically for the environmental niche inhabited by wBm. In order to predict this second set of genes, and reinforce the MHS based essential gene predictions, we identified genes with highly conserved orthologs across Wolbachia's parent order, Rickettsiales.
There are 27 complete genomes available within Rickettsiales. These include, 4 Wolbachia, including wBm, 3 genomes from the genus Anaplasma, 5 Ehrlichia, 11 Rickettsia, 1 Neorickettsia, 2 Orientia, and 1 Pelagibacter (Table 3). Of these genomes, all but Pelagibacter are obligate endosymbionts residing either in vacuoles or within the host cell cytoplasm. Of the endosymbionts, all but Wolbachia replicate within vertebrate hosts with most transmitted via an invertebrate vector. Wolbachia, on the other hand infects a diverse spectrum of arthropod hosts as well as filarial nematodes, many of which are themselves vertebrate parasites [37].
Table 3.
Genomes available within the order Rickettsiales
| Genus species Strain | Taxon ID |
|---|---|
| Anaplasma marginale St Maries | 234826 |
| Anaplasma phagocytophilum HZ | 212042 |
| Anaplasma marginale Florida | 320483 |
| Candidatus Pelagibacter ubique HTCC1062 | 335992 |
| Ehrlichia canis Jake | 269484 |
| Ehrlichia chaffeensis Arkansas | 205920 |
| Ehrlichia ruminantium Gardel | 302409 |
| Ehrlichia ruminantium Welgevonden UPSA | 254945 |
| Ehrlichia ruminantium Welgevonden CIRAD | 254945 |
| Orientia tsutsugamushi Boryong | 357244 |
| Orientia tsutsugamushi Ikeda | 334380 |
| Neorickettsia sennetsu Miyayama | 222891 |
| Rickettsia akari Hartford | 293614 |
| Rickettsia bellii OSU 85-389 | 391896 |
| Rickettsia bellii RML369-C | 336407 |
| Rickettsia canadensis McKiel | 293613 |
| Rickettsia conorii Malish 7 | 272944 |
| Rickettsia felis URRWXCal2 | 315456 |
| Rickettsia massiliae MTU5 | 416276 |
| Rickettsia prowazekii Madrid E | 272947 |
| Rickettsia rickettsii Iowa | 452659 |
| Rickettsia rickettsii Sheila Smith | 392021 |
| Rickettsia typhi wilmington | 257363 |
| Wolbachia Drosophila melanogaster | 163164 |
| Wolbachia Drosophila simulans | 66084 |
| Wolbachia Culex quinquefasciatus | 570417 |
| Wolbachia Brugia malayi TRS | 292805 |
Refseq protein sequences from the 27 available genomes (as of April 1, 2009) were retrieved from NCBI. The OrthoMCL package was used to predict clusters of orthologs among the genomes [38]. To gauge the extent of taxonomic diversity within each orthologous gene cluster, we initially tallied the number of taxa represented in the cluster. However, this measure inflated the phylogenetic diversity for groups containing multiple highly related taxa. To compensate, a minimum spanning tree (MST) was constructed using distances derived from aligned 16S rRNA gene sequences as edge weights between taxonomic nodes. A score for the MST was calculated by summing the distances between the connected taxonomic nodes. The MST was used to minimize the contributions from closely related taxa, while reflecting the overall taxonomic diversity. The MST distances for each cluster were incorporated into a metric we termed the gene conservation score (GCS), which represents both the extent of gene conservation across species, as well as the quality of that conservation. The integer portion of the GCS, from 0 to 100, is derived from the MST distances within an orthologous gene cluster. The decimal portion of the score represents the quality of alignments between the wBm gene and the other cluster members. Thus, within a group of clusters with the same MST, wBm genes are individually ranked based on the quality of their BLAST alignment to other genes within the cluster (see Materials and Methods). The distribution of GCS scores for the wBm genome is shown in Figure 4 [see also Additional file 1]. Approximately 300 wBm genes cluster with orthologs in all or nearly all Rickettsia members in the analysis and have a GCS of approximately 100. The next large group consists of 60 wBm genes that have a GCS of approximately 91 and orthologs in all members except for Pelagibacter ubique, the only free-living organism in the group. A third group of 60 genes has a GCS of approximately 29, and corresponds to clusters lacking orthologs to Orientia and most of the Rickettsia species. When picking an empirical threshold for prediction of gene essentiality we chose a GCS of 29 or higher, which includes the three groups described above and contains 544 genes. Though the third group of 60 genes has lost orthologs to most of the Rickettsia, it retains orthologs in the Anaplasma, Ehrlichia, Neorickettsia and the other Wolbachiae. As is illustrated by the distribution along the y-axis of Figure 5, however, there is a large break between groups with a GCS of 91 and 29, and a more conservative estimate could place a threshold significantly higher. From a practical standpoint, however, because the GCS value represents a prediction of the importance of a specific gene, a more useful approach is to sort the genome by GCS rather than picking a threshold. Manually assessing from the top of the ranking allows the identification of highly conserved genes which can be searched for favorable secondary protein properties; in our case, properties useful for entry into the rational drug design pipeline.
Figure 4.

Distribution of GCS in wBm. The X-axis indicates the 805 protein coding genes in the wBm genome, ranked by GCS. The Y-axis shows the value of the GCS for each protein.
Figure 5.

Comparison of the prediction of wBm gene essentiality by MHS and GCS. The X-axis shows normalized MHS on a log scale, while the Y-axis shows GCS. Grey lines indicate empirically determined thresholds for confidence in prediction of essentiality and are set at 7.3 × 10-3 for the MHS and 29 for the GCS. Therefore, the upper right quadrant contains genes with high confidence by both metrics. The upper left quadrant contains genes identified only by GCS, while the bottom right quadrant contains genes identified only by MHS. The numbers adjacent to the quadrant lines indicate gene counts in each quadrant. Red dots indicate Wolbachia genes which have significant protein sequence similarity to the targets of approved drugs and are predicted to be druggable.
Examination of the top 20 genes in the wBm genome ranked by GCS (Table 4) reveals important differences compared to ranking by MHS (Table 2). Many of the same genes or classes of genes which were ranked highly by MHS are also identified by GCS. RNA polymerase RpoB/C, topoisomerase, gyrase, and several tRNA synthetases all rank highly by both methods. However, several interesting genes not identified by MHS are placed at the top of the GCS ranking. For example, pyruvate phosphate dikinase, PPDK, has previously been identified by pathway analysis as a potential drug target [39]. By MHS, PPDK was ranked at position 309; GCS ranking placed it at position 3.
Table 4.
Top 20 wBm genes ranked by GCS. Annotations taken from the Refseq release of the wBm proteome.
| Rank | GCS | GI | Annotation |
|---|---|---|---|
| 1 | 101 | 58584652 | 2-oxoglutarate dehydrogenase complex, E1 component |
| 2 | 101 | 58584298 | Topoisomerase IA: TopA |
| 3 | 101 | 58584469 | Pyruvate phosphate dikinase |
| 4 | 101 | 58584904 | DNA-directed RNA polymerase: RpoB/RpoC |
| 5 | 101 | 58584952 | Ribonucleotide-diphosphate reductase alpha subunit |
| 6 | 101 | 58584808 | ATP-dependent Lon protease |
| 7 | 101 | 58584662 | DNA gyrase subunit A |
| 8 | 101 | 58584705 | Succinate dehydrogenase |
| 9 | 101 | 58584602 | Translation elongation factor, GT-Pase: FusA |
| 10 | 101 | 58584729 | Threonyl-tRNA synthetase |
| 11 | 101 | 58584633 | NADH dehydrogenase gamma sub-unit |
| 12 | 101 | 58584752 | Molecular chaperone: DnaK |
| 13 | 101 | 58584862 | Leucyl-tRNA synthetase |
| 14 | 101 | 58584524 | Translocase |
| 15 | 100.994 | 58585021 | DNA gyrase, topoisomerase II, B sub-unit: GyrB |
| 16 | 100.989 | 58584924 | GTP-binding protein: LepA |
| 17 | 100.987 | 58584410 | ATP-dependent Zn protease: HflB |
| 18 | 100.986 | 58584731 | NADH:ubiquinone oxidoreductase, NADH-binding, chain F |
| 19 | 100.974 | 58584620 | Isoleucyl-tRNA synthetase |
| 20 | 100.974 | 58584756 | DNA polymerase III alpha subunit |
Plotting MHS versus GCS demonstrates the identification of complementary sets of essential genes
The two methods of essential gene prediction used in this study identified complementary partially overlapping sets of wBm genes. Identification of a gene by both methods bolsters confidence in a prediction of essentiality. Genes uniquely identified by an individual method may represent, for MHS, genes essential to general bacterial processes; and for GCS, genes specifically important to the Rickettsiales order. To assess the distribution of essentiality prediction by both methods, the MHS and GCS for each wBm gene was graphed as a scatter plot (Figure 5). Lines indicating the empirically determined thresholds for the prediction of essentiality by each method produce four quadrants showing the classes of predicted essential genes. The upper-right quadrant contains 245 genes predicted essential by both methods. The upper-left quadrant contains 299 genes which are not similar to essential genes in more distantly related bacteria, but are still highly conserved across Rickettsiales. These genes represent a promising class of drug targets which are likely to be more specific to wBm. That there are only 8 outliers in the lower-right quadrant demonstrates that most genes which are predicted to be essential in multiple diverse bacteria are also highly conserved across Rickettsiales, as expected. Combined, we predict that 552 of 805 wBm genes--roughly 69%--have a high likelihood of being essential.
The ranked wBm genome as a tool for drug development
Our ranking of the wBm genome by predicted gene essentiality is designed as a tool to facilitate the manual exploration of viable new drug targets against the bacterium. Order within the list at a resolution of one or two positions is relatively uninformative; nearby rankings represent similar confidence in the prediction of gene essentiality. However, the quartile or decile in which a gene is placed strongly influences our confidence in its essentiality. In addition to predicting essential genes, each wBm gene can be further annotated to include protein or functional information useful in drug target prioritization, including similarity to human proteins, hydropathy predictions, or protein localization predictions. A similar strategy for prioritizing targets was used for B. malayi [9] and Mycobacterium tuberculosis [40]. One such annotation we chose to include is the potential for a protein to bind typical small molecule drugs, termed its druggability. There exist several purely sequence based methods of predicting druggability based on the identification of domains favorable to small molecule binding [41,42]. We also decided to take a more direct approach and identify wBm proteins with high sequence similarity to the targets of existing small molecule drugs and compounds. This allows us to not only identify proteins containing domains favorably structured to bind small molecules, but also proteins which are likely to have the localization and cellular kinetics important for a viable drug target.
We utilized the DrugBank database which is a comprehensive set of nearly 4,800 FDA-approved small molecule drugs, nutraceuticals and experimental compounds [43]. This database includes chemical, pharmacology, and mechanistic information for each compound, as well as protein target and pathway information for a large percentage of the entries. After downloading a local copy of the database, we used BLAST to align the wBm proteins to the list of drug targeted proteins from DrugBank, filtering for e-values more significant than 1 × 10-25. This method identified 198 wBm proteins highly similar to the binding partners of FDA approved drugs, experimental small molecule compounds, or nutraceutical compounds. In Figure 5 druggability is indicated by coloring predicted druggable wBm genes red. The prediction of druggability seems to correlate well with our predictions of potential drug targets by essentiality and gene conservation. In combination with essentiality predictions, the prediction of druggability can be used as a secondary screening criteria to identify genes for entry into the rational drug design pipeline.
Discussion
The overall goal of this work is to produce a result that can facilitate the selection of genes as drug target candidates. Sorting the full genome by prediction of essentiality then manually evaluating secondary protein properties attempts to avoid the issues related to developing a nuanced automated system capable of filtering down to a short list of candidate drug targets while still prioritizing the listing for high quality potential targets.
MHS predicted a slightly smaller number of essential genes than experimentally found in the individual genome surveys comprising DEG. In contrast, GCS predicted a slightly larger set (Figure 6). Because most of the entries within DEG represent genome wide surveys for essential genes we can compare the number of genes identified by our analysis to the number of essential genes in each DEG organism. Vibrio cholerae was removed as an outlier because it consists of 5 genes in DEG and does not represent a comprehensive genome survey. By MHS our analysis predicted approximately 250 genes or approximately 30% of the wBm genome as having reasonable confidence of essentiality. The raw number of predicted essential genes is lower than that for most of the DEG organisms, and under the mean for DEG of 392 genes. Mycoplasma genitalium and Mycoplasma pulmonis, which are also intracellular bacteria with genome sizes similar to wBm, have 381 and 310 genes within DEG, respectively. The relatively similar number of essential genes identified across DEG organisms suggests that these data are describing a common set of genes across a shared set of important pathways. It appears that we are able to predict a quite significant portion of these in wBm through the MHS, though it does appear that MHS alone may not be identifying the complete set. By GCS we identified 544 wBm genes as important within Rickettsiales, comprising approximately 69% of the wBm genome. This is greater than the Mycoplasmas and most other DEG organisms, but still less than Haemophilus influenzae (642), M. tuberculosis (614), or Escherichia coli (712) (Table 1). Overall, it appears that for prediction of essential genes both MHS and GCS score are effective. MHS is likely an incomplete survey. GCS prediction appears to identify a more complete set, encompassing all but 8 of the genes identified by MHS. However, the additional genes identified by GCS also probably include a number of genes that, while important, are not strictly essential. It is possible to overestimate the set of essential genes predicted by GCS as a result of using closely related organisms. Although we note that in the case of Rickettsiales, these organisms are in the process of reducing their genomes, adding significance to retained genes. Within the goals of this research, predicting essential genes as potential drug targets, our methods provide sufficient sensitivity and specificity as long as these caveats are recognized.
Figure 6.

Number of essential genes versus total number of Refseq genes. •-DEG organisms (V. cholerae omitted as an outlier). △-wBm essential gene prediction by MHS. ▽-wBm essential gene prediction by GCS score.
On the basis of the jackknife validation, MHS performs poorly on several organisms. M. genitalium represents a unique case; nearly 80% of its genes are essential. There is little difference between the AUC for the ideal sorting, the MHS sorting, and the random assortment. Even so, MHS produced a 38.8% sorting, with a p-value of 2 × 10-9 compared to random. It is unclear why H. influenzae and H. pylori and to a lesser extent E. coli performed poorly. This result suggests that these organisms may contain species specific essential genes. For H. pylori the authors of the initial essentiality screen note a surprising lack of overlap with the essential gene sets from other organisms [44]. As the number of essential genes in H. pylori is in the same range as most of the other organisms in DEG, this could suggest an alternative set of essential genes. In the case of E. coli, we note that the number of essential genes is nearly double the average for the other DEG organisms, which likely reflects its status as one of the most well-studied bacteria. This larger set may confound the E. coli jackknifing validation. Somewhat paradoxically, these features may be beneficial for this analysis. The outlier organisms may incorporate more diversity in our reference set of essential genes, increasing the likelihood of identification of diverse essential genes within wBm. This does come with the trade-off of increasing the false positive rate, however, this is mitigated by two factors. First, the design of the MHS assigns more confidence to genes conserved across multiple organisms, moving well supported essential gene predictions towards the top. Second, the pipeline for the rational drug design process utilizes the predictions of essential wBm genes to inform a manual selection of drug targets. A moderate false positive rate can be screened out based on manual analysis and pathway information. As an additional experiment, it could be informative to examine non-DEG genes predicted as essential in the jackknifing validation to identify essential genes missed by the knockout experiments. A gene conserved nearly universally across DEG but missing in a small number of organisms may be useful to investigate under alternative experimental conditions.
Genes identified by MHS are predicted to belong to a set of genes which are essential and broadly conserved across bacterial life. This set includes many targets of modern broad-spectrum antibiotics. A compound targeting genes from this class is more likely to produce antibiotics effective across a broad range of bacterial species. Though gene orthology does not specifically indicate drug cross-reactivity, the distribution of the targeted gene should be considered. While developing a novel broad-spectrum antibiotic would be advantageous, for this specific application such a compound may also come with negative side-effects. Ideally, a mass drug administration protocol against B. malayi would consist of a small number of high-dose antibiotic treatments. In this format, broad-spectrum antibiotics carry the risk of significant side-effects due to targeting mutualistic bacterial flora.
An alternative approach which attempts to avoid the issues surrounding broad-spectrum antibiotics is to select targets from the group of genes identified only by the GCS. These genes are highly conserved throughout the order Rickettsiales but have little similarity to essential genes in other bacteria. While it is quite possible that these wBm genes have orthologs throughout the bacterial kingdom, the experimental data available in DEG suggests that they would not be essential for the growth of bacteria in general. Druggability was predicted by identifying wBm proteins with sequence similarity to the targets of small molecule drugs. However, an intriguing secondary application exists. Comparison of wBm proteins to drug targeted proteins additionally produces a list of approved drug and drug-like compounds which bind proteins of similar sequences to wBm proteins. Protein sequence similarity does not guarantee identical structures or binding pockets, thus it is unlikely that a single turn-key compound will be identified through target similarity. However, it seems reasonable that careful filtering of this set could reveal a panel of potential binding compounds primed for optimization and derivatization using traditional medicinal chemistry. This opens the interesting possibility of applying bioinformatic analysis to bypass a portion of the arduous de novo drug development pipeline.
Conclusion
Through this analysis we were able to predict genes important for the survival of a biologically intractable organism using two complementary bioinformatic techniques. These predictions can then be used as a tool to facilitate the selection of genes to enter into the drug development process against this organism. Comparison of the two predictions revealed that different but overlapping sets of genes were predicted, stemming from the approaches applied. By MHS, 253 genes were predicted as having a high likelihood of being essential. All but 8 of those genes were also identified by the second method, GCS. An additional 299 genes were also identified by GCS alone as highly conserved in Wolbachia's parent order Rickettsiales. Overall, 552 wBm genes, approximately 69% of the genome, were identified as having a high confidence in a prediction of essentiality. The overlapping and uniquely identified sets of genes can facilitate alternative approaches for drug target selection.
Methods
BLAST against DEG
The 805 Refseq protein sequences for the Wolbachia endosymbiont of B. malayi strain TRS were downloaded from the NCBI ftp site ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria. The Database of Essential Genes (DEG) version 5.2 was provided by Dr. Ren Zhang at the Centre of BioInformatics, Tianjin University. The standalone release of the BLAST sequence alignment program version 2.2.19 was obtained from the NCBI BLAST website [45]. Using default parameters, blastp was used to align the wBm protein sequences against the protein sequences contained in DEG. To produce the multi-hit score, the negative log10 of the e-values of the highest scoring alignments to each of the DEG organisms were normalized between 0 and 1, squared, then averaged for all DEG organisms. E-values greater than 1 were truncated at 1.
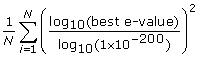 |
Where N = the number of DEG organisms and 1 × 10-200 is the smallest e-value reported by BLAST.
Jackknife Analysis
Complete Refseq protein sequences for the 15 organisms contained within DEG were downloaded from the NCBI Refseq ftp site ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria. For each organism, a filtered version of DEG was prepared, removing just the proteins from that organism. The full protein complement of that organism was then subjected to MHS analysis using the filtered version of DEG, and ranked based on MHS. Moving through the ranked genome from highest prediction of essentiality to lowest, the cumulative sum of DEG genes encountered was calculated. The area under the curve (AUC) of the cumulative sum describes the effectiveness of the ranking. The upper bound of the AUC is defined by an ideal sorting which places all DEG genes at the top of the list. The mean and standard deviation of the AUC for the null hypothesis of no sorting was determined by randomly permuting the genome sorting 1000 times. The AUCs for the random assortments was assumed to represent a normal distribution with the observed mean and standard deviation. The p-value of the MHS sorting versus the null hypothesis was calculated using the probability density for a normal distribution. For the calculation of percent sorting, the AUC for the unsorted diagonal was one-half of the total area of the graph, calculated as the total number genes in the genome multiplied by the number of DEG genes, divided by two.
Gene Conservation Across Rickettsiales
Refseq protein sequences were downloaded from the NCBI Refseq ftp site for the 27 sequenced organisms in the order Rickettsiales (Table 3). The standalone version 1.4 of the OrthoMCL ortholog prediction program was downloaded http://www.orthomcl.org/common/downloads/software/[38]. OrthoMCL was used with default settings and an inflation value of 1.5 to predict orthologs among the protein sequences of the 27 genomes. Briefly, OrthoMCL begins by using an all-versus-all BLAST search to identify reciprocal best BLAST hits among the genomes as putative orthologs, and reciprocal best BLAST hits within genomes as putative in-paralogs. These interconnections are used to form a similarity graph that is used by the MCL clustering algorithm to break mega-clusters into suitable sub-clusters of orthologs [46].
For each cluster of orthologous genes the minimum spanning tree (MST) distance was calculated based on the phylogenetic distances among the member genomes. The 16S rRNA gene was extracted from each of the complete genome sequences used in this study (Table 5). A multiple sequence alignment of the 16S genes was generated with Muscle v3.41 [47] using default values for maximum iterations and maximum time. A distance matrix was generated from the aligned sequences with the dnadist program from the Phylip suite v3.68 using the Kimura 2-parameter distance model. For each orthologous cluster, we extracted the taxon IDs of the taxa included in the cluster. Using the calculated distances between taxa based on aligned 16S sequences as edge weights between the taxon nodes, a minimum spanning tree (MST) was generated using Prim's algorithm [48]. Each MST was scored based on the sum of edge weights included in the tree.
Table 5.
16S rRNA gene sequence sources
| Refseq ID | Taxon | Coordinates | Species name |
|---|---|---|---|
| NC_012026.1 | 320483 | 246283-247795 | Anaplasma marginale str. Florida, complete genome |
| NC_004842.2 | 234826 | 247468-248989 | Anaplasma marginale str. St. Maries |
| NC_007797.1 | 212042 | 1057470-1058902 | Anaplasma phagocytophilum HZ |
| NC_007205.1 | 335992 | 511358-512831 | Candidatus Pelagibacter ubique HTCC1062 |
| NC_007354.1 | 269484 | 285955-287439 | Ehrlichia canis str. Jake |
| NC_007799.1 | 205920 | 942218-943726 | Ehrlichia chaffeensis str. Arkansas |
| NC_006831.1 | 302409 | 303748-305256 | Ehrlichia ruminantium str. Gardel |
| NC_006832.1 | 254945 | 306928-308437 | Ehrlichia ruminantium str. Welgevonden |
| NC_005295.2 | 254945 | 326964-328421 | Ehrlichia ruminantium str. Welgevonden |
| NC_007798.1 | 222891 | 36268-37765 | Neorickettsia sennetsu str. Miyayama |
| NC_009488.1 | 357244 | 1322598-1324120 | Orientia tsutsugamushi str. Boryong |
| NC_010793.1 | 334380 | 379135-380647 | Orientia tsutsugamushi str. Ikeda, complete genome |
| NC_009881.1 | 293614 | 864179-865686 | Rickettsia akari str. Hartford |
| NC_009883.1 | 391896 | 1008161-1009668 | Rickettsia bellii OSU 85-389 |
| NC_007940.1 | 336407 | 537796-539303 | Rickettsia bellii RML369-C |
| NC_009879.1 | 293613 | 385940-387447 | Rickettsia canadensis str. McKiel] |
| NC_003103.1 | 272944 | 884601-886108 | Rickettsia conorii str. Malish 7 |
| NC_007109.1 | 315456 | 456383-457890 | Rickettsia felis URRWXCal2 |
| NC_009900.1 | 416276 | 968391-969898 | Rickettsia massiliae MTU5 |
| NC_000963.1 | 272947 | 772263-773769 | Rickettsia prowazekii str. Madrid E |
| NC_009882.1 | 392021 | 876489-877996 | Rickettsia rickettsii str. 'Sheila Smith' |
| NC_010263.1 | 452659 | 887263-888750 | Rickettsia rickettsii str. Iowa |
| NC_006142.1 | 257363 | 779669-781167 | Rickettsia typhi str. Wilmington |
| NC_010981.1 | 570417 | 1136001-1137446 | Wolbachia endosymbiont of Culex quin-quefasciatus Pel, complete genome |
| NC_002978.6 | 163164 | 1167943-1169389 | Wolbachia endosymbiont of Drosophila melanogaster |
| NC_006833.1 | 292805 | 634569-636083 | Wolbachia endosymbiont strain TRS of Brugia malayi |
| NC_012416.1 | 66084 | 1289969-1291473 | Wolbachia sp. wRi complete genome |
MST distances for each cluster containing a wBm gene were rounded to 2 decimal places and scaled to integers between 0 and 100. The average negative log10 of the e-value for the BLAST alignments between the wBm gene and the other cluster members was scaled to between 0 and 1 and added to the MST integers. This resulted in a ranking score ranging from 0 to 101. The MST distances comprise the majority the score. Within-cluster e-values comprise the minority of the score, thus, for clusters with identical MST distances, the quality of alignments within each cluster determines order.
Drug Target Similarity
The contents of the DrugBank database containing target protein sequence information was downloaded from the DrugBank website http://www.drugbank.ca/[43]. Blastp with default parameters was used to align the 805 wBm protein sequences against the list of protein targets of compounds found within DrugBank. The BLAST results were filtered to remove alignments with e-values less significant than 1×10-25.
Authors' contributions
AH participated in the design of the study, carried out the analyses and drafted the manuscript. PD computed minimum spanning trees and helped to draft the manuscript. JF and CC contributed to the conception of the study and helped to draft the manuscript. SK contributed to the conception of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Supplementary Table. Contains complete MHS and GCS rankings and BLAST data for all wBm genes.
Contributor Information
Alexander G Holman, Email: holman@neb.com.
Paul J Davis, Email: davisp@neb.com.
Jeremy M Foster, Email: foster@neb.com.
Clotilde KS Carlow, Email: carlow@neb.com.
Sanjay Kumar, Email: kumar@neb.com.
Acknowledgements
This work was funded by New England Biolabs and, as part of the A-WOL consortium, by the Liverpool School of Tropical Medicine through a grant from the Bill and Melinda Gates Foundation. We wish to thank Dr. Donald Comb and New England Biolabs for long-standing generous and unwavering support of research aimed at alleviating filariasis. The Database of Essential Genes version 5.2 was kindly provided by Dr. Ren Zhang at the Centre of BioInformatics, Tianjin University.
References
- Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25(4):451–7. doi: 10.1093/bioinformatics/btp002. [DOI] [PubMed] [Google Scholar]
- Agüero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, Carmona S, Carruthers IM, Chan AW, Chen F, Crowther GJ, Doyle MA, Hertz-Fowler C, Hopkins AL, McAllister G, Nwaka S, Overington JP, Pain A, Paolini GV, Pieper U, Ralph SA, Riechers A, Roos DS, Sali A, Shanmugam D, Suzuki T, van Voorhis WC, Verlinde CL. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov. 2008;7(11):900–7. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Research. 2009. pp. D455–8. [DOI] [PMC free article] [PubMed]
- Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17(5):448–56. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Behm CA, Bendig MM, McCarter JP, Sluder AE. RNAi-based discovery and validation of new drug targets in filarial nematodes. Trends Parasitol. 2005;21(3):97–100. doi: 10.1016/j.pt.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Caffrey CR, Rohwer A, Oellien F, Marhöfer RJ, Braschi S, Oliveira G, Mckerrow JH, Selzer PM. A comparative chemogenomics strategy to predict potential drug targets in the metazoan pathogen, Schistosoma mansoni. PLoS ONE. 2009;4(2):e4413. doi: 10.1371/journal.pone.0004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JM, Zhang Y, Kumar S, Carlow CKS. Mining nematode genome data for novel drug targets. Trends Parasitol. 2005;21(3):101–4. doi: 10.1016/j.pt.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Krasky A, Rohwer A, Schroeder J, Selzer PM. A combined bioinformatics and chemoinformatics approach for the development of new antiparasitic drugs. Genomics. 2007;89:36–43. doi: 10.1016/j.ygeno.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Kumar S, Chaudhary K, Foster JM, Novelli JF, Zhang Y, Wang S, Spiro D, Ghedin E, Carlow CKS. Mining predicted essential genes of Brugia malayi for nematode drug targets. PLoS ONE. 2007;2(11):e1189. doi: 10.1371/journal.pone.0001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sim TB, Kim YS, Chang YT. Tools for target identification and validation. Curr Opin Chem Biol. 2004;8(4):371–7. doi: 10.1016/j.cbpa.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Arigoni F, Talabot F, Peitsch M, Edgerton MD, Meldrum E, Allet E, Fish R, Jamotte T, Curchod ML, Loferer H. A genome-based approach for the identification of essential bacterial genes. Nat Biotechnol. 1998;16(9):851–6. doi: 10.1038/nbt0998-851. [DOI] [PubMed] [Google Scholar]
- Carbone A. Computational prediction of genomic functional cores specific to different microbes. J Mol Evol. 2006;63(6):733–46. doi: 10.1007/s00239-005-0250-9. [DOI] [PubMed] [Google Scholar]
- Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA. 1996;93(19):10268–73. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Xu D. Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinformatics. 2005;21(5):575–81. doi: 10.1093/bioinformatics/bti058. [DOI] [PubMed] [Google Scholar]
- Joyce AR, Reed JL, White A, Edwards R, Osterman A, Baba T, Mori H, Lesely SA, Palsson BØ, Agarwalla S. Experimental and computational assessment of conditionally essential genes in Escherichia coli. J Bacteriol. 2006;188(23):8259–71. doi: 10.1128/JB.00740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson AM, Snitkin ES, Parker SCJ, DeLisi C, Kasif S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. Bmc Genomics. 2006;7:265. doi: 10.1186/1471-2164-7-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seringhaus M, Paccanaro A, Borneman A, Snyder M, Gerstein M. Predicting essential genes in fungal genomes. Genome Res. 2006;16(9):1126–35. doi: 10.1101/gr.5144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter JP. Genomic filtering: an approach to discovering novel antiparasitics. Trends Parasitol. 2004;20(10):462–8. doi: 10.1016/j.pt.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Odenwald WF, Rasband W, Kuzin A, Brody T. EVOPRINTER, a multigenomic comparative tool for rapid identification of functionally important DNA. Proc Natl Acad Sci USA. 2005;102(41):14700–5. doi: 10.1073/pnas.0506915102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, Ruby JG, Brennecke J. Harvard FlyBase Curators; Berkeley Drosophila Genome Project; Hodges E, Hinrichs AS, Caspi A, Paten B, Park SW, Han MV, Maeder ML, Polansky BJ, Robson BE, Aerts S, van Helden J, Hassan B, Gilbert DG, Eastman DA, Rice M, Weir M, Hahn MW, Park Y, Dewey CN, Pachter L, Kent WJ, Haussler D, Lai EC, Bartel DP, Hannon GJ, Kaufman TC, Eisen MB, Clark AG, Smith D, Celniker SE, Gelbart WM, Kellis M. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450(7167):219–32. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaglehole R, Irwin A, Prentice T. The world health report 2004: Changing history. World Health Organization. 2004. http://www.who.int/whr/2004/en/
- Hoerauf A. New strategies to combat filariasis. Expert review of anti-infective therapy. 2006;4(2):211–22. doi: 10.1586/14787210.4.2.211. [DOI] [PubMed] [Google Scholar]
- Osei-Atweneboana MY, Eng JKL, Boakye DA, Gyapong JO, Prichard RK. Prevalence and intensity of Onchocerca volvulus infection and efficacy of ivermectin in endemic communities in Ghana: a two-phase epidemiological study. Lancet. 2007;369(9578):2021–9. doi: 10.1016/S0140-6736(07)60942-8. [DOI] [PubMed] [Google Scholar]
- Schwab AE, Boakye DA, Kyelem D, Prichard RK. Detection of benzimidazole resistance-associated mutations in the filarial nematode Wuchereria bancrofti and evidence for selection by albendazole and ivermectin combination treatment. Am J Trop Med Hyg. 2005;73(2):234–8. [PubMed] [Google Scholar]
- Chirgwin SR, Coleman SU, Porthouse KH, Nowling JM, Punkosdy GA, Klei TR. Removal of Wolbachia from Brugia pahangi is closely linked to worm death and fecundity but does not result in altered lymphatic lesion formation in Mongolian gerbils (Meriones unguiculatus) Infect Immun. 2003;71(12):6986–94. doi: 10.1128/IAI.71.12.6986-6994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoerauf A, Nissen-Pähle K, Schmetz C, Henkle-Dührsen K, Blaxter ML, Büttner DW, Gallin MY, Al-Qaoud KM, Lucius R, Fleischer B. Tetracycline therapy targets intracellular bacteria in the filarial nematode Litomosoides sigmodontis and results in filarial infertility. J Clin Invest. 1999;103:11–8. doi: 10.1172/JCI4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra D, Angiuoli SV, Creasy T, Amedeo P, Haas B, El-Sayed NM, Wortman JR, Feldblyum T, Tallon L, Schatz M, Shumway M, Koo H, Salzberg SL, Schobel S, Pertea M, Pop M, White O, Barton GJ, Carlow CKS, Crawford MJ, Daub J, Dimmic MW, Estes CF, Foster JM, Ganatra M, Gregory WF, Johnson NM, Jin J, Komuniecki R, Korf I, Kumar S, Laney S, Li BW, Li W, Lindblom TH, Lustigman S, Ma D, Maina CV, Martin DMA, McCarter JP, McReynolds L, Mitreva M, Nutman TB, Parkinson J, Peregrín-Alvarez JM, Poole C, Ren Q, Saunders L, Sluder AE, Smith K, Stanke M, Unnasch TR, Ware J, Wei AD, Weil G, Williams DJ, Zhang Y, Williams SA, Fraser-Liggett C, Slatko B, Blaxter ML, Scott AL. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317(5845):1756–60. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, Bhattacharyya A, Kapatral V, Kumar S, Posfai J, Vincze T, Ingram J, Moran L, Lapidus A, Omelchenko M, Kyrpides N, Ghedin E, Wang S, Goltsman E, Joukov V, Ostrovskaya O, Tsukerman K, Mazur M, Comb D, Koonin E, Slatko B. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005;3(4):e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CE, Lim BS, Nathan S, Mohamed R. In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biol (Gedrukt) 2006;6(4):341–6. [PubMed] [Google Scholar]
- Sakharkar KR, Sakharkar MK, Chow VTK. Biocomputational strategies for microbial drug target identification. Methods Mol Med. 2008;142:1–9. doi: 10.1007/978-1-59745-246-5_1. full_text. [DOI] [PubMed] [Google Scholar]
- Korf I, Yandell M, Bedell J. BLAST. OŔeilly; 2003. [Google Scholar]
- Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61(3):377–92. doi: 10.1128/mmbr.61.3.377-392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich HA. Molecular biology of rifomycin. MSS Information Corp; 1973. [Google Scholar]
- Tanaka K, Tamaki M, Watanabe S. Effect of furanomycin on the synthesis of isoleucyl-tRNA. Biochim Biophys Acta. 1969;195:244–5. doi: 10.1016/0005-2787(69)90621-2. [DOI] [PubMed] [Google Scholar]
- Hughes J, Mellows G. Inhibition of isoleucyl-transfer ribonucleic acid synthetase in Escherichia coli by pseudomonic acid. Biochem J. 1978;176:305–18. doi: 10.1042/bj1760305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee SW, Choi EC, Choi SY. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl Microbiol Biotechnol. 2003;61(4):278–88. doi: 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and 'HGE agent' as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51(Pt 6):2145–65. doi: 10.1099/00207713-51-6-2145. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research. 2003;13(9):2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raverdy S, Foster JM, Roopenian E, Carlow CKS. The Wolbachia endosymbiont of Brugia malayi has an active pyruvate phosphate dikinase. Mol Biochem Parasitol. 2008;160(2):163–6. doi: 10.1016/j.molbiopara.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Hasan S, Daugelat S, Rao PSS, Schreiber M. Prioritizing genomic drug targets in pathogens: application to Mycobacterium tuberculosis. PLoS Comput Biol. 2006;2(6):e61. doi: 10.1371/journal.pcbi.0020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ AP, Lampel S. The druggable genome: an update. Drug Discov Today. 2005;10(23-24):1607–10. doi: 10.1016/S1359-6446(05)03666-4. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008. pp. D901–6. [DOI] [PMC free article] [PubMed]
- Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol. 2004;186(23):7926–35. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- van Dongen S. Graph clustering by flow simulation. PhD Thesis, Univ. of Utrecht, the Netherlands. 2000. http://micans.org/mcl/
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormen TH, Leiserson CE, L R, Stein C. Introduction to Algorithms. 2. Cambridge: MIT Press; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table. Contains complete MHS and GCS rankings and BLAST data for all wBm genes.