

Abstract
Normal human diploid fibroblasts have a finite replicative lifespan in vitro, which has been postulated to be a cellular manifestation of aging in vivo. Several studies have shown an inverse relationship between donor age and fibroblast culture replicative lifespan; however, in all cases, the correlation was weak, and, with few exceptions, the health status of the donors was unknown. We have determined the replicative lifespans of 124 skin fibroblast cell lines established from donors of different ages as part of the Baltimore Longitudinal Study of Aging. All of the donors were medically examined and were declared “healthy,” according to Baltimore Longitudinal Study of Aging protocols, at the time the biopsies were taken. Both long- and short-lived cell lines were observed in all age groups, but no significant correlation between the proliferative potential of the cell lines and donor age was found. A comparison of multiple cell lines established from the same donors at different ages also failed to reveal any significant trends between proliferative potential and donor age. The rate of [3H]thymidine incorporation and the initial rates of growth during the first few subcultivations were examined in a subset of cell lines and were found to be significantly greater in fetal lines than in postnatal lines. Cell lines established from adults did not vary significantly either in initial growth rate or in [3H]thymidine incorporation. These results clearly indicate that, if health status and biopsy conditions are controlled, the replicative lifespan of fibroblasts in culture does not correlate with donor age.
Keywords: cell proliferation/cell senescence/aging
It is well established that phenotypically and karyotypically normal human cells exhibit a limited capacity to proliferate in culture (1, 2). The finite replicative lifespan of normal cells in culture is thought to result from multiple environmental and genetic mechanisms (3) and is frequently used as one model of human aging. Supporting the usefulness of this model, several laboratories, by using different types of normal human cells maintained in culture, have presented evidence for a negative correlation between donor age and proliferative lifespan in vitro (4–11). The colony-forming capacity of individual cells has been reported to decline as a function of donor age (12, 13). Furthermore, cultures established from donors with various diseases, including diabetics (11, 14, 15) and Werner’s syndrome patients (6, 16–19), have been found to exhibit significantly shorter proliferative lifespans in culture. Cultures derived from individuals with Hutchinson–Gilford syndrome (17) and Down’s syndrome (17, 20) also exhibit decreased proliferative potential, although results with these cell lines are more variable (21). Collectively, these observations have been interpreted generally to support the view that the proliferative lifespan of cells in culture reflects the aging changes in the donor from which the cells were originally obtained. However, it has always been difficult to reconcile how the number of replications observed in culture (which for nonstem cells is far greater than the number of divisions that would be expected in vivo during a normal postnatal lifespan) could reflect aging events that may occur in organisms. A key question is whether aging in vivo has any counterpart to senescence in culture or, conversely, whether the pathway leading to senescence in vitro directly reflects any aspects of aging in vivo.
Other questions arise from the relatively weak correlations observed between donor age and proliferative lifespan, as well as from the large variability in proliferative potential observed in cell lines established from individuals of similar age (5, 6, 11, 22). Generally, investigators have assumed that the replicative lifespan was largely a function of the proliferative capacity of the most vigorous subpopulation of cells in the tissue explant, and they thus underestimated the extent of differences in the proliferative capacity of cells in vivo (22). Additionally, at least some of the variability within age groups was attributed to differences in donor health status and the conditions and timing of the biopsy (11, 22).
In this study, we report the replicative lifespans of 124 cell cultures established from human skin biopsies as part of the Baltimore Longitudinal Study of Aging (BLSA). At the time of biopsy, all of the donors were certified as “healthy,” according to the medical criteria used by BLSA. The cell lines we examined also included cells from six individuals sampled sequentially during their participation in the study and eight dermal cell lines established from apparently normal fetuses in the second trimester of development (12- to 20-wk gestational age). Initial growth rates and [3H]thymidine labeling were also analyzed. The results presented here indicate that when health status and biopsy conditions were controlled, there was high variability in the replicative lifespans of cells derived from all donor age groups; however, no correlation was observed between replicative lifespan and donor age.
MATERIALS AND METHODS
Cell Lines and Culture Procedures.
A total of 42 human fibroblast cultures, established from skin samples derived from fetal (12–20 wk gestational age), young (17–64 yr), and old donors (73–92 yr), were obtained from the National Institute on Aging cell repository at the Coriell Institute for Medical Research (CIMR), Camden, NJ, and were cultured until they reached the end of their replicative lifespan. All the lines were started from 2-mm punch biopsies taken from the mesial aspect of the upper arm. All the donors were members of the BLSA and were medically examined and diagnosed as “healthy” when they entered the study. The donor of cell line AG12851, who was part of the longitudinal study, was healthy when first enrolled in the study, but later developed diabetes. Several donors had aging-associated conditions, and several were reported to have unusual diagnoses. According to BLSA protocol, an unusual diagnosis is a nondiabetic-, noncardiovascular-, nondementia-, and nonarthritis-related illness. For example, the donor of cell line AG13077 was diagnosed with polymyalgia rheumatica, and the donors of AG05247 and AG9602 had hypothyroidism and osteoporosis. The individual from whom AG09557 was established later developed a problem with alcohol abuse. Five of the cell lines exhibited minor karyotypic anomalies; however, all were phenotypically normal with a limited replicative lifespan (23). All other cell lines used in this study had a normal karyotype, i.e., 46,XX or 46,XY. Cells were grown in MEM without antibiotics and were supplemented with 2 mM l-glutamine and 10% (vol/vol) fetal bovine serum, according to our standard procedures (24). Briefly, cells were seeded at 1 × 104 cells per cm2 of growth area. Culture medium was gassed with a mixture of 5% CO2 and 95% air and were grown at 37°C. Most of the cell lines were subcultured weekly; slow-growing cultures were subcultured biweekly, but received fresh medium on weeks when they were not subcultivated. A Coulter Counter was used to determine cell number, as described (24). Cumulative population doubling level at each subcultivation was calculated from the cell count by using the equation
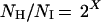 |
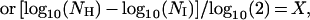 |
where NI = inoculum number, NH = cell harvest number, and X = population doublings. The population doubling increase that was calculated was then added to the previous population doubling level (PDL), to yield the cumulative population doubling level. The end of the replicative lifespan was defined by failure of the population to double after 4 wk in culture with 3 wk of consecutive refeeding.
An additional 82 cell lines derived from donors ranging in age from 30 to 90 yr were subcultivated to determine in vitro lifespan at CIMR, essentially as described above but with the following exceptions: (i) cells were counted by using a hemocytometer, (ii) a brief centrifugation of the cell suspension was included before seeding, (iii) 15% fetal bovine serum was used in growth medium, and 0.2% EDTA/0.25% trypsin was used to detach cell monolayers, and (iv) the end of the in vitro lifespan was determined as described, but with refeedings every 2–3 days over a 3-wk period.
Initial Slope.
Cells in early passage cultures tend to divide more rapidly than do those in late passage cultures. When the cumulative (PDL) was plotted against the time in culture, a clear decrease in the slope of the resulting line (change in growth rate) was observed. We have calculated this initial slope as a crude measure of maximal growth rate of the cell lines.
Labeling Index.
The incorporation of [3H]thymidine was used to determine the DNA synthesis activity of cultures during the initial rapid-growth phase of the in vitro lifespan. The percentage of labeled nuclei was determined by a previously described method (25). Briefly, 1.0 × 104 cells per cm2 were seeded in 60-mm Petri dishes containing 22-mm coverslips and were incubated at 37°C for 24 hr. [3H]Thymidine (2 Ci/mmol) was added to the medium to a final concentration of 0.1 μCi/ml. After an additional 30-hr incubation, the coverslips were washed twice with PBS and were fixed for 15 min in methanol. For autoradiography, the coverslips were dipped in Kodak NTB-2 emulsion. After a 4-day exposure period at 4°C, the slides were developed (Kodak D19 developer) for 5 min and were fixed (Kodak acid fixer) for 5 min. The slides were then stained lightly with Harris hematoxylin (Fisher Scientific), rinsed with cold running water, and air-dried. The percentage of labeled nuclei was determined by counting at least 400 cells in random fields throughout the coverslip. Typically, cells were either heavily labeled or were unlabeled. Cells with five grains or more were scored as labeled. Duplicate coverslips were counted for each line at each time point.
Statistics.
Means were compared by using standard ANOVA. Post hoc determinations were made by using least significant difference. The correlation coefficient (r) was determined by using least-squares regression line analysis, and level of probability of similarity (P) was calculated from the value of r and the number in the population (n), by using Pearson product moment correlation distribution.
RESULTS
As predicted by the Hayflick and Moorhead model (2), all the cell lines examined exhibited a finite proliferative lifespan (Fig. 1). An analysis of the proliferative potential of all postnatal skin fibroblast lines established from donors of different ages revealed no significant correlation between maximum PDL achieved and the age of the donor (correlation coefficient, r = −0.018; probability level, P = 0.85; population number, n = 116). The 95% confidence interval for this correlation reveals a lower limit of −0.2 and an upper limit of 0.16. With our sample size, neither the upper nor lower boundary of this interval is significant. Thus, even though variability is relatively high, it is highly improbable that it obscured any significant trend in our comparison of adult individuals. In fact, inclusion of the maximum PDL of cell lines derived from prenatal donors in the analysis (r = −0.17, P = 0.059, n = 124) or exclusion of short-lived postnatal cell lines (maximum PDL <10) failed to produce a significant change in the correlation (r = 0.061, P = 0.054, n = 102). Exclusion of the 82 lifespans collected by CIMR personnel (r = 0.0103, P = 0.95, n = 42) or the 42 lifespans determined by Center for Gerontological Research personnel (r = −0.055, P = 0.62, n = 82) failed to influence the level of significance. We observed both long- and short-lived cell lines in all age groups of donors (Fig. 1).
Figure 1.

Relationship between in vitro proliferative capacity of postnatal skin fibroblast cell lines and donor age (in years). A regression line (r = −0.018, P = 0.85, and n = 116) is shown. Males are represented by closed triangles and females by open circles.
The predicted lifespan reported in the CIMR catalogue was estimated by using the clone size distribution assay (13, 26); this assay differed significantly from the lifespans we determined for many of the lines. To determine whether the variation between actual and predicted lifespans was the result of technical variation, and to resolve the precision of our technique for quantifying replicative lifespans, the proliferative lifespan of nine of the cell lines was determined on each of two sublines by different operators. The results of this study are listed in Table 1. A mean difference of 4.5 ± 2.8 population doublings was obtained for the maximum lifespans between each of the subcultures derived from a single cell line. Thus, the reproducibility of lifespan determination in our laboratory is high in comparison to the mean difference of 13.8 ± 10.1 population doublings (data not shown) between the predicted replicative lifespan (stated in the CIMR catalogue) and the actual lifespan of any cell line, as determined in the study.
Table 1.
Replicative lifespans of cell lines determined in duplicate experiments
| Cell line | Replicative life span, PDL
|
Difference | |
|---|---|---|---|
| Exp. 1 | Exp. 2 | ||
| AG06234 | 22.2 | 25.9 | 3.7 |
| AG07306 | 31.8 | 24.3 | 7.5 |
| AG07719 | 34.1 | 37.4 | 3.3 |
| AG04441 | 38.3 | 36.8 | 1.5 |
| AG04062 | 33.8 | 44.6 | 10.8 |
| AG06285 | 28.9 | 26.3 | 2.6 |
| AG08817 | 25.7 | 30.7 | 5.0 |
| AG06291 | 19.1 | 19.2 | 0.1 |
| AG09602 | 47.9 | 41.8 | 6.1 |
Mean PDL difference = 4.5.
To examine further the relationship between donor age and lifespan in vitro, we determined the proliferative potential of cell lines derived from the same donors at different ages. As seen in Fig. 2, the proliferative capacity of cells from any single individual at different ages failed to yield a significant pattern with age. Indeed, cell lines established from individuals at older ages frequently exhibited a slightly greater proliferative potential than did the cell lines established from the same individuals at younger ages (Fig. 2).
Figure 2.

Proliferative potential of cell lines derived from the same donors at different ages. Shown are the maximum lifespans in vitro of two or more skin cultures established from each of six donors at the ages (in years) indicated in parentheses.
A subset of cell lines established from each donor age group was used to evaluate the relationship between growth kinetics and donor age. Known medical conditions that arose either before or after the biopsy date are summarized in Table 2. Other characteristics of the cell lines in this subset, including predicted and actual replicative lifespans, are presented in Table 2.
Table 2.
Summary of lines and cell donor medical information
| Line | Age, yr | Predicted lifespan | Actual lifespan | Date of biopsy | Diagnosis | Age at death, yr |
|---|---|---|---|---|---|---|
| AG04449 | F | 26 | 31–31 | 5/80 | Aborted | |
| AG04431* | F | >80 | 54 | 9/70 | Aborted | |
| AG04392* | F | >80 | 55 | 6/79 | Aborted | |
| AG04451* | F | 57 | 9/80 | Aborted | ||
| AG04525 | F | 27–36 | 5/78 | Aborted | ||
| AG04433 | 30–36 | 1/80 | Aborted | |||
| AG06559* | F | 30–31 | 10/82 | Aborted | ||
| AG06555 | F | 30–31 | 2/82 | Aborted | ||
| AG06234 | 17.2 | 40 | 22–26 | 6/80 | H | |
| AG07720* | 23.9 | 47 | 39 | 8/79 | H | |
| AG07719* | 27.9 | 36 | 34–37 | 10/82 | H | |
| AG07603* | 28.2 | 48 | 24–32 | 6/82 | H | |
| AG04441 | 29.4 | 47 | 37–38 | 8/78 | H | |
| AG13153 | 30.1 | — | 33 | 5/93 | H | |
| AG04062 | 31.1 | 31 | 34–44 | 2/78 | H | |
| AG06285A | 31.6 | 51 | 26–29 | 3/81 | H | |
| AG07471* | 32.3 | 42 | 44 | 7/82 | 1 | |
| AG11016* | 32.7 | 36 | 32 | 8/87 | H | |
| AG08817 | 33.8 | 39 | 26–31 | 5/79 | H | |
| AG12596* | 42.3 | — | 29 | 8/92 | 1 | |
| AG13156 | 44.2 | — | 42 | 5/93 | H | |
| AG04446 | 48.4 | 33 | 37 | 6/78 | 1, 3, 4, 6 | |
| AG12851 | 62.8 | — | 26 | 11/92 | 1, 3, 4, 6 | |
| AG04552 | 64.7 | 41 | 22 | 6/78 | 1, 4 | |
| AG06952* | 73.0 | 53 | 40 | 8/81 | 4, 6, 7, 11 | |
| AG05096 | 73.5 | 29 | 34 | 6/78 | 6, 7 | 90.7 |
| AG11246* | 77.6 | 26 | 31 | 4/87 | 4, 5, 6, 7 | |
| AG11081* | 77.9 | 44 | 45 | 10/87 | 2, 4, 6, 7 | |
| AG11020* | 78.7 | 26 | 67 | 4/87 | 4, 6, 7, 11 | |
| AG13152 | 79.6 | — | 26 | 5/93 | 1, 4 | |
| AG11240* | 81.2 | 21 | 26 | 2/87 | 4, 5, 7 | |
| AG09603* | 81.9 | — | 52 | 12/85 | 4, 5, 6, 7, 9 | 87.5 |
| AG05274 | 83.4 | 52 | 38 | 3/81 | 1, 4, 9 | 97.2 |
| AG09557 | 83.5 | 48 | 39 | 12/85 | 4, 5, 6, 7, 11 | |
| AG13077* | 84.6 | — | 47 | 4/93 | 4, 6, 7, 11 | |
| AG10884 | 86.3 | — | 33 | 3/84 | 1, 4, 9 | 97.2 |
| AG05247D* | 87 | 58 | 45 | 7/80 | 4, 5, 11 | 94.4 |
| AG13129 | 88.4 | — | 41 | 4/93 | 6, 7 | 90.7 |
| AG06291A | 89.4 | 44 | 19–19 | 3/81 | 1, 7 | 89.8 |
| AG09602* | 92.3 | — | 42–48 | 11/85 | 4, 5, 11 | 94.4 |
| AG04064 | 92.6 | 50 | 32 | 11/77 | 4, 6, 8 | 98.7 |
| AG08433 | 94.3 | 42 | 34 | 8/79 | 4, 6, 8 | 97.9 |
For age, F indicates fetal. For diagnosis, a boldface number indicates condition diagnosed before biopsy; an italic number indicates condition diagnosed at some time after biopsy. Diagnoses are as follows: 1, skin cancer; 2, other cancer; 3, diabetes; 4, arthritis; 5, coronary heart disease; 6, hypertension; 7, other cardiovascular-related disease; 8, consistent with dementia; 9, Parkinson’s disease with dementia; 10, probable Alzheimer’s disease; 11, unusual diagnosis (nature not defined); and H, healthy (none of the above disorders found).
Female.
Plotting the PDL of the cell lines at each subcultivation against the total time in culture revealed that in all cases the resulting line connecting the points changed from an early steep slope to a more gentle slope. We compared the initial steep slope obtained for the cell lines as a measure of early growth kinetics (Table 3). A significant difference was observed when cell lines in the three age groups were compared (ANOVA, P = 0.0002). However, subsequent post hoc analysis again revealed that postnatal lines did not vary significantly, and that the correlation observed arose entirely from differences between fetal and postnatal lines (Table 3). In fact, the initial growth rates of fetal lines were uniformly greater than those of postnatal lines, regardless of their proliferative lifespans.
Table 3.
Post hoc analysis* of initial growth rates of cell lines established from donors of different ages
| Group | Initial growth rate | Groups compared | P |
|---|---|---|---|
| Fetal | 2.09 ± 0.34 (13) | Fetal/young | 0.00008 |
| Young | 1.11 ± 0.49 (23) | Fetal/old | 0.0003 |
| Old | 1.26 ± 0.53 (20) | Young/old | 0.4 |
Total numbers of measurements are in parentheses. Initial growth rates (change in PDL per week) were determined in all lines and are presented as mean ± SD. Measurements were repeated in selected lines (chosen at random) from each age group to confirm sampling accuracy.
Total ANOVA, P = 0.0002.
Relative DNA synthesis activity in these cell lines was determined by calculating the percentage of labeled nuclei early in each culture’s proliferative lifespan. As described for WI-38 cells (25), the ability of skin fibroblast cells to incorporate [3H]thymidine diminished progressively with increasing proliferative age in all of the cell lines (data not shown). The percentage of labeled nuclei varied significantly between the different age groups (ANOVA, P = 0.018; Table 4). Post hoc analysis (least significant difference) revealed that the propensity of fetal skin fibroblasts to incorporate [3H]thymidine was significantly greater than was observed in either group of postnatal lines (Table 4). No significant difference was observed when only the two postnatal groups were compared (Table 4). Hence, the age-related trend observed for a decline in labeling index was significant only as a result of differences between fetal and postnatal groups.
Table 4.
Post hoc analysis* of initial labeling indices of cell lines established from donors of different ages
| Group | % Labeled nuclei | Groups compared | P |
|---|---|---|---|
| Fetal | 79.15 ± 13.7 (13) | Fetal/young | 0.01 |
| Young | 63.73 ± 12.9 (23) | Fetal/old | 0.01 |
| Old | 63.55 ± 20.5 (20) | Young/old | 0.9 |
Total numbers of measurements are in parentheses. Initial labeling indices were determined in all lines and are presented as mean ± SD. Measurements were repeated in selected lines (chosen at random) from each age group to confirm sampling accuracy.
Total ANOVA, P = 0.018.
DISCUSSION
It is generally accepted that there is a reduction in proliferative activity of cells in vivo as organisms age (recently reviewed by Rubin in ref. 27). Support for the view that the mechanisms responsible for aging in vitro are also active in vivo is found in the fact that normal cells, serially transplanted to compatible hosts, exhibit a proliferative decline (28–38). A number of investigators have presented data suggesting that this loss of proliferative activity is expressed in vitro as decreased outgrowth of cells from biopsies, reduced clone size distribution, and decreased replicative lifespan (4, 5, 10, 11, 22). However, the interpretation of replicative lifespan data has been plagued by large individual variations and relatively low correlations. For example, in the studies reported by Martin et al. (6, 22), replicative lifespan was determined in more than 100 cell lines, yet a correlation coefficient of only −0.33 was obtained. Although these correlations and correlations reported by others are statistically significant (10), donor age-dependent declines in replicative lifespans are usually small, and hence it is difficult to assess whether they compromise the physiology of the organism or its proliferative homeostasis (22, 27).
In at least one study, the inverse relationship between donor age and proliferative lifespan observed in human skin fibroblasts (6) was reported to become insignificant when cultures of cells from newborns were excluded from the analysis (39), suggesting that no difference existed between young and old donors. Another study of skin fibroblasts (11), frequently cited to support the existence of a donor age effect on proliferative capacity, actually showed that such a relationship existed only when cell lines derived from diabetics and prediabetics were included (see also ref. 15). Furthermore, no donor age-dependent correlation is observed when earlier studies (6, 22) are re-analyzed to exclude data from lines established from cadaver material; however, fibroblasts derived from autopsy skin did reveal significant declines in replicative potential with increasing donor age (G. Martin, personal communication).
The fact that we observed no relationship between donor age and proliferative lifespan in culture contrasts with results previously reported by several other groups (see above and refs. 4–6, 10). It seems likely that the health state of the cell donors partly accounted for differences in our results and those reported by others. As noted above, the cell lines used in this study were all established from healthy donors. The health status of the donors was determined at the time the biopsy was taken. The biopsies were taken from a uniform site (mesial aspect of the mid-upper left arm), which was chosen for minimal sun damage. Pathologies that developed after the biopsies were taken were also monitored. Cell lines established from donors that were not screened thoroughly for disease, as well as cell lines derived from cadavers, have been used to determine the effects of donor age on proliferative potential in most other studies. Variations in the biopsy site may also have influenced the results obtained in other studies. One previously reported study (5) also used cell lines from the BLSA population; however, the authors do not report a significant correlation between replicative lifespan and donor age. Instead, they report a small but significant (P < 0.05) difference in replicative lifespan when two groups arbitrarily designated “young donors” and “old donors” were compared. A similar comparison of groups in our study failed to reveal any significant differences (ANOVA, P = 0.34).
Several studies of rodent skin fibroblasts appear to support the existence of a small, though significant, inverse correlation between donor age and replicative lifespan (9, 40, 41). It has also been observed that treatment of hamster skin fibroblasts with growth promoters can extend the proliferative life of cultures established from young organisms but has negligible effects on cultures established from older donors (42). Aside from the inherent species differences and the effects of inbreeding that may influence these results, it is also apparent that rodent skin is better protected from some types of environmental injury, such as light exposure. However, even in rodents the relationship between donor age and proliferative potential is not entirely clear. For example, an examination of hamster skin fibroblast cultures established from the same donors at different ages reveals no age-associated changes in proliferative potential in animals older than 12 mo (41).
A related question is whether replicative senescence has a direct counterpart in vivo. A number of studies have addressed this question in different ways. In terms of morphological characteristics, Robbins et al. (43) reported that fibroblast senescence in tissue culture and in the intact organism are not homologous. They observed that progressive morphological changes began to develop in diploid cell cultures shortly after they were established, regardless of the donor age. These authors also reported that no cells were found in vivo at any age that exhibited the morphological phenotype of cells in vitro at the end of their replicative lifespan (43). Diminished expression of genes that control cell cycle is a phenomenon tightly linked to replicative senescence, and yet it is not evident in centenarian humans (44). Similarly, reports of donor age-dependent increases in β-galactosidase staining (45) really reflect less than 1 in 104 cells stained even in the oldest individuals and are of questionable significance in vivo (V.J.C., unpublished work). Rubin (27) suggests that the limited replicative lifespan in vitro may be an artifact that reflects the failure of diploid cells to adapt to the trauma of dissociation and to the radically foreign environment of cell culture.
There are also a number of differences reported between fetal- and adult-derived cell lines related to growth factor requirements for proliferation and migration (46–49) that remain even as these cultures become senescent. For example, Wharton (46) has shown that fetal dermal fibroblasts will proliferate in plasma or serum, while adult-derived dermal fibroblasts require serum for growth. Throughout the replicative lifespan of the fetal cells, they never lose the ability to grow on plasma. If the pathway to senescence in vitro were similar to aging in vivo, fetal cells in culture would be expected to acquire a postnatal phenotype as they senesce; this is clearly not the case in the above examples. Alternatively, these observations might be expected if fetal cell lines in a culture environment “differentiate” along another pathway for which there is no direct counterpart in vivo. It is also possible that, when cells are placed in culture, important though as yet unknown signals are missing from the fetal bovine serum that prevent cells from utilizing normal sequences of developmental pathways.
Bayreuther et al. (50) have argued that aging of human dermal fibroblasts in vivo cannot result from depletion of cells with relatively high proliferative capacity because they observed >60% mitotic fibroblasts in primary populations of cells taken from individuals between the ages of 60 and 80 years. Nevertheless, serial transplantation studies have clearly established that the proliferative capacity of cells can be diminished in vivo (28–38). If replicative potential in vivo is thus limited, and cells in vivo are at least occasionally stimulated to divide, it is reasonable to expect that the proliferative ages of cells in vivo will vary depending on their proliferative history. In fact, cell lines established from biopsies taken from the same individual at multiple sites exhibit a wide variation in proliferative potential (22).
Because the process of establishing cell cultures ultimately selects for the hardiest cells from a biopsy (those best able to grow), the extent of replicative senescence in vivo is grossly underestimated. Nevertheless, the high variability in replicative lifespan observed in cell lines established from donors of similar age is inconsistent with the hypothesis that aging in vivo results in a uniform loss of proliferative potential. Similarly, the absence of any donor age-dependent decrease in proliferative potential in well controlled studies of healthy individuals, and the failure of the replicative lifespan of cell lines established sequentially from the same donor at different ages to correlate inversely with donor age, both fail to support the hypothesis that replicative aging occurs uniformly in vivo. It appears far more probable that loss of proliferative potential in vivo occurs as a mosaic in which both long- and short-lived cells may lie in close proximity. Those variations in proliferative potential that do exist probably stem mostly from differences in mitotic history. Studies of cultures maintained in a nondividing state for long periods clearly demonstrate that replicative potential is not diminished (51–53). However, other as yet unknown microenvironmental factors may also account for differences in proliferative lifespan observed in cell lines established from a single individual. For example, the individual cells of a single subclone exhibit markedly different proliferative potentials (54), indicating that microenvironmental factors may influence proliferative lifespan even after cells are placed in a culture environment.
Because we found no significant relationship between donor age and replicative potential in vitro, we examined other growth parameters in a subgroup of cell lines. Both the initial growth rates and the labeling indices of the fetal fibroblast lines were higher than those observed in adults. This difference indicates that cell lines established from fetal skin divided more frequently initially, even though the replicative lifespan of these cell lines did not exceed that observed in the postnatal cell lines. Indeed, the initial growth rates of fetal lines were uniformly greater than were those of postnatal lines, even in those cases where the proliferative lifespan of the postnatal lines was greater. It would seem equally relevant that the labeling indices and initial growth rates of the two postnatal groups did not vary with respect to age. In view of differences in initial growth rates and in intraclonal variations in the proliferative potential of single cells (54), it seems probable that clone size distribution and replicative lifespan measure different things and that the use of clone size distribution to estimate replicative lifespan (12, 13) could lead to significant variation when compared with actual lifespan.
The reproducible loss of proliferative potential in vitro may not reflect changes in replicative capacity that occur in vivo; however, fibroblast cultures remain a powerful model for a variety of aging-related studies. Among these are studies of heritable damage to cell populations that simulate the effects of aging in vivo (27), a variety of chemical and molecular manipulations used to induce a senescence phenotype, and the effects of stress (27, 55–59). This system can also be used to study abnormal growth or quiescence (3). Loss of capacity for senescence is, of course, a necessary step for immortalization and, ultimately, transformation to a malignant phenotype. The model may also prove useful in studies of the relationship between differentiation and replicative aging (46–49).
The results of this study clearly demonstrate that there is no significant donor age-dependent change in proliferative lifespan when cell lines are established from healthy individuals. The study presented here uniquely includes a longitudinal analysis. We also show that the large difference in initial growth rates, as well as the [3H]thymidine incorporation observed in fetal and postnatal lines, occur independently of proliferative potential. These results also suggest that aging in vivo may occur in mosaic patterns rather than uniformly.
Acknowledgments
We thank Adrianna Chernack for her excellent technical assistance and Dr. Ed Gracely (Allegheny University of the Health Sciences, Philadelphia, PA) for his assistance in performing some of the statistics presented here. We gratefully acknowledge Dr. James Fozard and Dr. Jeffrey Metter of the National Institute on Aging Gerontology Research Center, and Dr. Claudia Kawas of John Hopkins University (Baltimore, MD) for their help and advice on the health status of the donor subjects used in this investigation. This work was supported by National Institutes of Health Grants AG00378 and AG00131 (V.J.C.), an Allegheny-Singer Research Institute grant (R.G.A.), and National Institutes of Health Contract N01-AG-1-2101.
ABBREVIATIONS
- BLSA
Baltimore Longitudinal Study of Aging
- CIMR
Coriell Institute for Medical Research
- PDL
population doubling level
References
- 1.Swim H E, Parker R F. Am J Hyg. 1957;66:235–243. doi: 10.1093/oxfordjournals.aje.a119897. [DOI] [PubMed] [Google Scholar]
- 2.Hayflick L, Moorhead P S. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 3.Cristofalo V J, Pignolo R J. Physiol Rev. 1993;73:617–638. doi: 10.1152/physrev.1993.73.3.617. [DOI] [PubMed] [Google Scholar]
- 4.Hayflick L. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 5.Schneider E L, Mitsui Y. Proc Natl Acad Sci USA. 1976;73:3584–3588. doi: 10.1073/pnas.73.10.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin G M, Sprague C A, Epstein C J. Lab Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- 7.Guilly Y L, Simon M, Lenoir P, Bourel M. Gerontologia (Basel) 1973;19:303–313. doi: 10.1159/000211984. [DOI] [PubMed] [Google Scholar]
- 8.Bierman E L. In Vitro. 1978;14:951–955. doi: 10.1007/BF02616126. [DOI] [PubMed] [Google Scholar]
- 9.Bruce S A, Deamond S F, Ts’o P O P. Mech Ageing Dev. 1986;34:151–173. doi: 10.1016/0047-6374(86)90032-1. [DOI] [PubMed] [Google Scholar]
- 10.Allsopp R C, Vaziri H, Patterson C, Goldstein S, Younglai E V, Futcher A B, Greider C W, Harley C B. Proc Natl Acad Sci USA. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein S, Moerman E J, Soeldner J S, Gleason R E, Barnett D M. Science. 1978;199:781–782. doi: 10.1126/science.622567. [DOI] [PubMed] [Google Scholar]
- 12.Schneider E L. Fed Proc. 1979;38:1857–1861. [PubMed] [Google Scholar]
- 13.Smith J R, Pereira-Smith O M, Schneider E L. Proc Natl Acad Sci USA. 1978;75:1353–1356. doi: 10.1073/pnas.75.3.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein S, Littlefield J W, Soeldner J S. Proc Natl Acad Sci USA. 1969;64:155–160. doi: 10.1073/pnas.64.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein S, Moerman E J, Soeldner J S, Gleason R E, Barnett D M. J Clin Invest. 1979;63:358–370. doi: 10.1172/JCI109311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oshima J, Campisi J, Tannock C A, Martin G M. J Cell Physiol. 1995;162:277–283. doi: 10.1002/jcp.1041620213. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein S, Harley C B. Fed Proc. 1979;38:1862–1867. [PubMed] [Google Scholar]
- 18.Danes B S. J Clin Invest. 1971;50:2000–2003. doi: 10.1172/JCI106692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin G M. In: Genetic Effects on Aging. Bergsma D, Harrison D E, editors. New York: Liss; 1978. pp. 5–39. [Google Scholar]
- 20.Schneider E L, Epstein C J. Proc Soc Exp Biol Med. 1972;141:1092–1094. doi: 10.3181/00379727-141-36940. [DOI] [PubMed] [Google Scholar]
- 21.Brown W T. Annual Review of Gerontology and Geriatrics. 1990;10:23–52. doi: 10.1007/978-3-662-38445-9_2. [DOI] [PubMed] [Google Scholar]
- 22.Martin G M, Ogburn C E, Sprague C A. In: Aging: A Challenge to Science and Society. Danon D, Shock N W, Marios M, editors. Vol. 1. Oxford: Oxford Univ. Press; 1981. pp. 124–135. [Google Scholar]
- 23.Allen R G, Keogh B P, Gerhard G, Pignolo R, Horton J, Cristofalo V J. J Cell Physiol. 1995;165:576–587. doi: 10.1002/jcp.1041650316. [DOI] [PubMed] [Google Scholar]
- 24.Cristofalo V J, Charpentier R. J Tissue Cult Methods. 1980;6:117–121. [Google Scholar]
- 25.Cristofalo V J, Sharf B B. Exp Cell Res. 1973;76:419–427. doi: 10.1016/0014-4827(73)90394-7. [DOI] [PubMed] [Google Scholar]
- 26.Schneider E L, Monticone R, Smith J, Braunschweiger K, Roberts T. Cytogenet Cell Genet. 1981;31:40–46. doi: 10.1159/000131624. [DOI] [PubMed] [Google Scholar]
- 27.Rubin H. Mech Ageing Dev. 1997;98:1–35. doi: 10.1016/s0047-6374(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 28.Krohn P L. Proc R Soc London Ser B. 1962;157:128–147. doi: 10.1098/rspb.1962.0066. [DOI] [PubMed] [Google Scholar]
- 29.Krohn P L. In: Topics in the Biology of Aging. Krohn P L, editor. New York: Wiley; 1962. pp. 125–148. [Google Scholar]
- 30.Daniel C W, De Ome K B, Young J T, Blair P B, Faulkin L J., Jr Proc Natl Acad Sci USA. 1968;61:53–60. doi: 10.1073/pnas.61.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison D E. Proc Natl Acad Sci USA. 1973;70:3184–3188. doi: 10.1073/pnas.70.11.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison D E. Nat New Biol. 1972;237:220–222. doi: 10.1038/newbio237220a0. [DOI] [PubMed] [Google Scholar]
- 33.Harrison D E. J Gerontol. 1975;30:279–285. doi: 10.1093/geronj/30.3.279. [DOI] [PubMed] [Google Scholar]
- 34.Harrison D E, Doubleday J W. J Immunol. 1975;114:1314–1317. [PubMed] [Google Scholar]
- 35.Price G B, Makinodan T. J Immunol. 1972;108:413–417. [PubMed] [Google Scholar]
- 36.Rosendaal M, Hodgson G S, Bradley T R. Nature (London) 1976;264:68–69. doi: 10.1038/264068a0. [DOI] [PubMed] [Google Scholar]
- 37.Cudkowicz G, Upton A C, Shearer G M, Hughes W L. Nature (London) 1964;201:165–167. doi: 10.1038/201165a0. [DOI] [PubMed] [Google Scholar]
- 38.Siminovitch L, Till J E, McCulloch E A. J Cell Physiol. 1964;64:23–31. doi: 10.1002/jcp.1030640104. [DOI] [PubMed] [Google Scholar]
- 39.Kohn R R. Science. 1975;188:203–204. doi: 10.1126/science.188.4185.203-b. [DOI] [PubMed] [Google Scholar]
- 40.Pignolo R J, Masoro E J, Nichols W W, Brandt C I, Cristofalo V J. Exp Cell Res. 1992;201:16–22. doi: 10.1016/0014-4827(92)90343-7. [DOI] [PubMed] [Google Scholar]
- 41.Bruce S A, Deamond S F. Exp Geront. 1991;26:17–27. doi: 10.1016/0531-5565(91)90058-t. [DOI] [PubMed] [Google Scholar]
- 42.Deamond S F, Bruce S A. Mech Ageing Dev. 1991;60:143–152. doi: 10.1016/0047-6374(91)90127-l. [DOI] [PubMed] [Google Scholar]
- 43.Robbins E, Levine E M, Eagle H. J Exp Med. 1970;131:1211–1222. doi: 10.1084/jem.131.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grassilli E, Bellesia E, Salomoni P, Croce M A, Sikora E, Radziszewska E, Tesco G, Vergelli M, Latorraca S, Babieri D, et al. Biochem Biophys Res Commun. 1996;226:517–523. doi: 10.1006/bbrc.1996.1387. [DOI] [PubMed] [Google Scholar]
- 45.Dimri G P, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano E E, Linskens M, Rubelj I, Pereira-Smith O, et al. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wharton W. Exp Cell Res. 1984;154:310–314. doi: 10.1016/0014-4827(84)90691-8. [DOI] [PubMed] [Google Scholar]
- 47.Slayback J R, Cheung L W, Geyer R P. Exp Cell Res. 1977;110:462–466. doi: 10.1016/0014-4827(77)90313-5. [DOI] [PubMed] [Google Scholar]
- 48.Clemmons D R. J Cell Physiol. 1983;114:61–67. doi: 10.1002/jcp.1041140110. [DOI] [PubMed] [Google Scholar]
- 49.Kondo H, Yonezawa Y. Exp Cell Res. 1995;220:501–504. doi: 10.1006/excr.1995.1342. [DOI] [PubMed] [Google Scholar]
- 50.Bayreuther K, Rodemann H P, Hommel R, Dittamann K, Albiez M, Francz P I. Proc Natl Acad Sci USA. 1988;85:5112–5116. doi: 10.1073/pnas.85.14.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dell’Orco R T, Mertens G B, Kruze P F. Exp Cell Res. 1974;84:363–366. doi: 10.1016/0014-4827(74)90416-9. [DOI] [PubMed] [Google Scholar]
- 52.Goldstein S, Singal D P. Exp Cell Res. 1974;88:359–364. doi: 10.1016/0014-4827(74)90252-3. [DOI] [PubMed] [Google Scholar]
- 53.Cristofalo V J, Palazzo R, Charpentier R. In: Neural Regulatory Mechanisms During Aging. Adelman R C, Roberts J, Baker G T, Baskin S I, Cristofalo V J, editors. New York: Liss; 1980. pp. 203–206. [Google Scholar]
- 54.Smith J R, Whitney R G. Science. 1980;207:82–84. doi: 10.1126/science.7350644. [DOI] [PubMed] [Google Scholar]
- 55.Chen Q, Fisher A, Reagan J D, Yan L J, Ames B N. Proc Natl Acad Sci USA. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venable M E, Lee J Y, Smyth M J, Bielawska A, Obeid L M. J Biol Chem. 1995;270:30701–30708. doi: 10.1074/jbc.270.51.30701. [DOI] [PubMed] [Google Scholar]
- 57.Tresini M, Mawaldewan M, Cristofalo V J, Sell C. Cancer Res. 1998;58:1–4. [PubMed] [Google Scholar]
- 58.Ogryzko V V, Hirai T H, Russanova V R, Barbie D A, Howard B H. Mol Cell Biol. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serrano M, Lin A W, McCurrach M E, Beach D, Lowe S W. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]