

Abstract.
Post-translational modification of chromatin is emerging as an increasingly important regulator of chromosomal processes. In particular, histone lysine and arginine methylation play important roles in regulating transcription, maintaining genomic integrity, and contributing to epigenetic memory. Recently, the use of new approaches to analyse histone methylation, the generation of genetic model systems, and the ability to interrogate genome wide histone modification profiles has aided in defining how histone methylation contributes to these processes. Here we focus on the recent advances in our understanding of the histone methylation system and examine how dynamic histone methylation contributes to normal cellular function in mammals.
Keywords. Chromatin, methylation, demethylation, histone, epigenetics
Introduction
Since histones were first discovered over one hundred years ago a great deal has been learned about the structure and function of these small basic proteins [1]. Importantly, histone molecules interact to form a scaffolding unit capable of compacting DNA within the relatively small confines of the nucleus [2]. Compaction is achieved by wrapping DNA around a globular histone octamer composed of a tetramer of histone H3/H4 and two dimers of H2A/H2B. Together, this DNA-protein structure is called a nucleosome and forms the basic repeated unit of chromatin. This classical view of chromatin as a simple structural entity has recently been superseded by a wide body of evidence demonstrating that nucleosome deposition, subunit composition, and post-translational modification can profoundly affect how chromatin function is regulated. For example, nucleosome placement over defined regulatory elements can impact how transcription factors recognize DNA and regulate transcription [3], incorporation of histone variants into the octamer can define how the nucleosome functions [4], and post-translational modification of histone tails can act as nucleation sites for factors involved in chromatin regulation and metabolism [5, 6].
Over the past decade it has become increasingly clear that post-translational modification of histones plays a central role in regulating information that is stored within DNA. Many of these modifications are found on the N-terminal histone tails that protrude from the globular core of the nucleosome and include: phosphorylation, acetylation, ribosylation, ubiquitylation, and methylation. Of particular interest are the recent advances in our understanding of the histone arginine and lysine methylation systems, which will be discussed in this review. These modifications have been shown to play important roles in maintaining genome integrity, regulating transcription, and contributing to epigenetic memory [7, 8]. Unlike some histone modifications that can affect chromatin function by altering the charge and structural interactions within the nucleosome, histone methylation is a relatively inert modification that appears to function as a nucleation site for ‘effector’ proteins [5]. These effector proteins often elicit their function by recruiting other factors that directly impact chromatin. To regulate post-translational histone modifications, a corresponding class of enzymes has evolved that can directly remove these modifications. For example, a class of histone demethylase enzymes has recently been identified [9] that appear to play central roles in regulating chromatin function.
The emergence of the histone methylation and chromatin modification systems as important regulators of cellular information has garnered much attention and an excellent series of detailed review articles covering this topic have been published [7, 10–13]. Therefore, in this article we will provide a more focussed discussion of the recent advances in our understanding of how histone methylation is placed, interpreted, and dynamically regulated in mammals. We will also highlight a series of recent studies that suggest histone methyltransferase/demethylase systems modify non-histone proteins, many of which have important roles in transcription and cancer.
Histone methylation
Methylation of lysine or arginine residues can occur in several modification states. Lysine residues can house either one (me1), two (me2) or three (me3) methyl moieties on their amine group, whereas arginine residues can carry one (me1) or two (me2) methyl groups on their guanidinyl group. The di-methyl arginine state is further defined by whether the modification exists in the symmetric (me2s) or the asymmetric (me2a) configuration [14]. As will be discussed inmore detail in later sections, these defined modification states can have different and profound implications on the function of chromatin. The most thoroughly studied histone lysine methylation marks are found on H3K4, K9, K27, K36, K79 and H4K20. In general, H3K4, K36, and K79 methylation are found near active or poised transcriptional units and H3K9 and H4K20 modifications are hallmarks of silenced or heterochromatic regions. Histone arginine methylation occurs on H3R2, R8, R17 and R26 and H4R3 and has roles in defining both active and repressed chromatin states. Our understanding of the histone residues that are modified by methylation has largely been achieved by examining the in vitro substrate specificity of histone methyltransferase enzymes and by the generation of antibody reagents that specifically recognize modified lysine and arginine residues. Although simple and robust, these two approaches have clear limitations in that they either rely on the knowledge of the enzyme that places the modification to identify the site or are essentially predictive and rely on the costly approach of modification specific antibodies.
A coming of age for mass spectrometry based histone methylation analysis
In an attempt to identify new histone methylation marks and to study the combinatorial state of histone modifications, a great deal of effort has been invested in refining mass spectrometry based analysis techniques. One of the goals of using mass spectrometry for histone modification analysis is to apply relatively unbiased methodologies in gauging and defining cellular chromatin modification patterns, thus eliminating some of the limitations inherent to enzyme and antibody based approaches. Initially, ‘bottom up’ mass spectrometry approaches were used in which histones are fragmented into small peptides prior to ionization and mass spectrometry analysis (Fig. 1, left hand side). This proved an effective tool in characterizing histone methylation marks, some of which were not previously identified using traditional techniques. For example, bottom up sequencing yielded evidence that H3K79[15–17] and H4R3[18, 19] were methylated and permitted the characterization of the enzymes responsible for placing these modifications. Subsequently, more in depth histone modification analysis has suggested methylation also occurs on: H2BK47 [20], H2BK57 [20], H2BK108 [20], H3K18 [21, 22], H3K23 [21], H3K56 [22], H3K64 [21, 22], H3K122 [21–23], H4K59 [24], H4K31 [20, 21], H4R55 [20], and H4K77 [20]. Although the biological relevance and the histone methyltransferase enzymes that place these additional modifications remain to be fully established, bottom up strategies have proven successful in yielding novel and potentially interesting methylation sites.
Figure 1.

Mass spectrometry based approaches for studying histone modifications. Histones are extracted from tissue or cultured cells and biochemically purified, usually by means of reverse phase high performance chromatography. As the histones elute from the column they are fractionated and individual histones are isolated (i.e. Histone H4 in the schematic). The purified histone is then analysed using bottom up or top down approaches. Bottom up analysis involves enzymatic digestion of the histone prior to ionization, whereas top down analysis relies on ionization the intact histone. Both approaches permit interrogation of histone modifications, but top down approaches have the advantage of retaining information about modifications that occur on the same histone. MS - mass spectrometry.
One clear disadvantage of ‘bottom up’ mass spectrometry approaches is that information about modifications that occur on the same histone tail can be lost during the digestion step prior to ionization and mass analysis. To overcome this limitation ‘top down’ approaches have been pioneered where larger fragments of the histone are directly ionized prior to mass analysis (Fig. 1, right hand side). This was recently employed to analyse the 50 amino acid N-terminal tail fragment of histone H3.2 [25]. The results of this analysis demonstrated that the majority of histone H3.2 modification occurs in the form of H3K9, K36, and K27 methylation without an apparent modification hierarchy between these sites and suggesting they are independently regulated. Interestingly, the same work also revealed that H3K4 methylation, which is generally considered a mark of active chromatin, is predominantly found on hyper-acetylated H3 and is never found together with H3K9me3, a hallmark of repressed chromatin. These molecular observations corroborate many years of cell-based work that had suggested this type of arrangement of histone modifications in active chromatin [26–29]. As advanced techniques in mass spectrometry are more routinely applied to understanding histone methylation, their utility is becoming more obvious and applicable. Recently top down techniques were applied to study H4K20 methylation during the cell cycle [30]. This work demonstrated that newly synthesized H4 is progressively methylated to the me2 modification state with a subset of H4K20 being methylated to the me3 state. Furthermore, this study demonstrated that once in place, H4K20 methylation is not dynamically regulated suggesting its levels are not controlled at the global level by histone demethylase activities. In contrast to previous suggestions that H4K20 methylation and H4K16 acetylation are antagonistic modifications, mass spectrometry analysis demonstrates that they co-exist on the same histone tail and are independently regulated. The utility of the top down mass spectrometry approach has been further highlighted by a recent study that defined the dynamics of histone H4 modification during human embryonic stem cell differentiation [31]. Although in its infancy as a tool for studying histone modification, the use of top down mass spectrometry based approaches to understand histone methylation, the dynamics of these modifications and their combinatorial states has proven a powerful tool. Looking to the future, mass spectrometry approaches will become a common addition to the chromatin biologist repertoire of experimental tools.
Histone methyltransferases
Since the discovery of the first histone lysine methyltransferase in 2000 [32], an extended family of histone lysine methyltransferases have been identified (reviewed in [7, 12]). The majority of these methyltransferases share a SET domain as their catalytic core and have very defined residue and modification state specificity. With the exception of a few lysine methylation sites that are of low abundance in chromatin, the enzymes that place lysine methylation are known and their catalytic activity are characterized. In contrast, histone arginine methylation is placed by the protein arginine N-methyltransferase (PRMT) class of histone methyltransferase enzymes. Although the histone residue specificity of the PRMT enzymes is not as well characterized as the lysine methyltransferase enzymes, these enzymes are typically more promiscuous and often target multiple arginine residues on the N-terminal tails of histone H3 and H4. Furthermore, PRMT enzymes also target a broad range of other cellular proteins suggesting they contribute to regulation of additional non-chromatin based processes [8].
Amore complexmethylation system in mammals with a division of labour
In budding yeast there is a simple histone lysine methylation system with three prominent histone lysine methylation marks that occur on histone H3 in position K4, K36 and K79. The KMT2/SET1(H3K4), KMT3/SET2(H3K36), and KMT4/DOT1(H3K79) methyltransferase enzymes place all three methylation states (me1, me3, me3) on their respective target residues [33]. In stark contrast to budding yeast, mammals have a greatly expanded genomic histone lysine methylation site modification profile, produced by a larger family of enzymes. For example, there are at least three proteins that place H3K36 methylation [34–37]. Among these H3K36 methyltransferases, individual enzymes have evolved to recognize and target defined modification states. This is exemplified in a recent study that demonstrated the human KMT3A(Setd2) enzyme catalyses H3K36me3 and its depletion in cultured cells causes an acute loss of the me3 state but does not affect me2/1 levels [37]. This observation suggests that H3K36me1 and me2 modification states are maintained by additional methyltransferases. Asimilar division of labour was observed when H3K36 methyltransferases were depleted in Drosophila cells [38], suggesting this diversification in modification state specificity is a feature of higher eukaryotes.
This trend of expansion and diversification among the histone lysine methylation system also applies to enzymes that place other histone methylation marks. For example, H3K4 and H3K9 methylation are catalysed by an extended group of methyltransferase enzymes most of which have distinct modification state preferences in higher eukaryotes [7]. Although there are mechanisms in budding yeast to regulate to some extent how individual methylation states are defined [39–42], the sheer increase in complexity of the enzymes that place histone methylation and their unique modification state specificity in mammals suggest a more important role for this modification system in higher eukaryotes. This may represent an evolutionary adaptation that allows increased capacity for information storage through strict placement and recognition of defined modification states by individual methyltransferase enzymes and effector proteins. Perhaps this expansion and specialization of the histone methylation system helps multicellular organisms to cope with more complex requirements for long term chromatin based memory in controlling gene expression patterns and tissue specificity during development.
Histone methyltransferases as pharmacological targets in cancer and disease
With a firmgrasp of the identity ofmany of the enzymes that place histone methylation, a significant amount of effort has been applied to understand how these enzymes are involved in regulating important nuclear processes. Mouse knockout models for enzymes that place H3K4, H3K9, and H3K27methylation have been informative in demonstrating that methyltransferases play important roles in regulating transcription, maintaining genome integrity, and development [36, 43–49]. Furthermore, enzymes involved in placing histone methylation are targets of translocations that produce oncogenic fusion proteins [50, 51]. The realization that histone methyltransferases impact so many important aspects of cellular biology has spawned a huge effort both in the academic and industrial setting to identify small molecule compounds that directly and specifically regulate individual histone methyltransferase enzymes. An initial attempt to identify inhibitory molecules used a library of natural compounds to search formolecules that target the KMT1A(Suv39H1) H3K9me3 methyltransferase enzymes [52]. One of the compounds isolated in this screen, a fungal toxin chaetocin, inhibited KMT1A in addition to a related H3K9me1/2 methyltransferase, KMT1C(G9a). Chaetocin also functionally inhibited H3K9 methylation in cell based assays, but its use as a molecular probe appears limited due to complications with cellular toxicity. Furthermore, from a preclinical standpoint the chemical properties of chaetocin make it a poor lead compound for further inhibitor design.
A more recent study took advantage of a high throughput activity based assay to screen a library of 125 000 chemical compounds for their ability to inhibit the histone H3K9me1/me2 methyltransferase enzyme KMT1C [53]. This screen resulted in seven positive hits based on four distinct molecular scaffolds. Of these initial hits one compound, BIX-01294, was shown to specifically inhibit KMT1C. The functionality of this compound was then tested in cell-based assays and efficiently inhibited KMT1C mediated H3K9me1/me2 methylation at known KMT1C target loci. Furthermore, cells treated with BIX-01294 largely recapitulated global changes in histone H3K9 methylation levels observed in KMT1C deficient cells. Although the IC50 of BIX-01294 is relatively modest at 1.7 µM, this base compound will provide an opportunity for lead optimization and synthesis of more potent methyltransferase inhibitors. Clearly from these initial reports reasonable scope exists for the development of enzyme specific inhibitors of the histone methylation system. Compounds that are nontoxic and cell permeable will clearly provide important molecular probes for studying histone methylation in the laboratory and provide a basis for designing compounds that are useful for pharmacological intervention in human disease where the histone methylation system is perturbed.
Effectors
Unlike acetylation and phosphorylation that alter the charge properties of a modified residue, methylation of lysine and arginine residues is comparatively inert and does not affect residue charge. Instead, histone methylation acts as a nucleation site for effector proteins that elicit functional outcome. For example, the plant homeodomain (PHD) of the bromodomain PHD transcription factor (BPTF) protein binds H3K4me2/me3 (Fig. 2D) and recruits the NURF chromatin-remodelling complex to target genes in concert with transcriptional activation [54, 55]. In contrast, the chromo domain of heterochromatin protein 1 (HP1) proteins recognizes H3K9 (Fig. 2B) methylation and helps to reinforce the repressed state at silenced genes and heterochromatic regions of the genome [56–58]. As we learn more about the specificity and function of methyl-lysine binding effector proteins it is becoming clear that these proteins also have the capacity to preferentially recognize defined modification states. This molecular selectively provides the histone methylation system with the ability to encode additional information within each modified residue depending on the modification state specificity of the methyltransferase placing the modification and the effector protein that recognizes it. Furthermore, several PHD domain containing proteins were recently shown to have the unique ability to recognize histone lysine residues only when they lack methylation [59, 60]. Therefore it appears that methylated residues act as modules capable of encoding information that can be preferentially recognized by site and state specific effector proteins (reviewed in [5, 61]). Although the majority of well studied histone lysine methylation marks have corresponding effector proteins, some of the more recently identified histone lysine modifications occur in the core of the nucleosome and this could sterically limit accessibility by effector proteins. It remains possible that these methylation marks may have effector independent roles in regulating nucleosome structure and function. Nevertheless, over the past year several important structural studies have highlighted unique properties of certain methyl-lysine binding effector proteins (Fig. 2A–C) demonstrating a unique complexity in chromatin recognition by effector proteins.
Figure 2.

Effector proteins form an aromatic cage that recognizes methylated lysine residues (A–E). Cartoon representations corresponding to the three dimensional structure of effector protein methyl-lysine binding domains (top half of each section) with a close up view of the aromatic cage in association with a methylated ligand (bottom half of each section). (A) The KMT1D ankyrin repeat in complex with H3K9me2 (PDB 3b95), repeats 3, 4 and 5 shown only. (B) The chromodomain of heterochromatin protein 1 (HP1) in complex with H3K9me2 (PDB 1kna). (C) The MBT repeats of L3MBTL1 in complex with H4K20me2 (PDB 2pqw), repeat 2 shown only. (D) The PHD finger of bromodomain PHD transcription factor (BPTF) in complex with H3K4me3 (PDB 2f6j), PHD finger shown only. (E) The double tudor domain of KDM4A in complex with H4K20me3 (PDB 2qqs).
One cage, two binding modes
The crystal structures of effector proteins bearing PHD, chromo, and tudor domains have been solved (Fig. 2A–E). A recognition motif consisting of two to four aromatic residues that create a cage capable of accommodating the lysine methyl ammonium group appears to be a common chromatin recognition interface for these domains. The KDM4A (JHDM3A/JMJD2A) histone demethylase enzymes contain a double tudor domain that recognizes either H3K4 or H4K20 when modified in the me2 and me3 modification state [62]. Interestingly, the amino acid sequence surrounding the H3K4 and H4K20 share no significant sequence homology suggesting that modification site specificity is inherent to the tudor domain. Whilst the structural basis for H3K4 binding has been elucidated [62], the molecular mechanism by which the same effector could also bind H4K20 was unknown. This was recently clarified by a study reporting the structure of the KDM4A double tudor domain in complex with an H4K20me3 peptide [63](Fig. 3A). In both structures, the aromatic cage engages the methylated H3K4 and H4K20 residues endowing methylation dependent substrate recognition. In contrast, the bi-functional binding site specificity of the hybrid double tudor domain appears to be due to the usage of two binding interfaces. The H3K4 peptide traverses the intra-domain boundary of the two tudor domains, whereas the H4K20 peptide makes contacts with only one of the tudor domains. Therefore the two peptides bind the effector surface in opposite orientations and engage in different binding interactions (Fig. 3A). Despite the clear specificity of the KDM4A tudor domain for methylated histones it remains to be determined whether this chromatin binding motif is required to target histone demethylase activity to genomic loci that also contain methylated histones.
Figure 3.

Unique substrate recognition properties of two methyl-lysine recognition domains. (A) Space filling representation of the three dimensional structure of the KDM4A tandem-tudor domain in complex with H3K4me3 (magenta) and H4K20me3 (yellow) histone substrates. The N- and C- terminus of the of histone peptides are labelled and the lysine side chain is depicted projecting into the aromatic methyl-lysine recognition cage (cyan). The two unique binding faces of KDM4a appear to permit the dual substrate recognition properties of this methyl lysine binding effector protein. (B) Space filling representation of the RAG2-PHD domain in association with an H3 peptide (green) containing H3K4me3 and H3R2me2s modifications. The methyl-lysine binding aromatic cage is coloured cyan and the Tyr445 residue that interacts with the symmetric dimethyl-arginine is coloured magenta.
RAG2 PHD — a dual-mark reader?
The proximity of lysines and arginines within the histone tails suggest there may be synergistic or antagonistic cross-talk between adjacent modifications. Two recent studies have addressed the relationship between H3K4 trimethylation, a transcription activation mark, and H3R2 methylation. Using in vitro peptide pull-downs, it was demonstrated that H3R2 methylation abrogates H3K4me3 recognition by many known effector proteins including the double tudor domain of KDM4A and the PHD domains of tumour suppressor protein ING2 (inhibitor of growth 2) and BPTF. In contrast, the PHD domain of recombination activation gene RAG2, a component of the VDJ recombinase, binds more efficiently to H3K4me3 when the adjacent H3R2 residue is dimethylated [64, 65]. The structure of RAG2-PHD bound to a dual-modified H3R2me2/H3K4me3 peptide revealed two significant differences when compared to other H3K4me3 binding PHD domains (Fig. 3B). Firstly, the H3K4me3 binding site in RAG2 does not form the typical aromatic cage, but instead has a channel lined by two aromatic residues. Secondly, and more interestingly, RAG2 lacks an acidic residue that forms salt bridges with the unmodified H3R2 residue in other H3K4me3 recognition domains. In the RAG2 PHD domain this acidic residue is replaced by a tyrosine that favours an interaction with symmetrically di-methylated H3R2. Therefore, the RAG2 PHD domain can uniquely bind H3K4me3 in the context of H3R2me2s and may represent the first example of a dual-mark reader.
Ankyrin and MBT repeats — lessons for recognizing lower methylation states
The ankyrin repeats of histone methyltransferases KMT1C and KMT1C-like protein (KMT1D) preferentially bind H3K9me1/me2 and hence represent the newest addition to the already diverse class of methyllysine effectors [66]. The structure of KMT1D bound to an H3K9me2 peptide demonstrates that the ankyrin repeats recognize H3K9me2 using a partially aromatic pocket lined by tryptophans on three faces (Trp839, 844, 877) and an acidic residue on the fourth face (Glu847) (Fig. 2A). Importantly, the acidic residue forms a salt bridge with the methylammonium group sterically constricting the cavity and preventing recognition of the me3 methylation state. The biological implications for the presence of domains in KMT1C and KMT1D which recognize the same modification that their enzymatic domain catalyses is interesting and merits further study, but such an arrangement could represent a mechanism for spreading of H3K9me1/2 from nucleation sites in silenced chromatin.
The KMT1C and KMT1D ankyrin repeats are not alone in preferentially recognizing lower lysine methylation states. Other examples include the 53BP1 tandem tudor domain that selectively recognizes H4K20me1 and me2 [67] and the lethal (3) malignant brain tumour-like protein 1 (L3MBTL1), which binds various me1 and me2 modifications [68, 69](Fig. 2C). Structures of these two effector molecules in complex with methyl-lysine ligands reveal a striking similarity with KMT1C/D ankyrin repeats in the arrangement of the binding pocket. Methyl-lysine recognition occurs in a constricted pocket with three to four aromatic residues and a critical acidic residue. The acidic residue again interacts with the methyl ammonium group precluding recognition of the me3 modification state suggesting this is the determining factor in constricting this group of effectors to the me1 and me2 methylation states. In support of this possibility, if a tyrosine residue in the aromatic cage of the BPTF PHD finger is substituted with glutamic acid, the PHD domain is converted into a me2 recognition domain from its usual preference for the me3 modification state.
WD40 repeats
The WD40 repeat of the WDR5 protein was originally identified as a potential binding domain for methylated H3K4. Subsequently, several structural studies have revealed that histone binding by WDR5 occurs through several peptide side-chain contacts to the WD40 domain without creating a specific hydrophobic or aromatic cage as described for the other methylated lysine binding modules [70–73]. Instead, in the case of WDR5, the methylated lysyl side-chain might be exposed to the solvent, thus allowing further modification by histone methyltransferases [72].
Histone Demethylases
Despite the fact that global histone methylation turnover is relatively low [74, 75] it has recently been demonstrated that histone lysine methylation can be dynamically regulated by histone demethylases [76, 77]. These enzymes are of two general classes. The fist class of enzymes are amine oxidase enzymes which are typified by the mammalian lysine specific demethylase 1 (KDM1/LSD1) which uses FAD as a co-factor and targets removal of the me1 and me2 modification states (Fig. 4A, top)[76]. The second class of enzymes belongs to a large family of proteins that contain a Jumonji-C (JmjC) domain as their catalytic core. The JmjC domain-containing proteins are iron and alpha-ketoglutarate dependent oxygenases that target removal of all three histone lysine methylation states (Fig. 4A, bottom). The structure of KDM1 [78–80] (Fig. 4A, top) and KDM4A [81–84](Fig. 4A, bottom) histone demethylases have been solved and provided important insight into the substrate recognition and regulatory properties of these proteins. Interestingly, the crystal structures of peptide substrate complexes from both types of demethylases reveal that the active sites are predominantly polar as opposed to the largely hydrophobic recognition sites described above for the effector domains [81, 85]. Not surprisingly, the identification and characterization of histone lysine demethylases has demonstrated that these enzymes regulate many of the cellular processes that histone methylation has been implicated in including transcriptional regulation, maintenance of genome integrity, and regulation of epigenetic memory [9, 86].
Figure 4.

Proposed lysine and arginine demethylase reaction mechanisms. (A) A schematic indicating potential lysine demethylation reaction mechanisms. (Top) Mono-methyl lysine demethylation catalyzed by JmjC domain-containing proteins using 2OG and Fe(II) as cofactors. The reaction produces succinate, CO2, and formaldehyde as by-products. A ribbon structure of the catalytic domain of the KDM4A demethylase enzymes is depicted to the left (PDB 2oq6). (Bottom) The amine oxidase histone demethylase, KDM1, uses FAD as cofactor to demethylate mono-methyl lysine. The reaction produces H2O2, FADH2, and formaldehyde as by products. The ribbon structure of the KDM1 catalytic domain is depicted to the left (PDB 2v1d). (B) A schematic indicating potential arginine deimination and demethylation reaction mechanisms. (Top) Deimination of mono-methyl arginine catalysed by the peptidylarginine PADI4 enzyme. The ribbon structure of the PADI4 enzyme is depicted to the right (PDB 2dex). This reaction antagonizes histone arginine methylation by converting it to citrulline. (Bottom) Demethylation of mono-methyl arginine by the JmjC domain-containing protein JMJD6. The atomic structure of JMJD6 remains to be solved.
Identification of a histone arginine demethylase
The JmjC family of proteins have been extensively analysed for enzymatic activities that target histone lysine methylation(reviewed in [9, 87–89]). Recently, a member of this family of proteins, JMJD6, was shown to apparently reverse histone arginine methylation [90]. In contrast to previously identified histone deiminase enzymes that convert methyl arginine to citrulline and simply antagonize arginine methylation [91, 92] (Fig. 4B, top), JMJD6 utilizes a hydroxylation based reaction to directly reverse arginine methylation. JMJD6 appears to remove both H3R3 and H4R3 methylation and targets both the me1 and me2 modification states. This important discovery suggests that both arginine and histone methylation can be dynamically regulated, but how JMJD6 contributes to regulation of arginine methylation dependent process in vivo remains to be examined. Interestingly, mass spectrometry analysis of JMJD6 reacted histone substrate revealed that JMJD6 also catalyzes oxidation of several non-modified lysine residues. The biological relevance of this additional activity remains elusive and warrants further examination.
The by-product of histone demethylation mediates DNA damage dependent gene activation
Histone demethylation can produce reactive by-products including formaldehyde and H2O2 that may be damaging to chromatin and nuclear proteins. Currently, it remains unclear if these by-products are further metabolized to make them less harmful to the cell or whether they have adverse affects in the nucleus due to their reactive nature. In addressing this question, a recent study has revealed that H2O2 produced as a by-product during KDM1 mediated demethylation at estrogen receptor (ER) target genes results in production of 8-oxo-guanine lesions [93] (Fig. 5A–B). This DNA damage event resulted in the mobilization of the 8-oxo-guanine DNA glycosylase-1 (OGG1) and topoisomerase IIb repair enzymes to the regulatory regions of the gene (Fig. 5C). Surprisingly, this DNA damage event appears to be required for efficient transcription of the ER target genes. The authors propose that single stranded breaks induced during the DNA repair process may help to facilitate DNA bending permitting more efficient RNApolII loading onto the promoter during gene activation (Fig. 5D). Interestingly, a recent report has also implicated a gylcosylase dependent process in mediating removal of methylated DNA bases during the same estrogen receptor mediated gene activation event [94, 95]. Clearly, DNA breaks induced by this system could also result in similar transcriptional outcomes as are proposed for an H2O2 damage dependent mechanism. In light of these two independent mechanisms for inducing single stranded DNA breaks during ER induced gene activation, it remains to be determined what the relative contribution of each pathway is to transcriptional activation. Nevertheless, it appears that in some instances the reactive by-products of the demethylation process may contribute in an unexpected way to chromatin regulation. Given that formaldehyde is produced during JmjC-mediated demethylation reactions and can cause adverse DNA and protein adducts, it is likely that a formaldehyde scavenging system is used to inactivate this reactive by-product. A possible candidate for this type of reaction is the class III alcohol dehydrogenase that uses GSH and NAD to inactivate formaldehyde [96, 97]. Since this enzyme is also found in the nucleus [98], it will be important to determine if this system is targeted to genomic sites undergoing histone demethylation to inactivate formaldehyde.
Figure 5.

Hydrogen peroxide produced by LSD1 during demethylation contributes to transcriptional activation. (A) During transcriptional activation of estrogen receptor target genes, KDM1 removes repressive H3K9me2 marks. (B) A by-product of this demethylation reaction is hydrogen peroxide (H2O2) which is reactive and can cause 8-oxo-guanine (8-OG) lesions on DNA. (C) LSD1 mediated 8-OG is targeted for removal by the 8-oxo-guanine DNA glycosylase-1 (OGG1) and perhaps other components of the base excision repair system. This repair process leads to single stranded DNA nicks that are a substrate for topoisomersase IIb (TOPO). Recruitment of topoisomerase IIb can lead to alterations in DNA architecture. (D) Changes in DNA architecture may aid in RNApolII loading onto target genes by promoting chromatin accessibility or DNA bending and therefore contribute to transcriptional activation. ER — estrogen receptor.
Histone demethylases play important roles in germ cell development and embryonic stem cell function
Although only recently JmjC domain-containing histone demethylases were identified, there has been a rapid advancement of our understanding of the function of this large family of enzymes through the use of gene knockdown and knockout strategies [99–112]. A recent study has determined that two histone demethylase enzymes, KDM3A (JMJD1A) and KMD4C (JHDM3C/JMJD2C), are direct transcriptional targets of the pluripotency promoting transcription factor Oct-4[113]. Based on the central role of Oct-4 in regulating embryonic stem cell (ES) pluripotency, it was proposed that regulation of these demethylase enzymes by Oct-4 may contribute to downstream maintenance of stemness through histone demethylation. To test this hypothesis KDM3A (an H3K9me2 demethylase) and KDM4C (an H3K9/36me3 demethylase) enzymes were knocked-down in mouse ES cells using RNAi mediated approaches. Interestingly, when these enzymes were depleted, mouse ES cells lost their characteristic morphology and self renewal was inhibited. Loss of ES cell characteristics appeared to correlate with reduction in H3K9me levels and changes in gene expression. For example, KDM3A knockdown caused reduced expression of the Tcl-1 gene, a known regulator of self renewal in ES cells, and the promoter region of the gene displayed increased levels of H3K9me2. Furthermore, knockdown studies revealed that KDM4C binds to the Nanog gene and potentiates gene expression while counteracting H3K9me3 and HP1 effector protein binding. Together, these observations suggest a central role for H3K9 demethylation in maintaining stem cell function. Surprisingly, it has subsequently been demonstrated that KDM3A hypomorphic and knockout mice are viable and grossly normal with the exception thatmalemice are infertile [114]. This suggests that although KDM3A is important for ES cell pluripotency in cell culture, it is not essential for normal mouse development. Nevertheless, in KDM3A deficient mice male sterility appears to be caused by a lack of expression of genes central to the histone to protamine transition during sperm development. Loss of normal gene expression correlated with increases in H3K9me at promoter regions suggesting that histone demethylation contributes to regulated gene expression during germ cell development in males. Given the complex gene expression patterns of demethylase enzymes in mammals, it will be important to examine in more detail how developmental and tissue specific regulation of these interesting enzymes contributes to normal epigenetic regulation of gene expression.
Reversible methylation of non-histone proteins
Although studies on protein methylation have most recently focussed on lysyl- and arginyl methylation of histones, it should be emphasized that methylation of non-histone proteins also occurs to a significant extent (reviewed in [115]). Many important proteins have been shown to be subject to lysine methylation, including the tumour suppressor protein p53, a kinetochore protein (DAM1)[116], the retinoic acid receptor alpha [117], components of the transcriptional machinery such as TAF10 [118], as well as chloroplast (Rubisco)[119] and mitochondrial proteins (cytochrome c)[120]. Furthermore, many proteins are arginine methylated, including RNA-binding proteins, splicing factors, coactivator p300/CBP, and DNA polymerase β [8]. Given the importance of histone methylation in regulation of important chromatin based processes, it seems likely that protein methylation in general has important regulatory functions and is subject to regulation by demethylase enzymes.
Dynamic and modification state specific methylation of p53
Lysine methylation has recently been identified on the tumor suppressor p53 at three particular sites in the Cterminal domain [45, 121–124](Fig. 6). Methylation of p53 by the KMT7(SET7/9) methyltransferase enzyme on Lys372 in the C-terminal region of the protein following DNA damage results in p53 stabilization and trans-activation of p53 target genes like p21 [124]. Conversely, mono-methylation of Lys370 by KMT3C(Smyd2) antagonizes association of p53 with target genes and therefore inhibits p53 mediated trans-activation [121]. Interestingly, when p53 is dimethylated on Lys370 this creates a binding site for the tudor domain containing co-activator protein 53BP1 and permits activation of p53 target genes. These observations suggest that differing methylation states on the same residue of p53 can have diverse functional outcomes. In analogy to histone methylation, it was recently demonstrated that p53 Lys370me2 methylation can be dynamically regulated by the LSD1 histone demethylase enzyme; removal of the Lys370me2 inhibited association of p53 with 53BP1 and repressed p53 function [125]. To further the analogy to histone modification, the same p53 lysine residues that are methylated are also subject to other modifications like acetylation, sumoylation and ubiquitylation [126]. This suggests that cross talk or interference between different protein modification pathways can also occur in the post-translational modification of p53.
Figure 6.
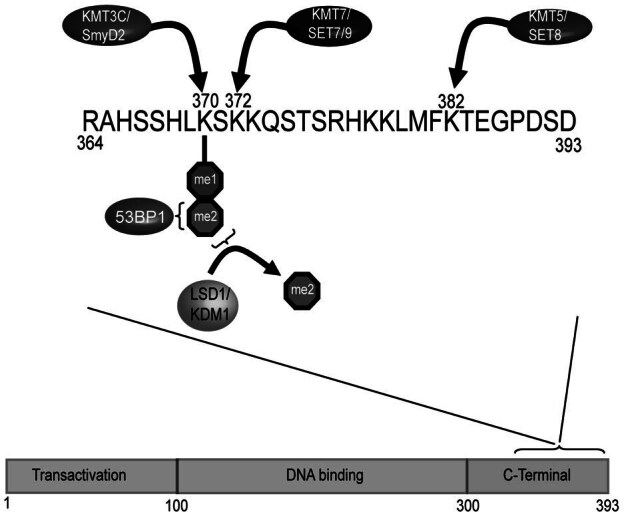
p53 is regulated by protein methylation and demethylation. The p53 protein is made up of an N-terminal transcriptional activation domain (TAD), a central DNA binding domain (DBD), and a C-terminal domain (CTD). Methylation of the CTD on Lys372 by SET7/9 occurs following DNA damage and stabilizes p53 leading to transactivation of p53 target genes. Conversely, mono-methylation of Lys370 by KMT3C/Smyd2 antagonizes binding of p53 to target genes and inhibits p53 mediated transactivation. Surprisingly, di-methylation of the same Lys370 residue creates a binding site for 53BP1 leading to transactivation of p53 target genes. Di-methylation of p53 is enzymatically reversed by the LSD1 histone demethylase indicating that this post-translational modification and its effects on p53 function are regulated. Recent evidence also indicates the KMT5/Set8 enzyme methylates Lys382 in the CTD, indicating that p53 is even more broadly regulated by lysine methylation than previously realized.
A complicated and dynamic pattern of histone methylation
Although we now know many of the enzymes that place and remove histone methylation, it is not completely clear how the action of these enzymatic activities translates into genomic histone methylation profiles in mammals. In many ways this understanding has been limited by the technology used to interrogate histone methylation profiles. Chromatin immunoprecipitation (ChIP) is the most widely used technique to define histone methylation profiles. ChIP relies on crosslinking histones to DNA followed by immunoprecipitation using antibodies that recognize specific methylation marks. This DNA is then analysed using quantitative PCR or by hybridization to microarrays to define the relative enrichment of a modification at a given locus. The relatively small size of the yeast genome has made array hybridization techniques feasible as well as cost effective to obtain genome wide profiles of histone methylation in this organism [127, 128]. In mammalian systems, this type of approach has been limited due to costs associated with arrays that interrogate the entire genome. Recent advances in massively parallel sequencing technologies have provided a cost-effective alternative to analysing ChIP DNA (ChIP-Seq) and provided the first comprehensive glance at the genome wide histone methylation profiles [129, 130].
‘ChIPing away’ at histone methylation profiles in mammals
Several important revelations regarding the methylation state of histone in mammals have arisen from studies using ChIP-array and ChIP-seq technologies. Firstly, histone methylation marks appear in a highly co-ordinated fashion over genic and non-genic regions. Although some of these profiles were inferred from studies analysing isolated genomic regions in mammals and histone methylation in model organisms like budding yeast, the genome-wide view of mammalian histone methylation indicates that there are very specific modification patterns that share uniformity over similar genomic elements. For example, H3K4me3 methylation is a hallmark of regulatory elements at the 5′ end of transcriptionally active genes or of genes poised for transcriptional activation, whereas H3K36me3 methylation is largely restricted to the body and 3′ end of the gene [129–131]. These observations are in close agreement with the targeting properties of the enzymes that place these modifications. For example, it is known that H3K4 methyltransferases are associated with transcriptional regulators that recognize promoters, whereas enzymes that place H3K36 methylation can associate with the elongating form of RNA pol II over the body of genes [132]. Since the profiles of H3K4me3 and H3K36me3 appear to be highly correlated with the initiation and elongation of transcription, these landmarks appear to provide a general tool in identifying novel transcriptional units. In particular microRNA and non-coding RNAs may be rapidly processed from longer precursors and not readily detectable at the mRNA level, but histone modifications over their regulatory and genic regions highlight their existence on chromatin [129, 130]. In non-genic regions histone H3K9me3 and H4K20me3 silencing marks are associated with repetitive or transposable DNA elements including satellite sequences and long terminal repeats. It is thought that these marks may be placed in response to the production of double stranded RNAs through mobilization of the RNA interference based transcriptional silencing pathway as has been reported for H3K9 methylation in other organisms.
A second important observation regarding mammalian histone methylation profiles is the existence of a specific bivalent modification pattern containing H3K4me3 and H3K27me3 at the regulatory elements of certain genes in ES cells [129, 133–135]. This observation was initially very puzzling given that H3K4me3 methylation was known to be involved in transcriptional activation whereas the H3K27me3 modification is a part of the polycomb mediated silencing system [136–138]. It turns out that these bivalent domains are largely found at genes with more complex expression patterns including those key developmental transcription factors [129, 135]. In ES cells, genes with bivalent domains are largely silenced or expressed at low levels, but during differentiation into defined cell lineages their bivalent modification profile is often resolved to contain either H3K4me3 methylation in concert with transcriptional activation, or to contain H3K27me3 and remain strongly silenced. Interestingly, the recently discovered KDM6 class of histone demethylase enzymes, that remove the H3K27me3, were found to be part of an H3K4 methyltransferase complex [103–105, 110, 139, 140]. This suggests that bivalent domains may be resolved by reinforcing the H3K4 methylation state while actively removing the H3K27me3 modification. Based on these observations, bivalent modifications in ES cells appear to play an important role in maintaining genes in a poised epigenetic state from which they can be resolved to either the activated or permanently silenced state following cellular differentiation.
Although histone arginine methylation is known to play important roles in transcription regulation, the genomic profile in mammalian chromatin is poorly understood. In a recent study the profiles of H3R2me2a at a cohort of genes was analysed by ChIP and quantitative PCR [141]. This study revealed that H3R2me2a is generally found over silenced genes and inhibits recruitment of H3K4 methyltransferase enzymes by blocking effector protein recognition of the adjacent H3K4 methylation site. Conversely, H3K4 methylation at active genes appears to block PRMT6 from modifying H3R2me2a [141, 142]. This interesting observation suggests that there is a specific interplay between methylation at H3R2 and H3K4, with a unique interdependence between histone lysine and arginine methylation. A future challenge will be to profile histone arginine methylation on a genomewide scale and to understand the functional interplay between these modifications and other covalent histone modifications.
Conclusions and future directions
The importance of the histone methylation system in regulating chromatin based processes is becoming increasingly clear as we understand more about the enzymes which place methylation, the proteins which interpret it, and the enzymes which counteract this modification. A clear challenge for the future is to understand and integrate how additional modifications on the same histone, within the same nucleosome, and in contiguous chromatin domains interact to define local and global epigenetic histone modification patterns. In part this will rely on mass spectrometry based approaches to interrogate combinatorial modifications on individual histones and comprehensive genome location analysis of histone modifications using ChIP-array or ChIP-seq technologies to understand genomic modification patterns. Armed with a more defined understanding of the histone modification pattern, genetic manipulation of factors involved in defining these arrangements will help to elucidate how modifications like histone lysine and arginine methylation dictate or respond to epigenetic states involved in transcription and DNA repair. A more holistic and integrated view of the histone modification system will be essential in defining how these processes are perturbed and exploited in human disease and provide potentially novel avenues for pharmacological intervention. Clearly, with the pace that the histone methylation and chromatin modification fields are advancing and the new technologies being applied to their study, more secrets of this important epigenetic modification system will soon to be revealed.
Acknowledgment
We would like to thank Dr. Emma Klose for critical reading of the manuscript. The Structural Genomics Consortium is a registered charity (number 1097737) and receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck and Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. Work in R.J.K’s lab is supported by the Wellcome Trust.
Footnotes
Received 29 May 2008; received after revision 04 September; accepted 09 September 2008
Contributor Information
U. Oppermann, FAX: + 44 1865 617575, Email: udo.oppermann@sgc.ox.ac.uk
R. J. Klose, Email: rob.klose@bioch.ox.ac.uk
References
- 1.Kossel A. Ueber einen peptoartigen bestandheil des zellkerns. Z. Physiol. Chem. 1884;8:511–515. [Google Scholar]
- 2.Kornberg R.D., Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 3.Agalioti T., Lomvardas S., Parekh B., Yie J., Maniatis T., Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarthy S., Park Y.J., Chodaparambil J., Edayathumangalam R.S., Luger K. Structure and dynamic properties of nucleosome core particles. FEBS Lett. 2005;579:895–898. doi: 10.1016/j.febslet.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Taverna S.D., Li H., Ruthenburg A.J., Allis C.D., Patel D.J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Martin C., Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell. Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 8.Bedford M.T. Arginine methylation at a glance. J. Cell. Sci. 2007;120:4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- 9.Klose R.J., Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell. Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 10.Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein B.E., Meissner A., Lander E.S. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 12.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 14.Turner B.M. Reading signals on the nucleosomewith a new nomenclature for modified histones. Nat. Struct. Mol. Biol. 2005;12:110–112. doi: 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
- 15.Feng Q., Wang H., Ng H.H., Erdjument-Bromage H., Tempst P., Struhl K., Zhang Y. Methylation of H3-lysine 79 is mediated by a new family ofHMTases without a SET domain. Curr. Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 16.Ng H.H., Feng Q., Wang H., Erdjument-Bromage H., Tempst P., Zhang Y., Struhl K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Leeuwen F., Gafken P.R., Gottschling D.E. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 18.Strahl B.D., Briggs S.D., Brame C.J., Caldwell J.A., Koh S.S., Ma H., Cook R.G., Shabanowitz J., Hunt D.F., Stallcup M.R., Allis C.D. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 19.Wang H., Huang Z.Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B.D., Briggs S.D., Allis C.D., Wong J., Tempst P., Zhang Y. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 20.Beck H.C., Nielsen E.C., Matthiesen R., Jensen L.H., Sehested M., Finn P., Grauslund M., Hansen A.M., Jensen O.N. Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics. 2006;5:1314–1325. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Garcia B.A., Hake S.B., Diaz R.L., Kauer M., Morris S.A., Recht J., Shabanowitz J., Mishra N., Strahl B.D., Allis C.D., Hunt D.F. Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 22.Peters A.H., Kubicek S., Mechtler K., O’Sullivan R.J., Derijck A.A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., Martens J.H., Jenuwein T. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 23.Cocklin R.R., Wang M. Identification of methylation and acetylation sites on mouse histone H3 using matrixassisted laser desorption/ionization time-of-flight and nanoelectrospray ionization tandem mass spectrometry. J. Protein. Chem. 2003;22:327–334. doi: 10.1023/a:1025334006014. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L., Eugeni E.E., Parthun M.R., Freitas M.A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 2003;112:77–86. doi: 10.1007/s00412-003-0244-6. [DOI] [PubMed] [Google Scholar]
- 25.Garcia B.A., Pesavento J.J., Mizzen C.A., Kelleher N.L. Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 26.Hazzalin C.A., Mahadevan L.C. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:e393. doi: 10.1371/journal.pbio.0030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strahl B.D., Ohba R., Cook R.G., Allis C.D. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei inTetrahymena. Proc. Natl. Acad. Sci. USA. 1999;96:14967–14972. doi: 10.1073/pnas.96.26.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang G., Lin J.C., Wei V., Yoo C., Cheng J.C., Nguyen C.T., Weisenberger D.J., Egger G., Takai D., Gonzales F.A., Jones P.A. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. USA. 2004;101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nightingale K.P., Gendreizig S., White D.A., Bradbury C., Hollfelder F., Turner B.M. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J. Biol. Chem. 2007;282:4408–4416. doi: 10.1074/jbc.M606773200. [DOI] [PubMed] [Google Scholar]
- 30.Pesavento J.J., Yang H., Kelleher N.L., Mizzen C.A. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 2008;28:468–486. doi: 10.1128/MCB.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phanstiel D., Brumbaugh J., Berggren W.T., Conard K., Feng X., Levenstein M.E., McAlister G.C., Thomson J.A., Coon J.J. Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2008;105:4093–4098. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rea S., Eisenhaber F., O’Carroll D., Strahl B.D., Sun Z.W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D., Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 33.Allis C.D., Berger S.L., Cote J., Dent S., Jenuwien T., Kouzarides T., Pillus L., Reinberg D., Shi Y., Shiekhattar R., Shilatifard A., Workman J., Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka Y., Katagiri Z., Kawahashi K., Kioussis D., Kitajima S. Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene. 2007;397:161–168. doi: 10.1016/j.gene.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 35.Sun X.J., Wei J., Wu X.Y., Hu M., Wang L., Wang H.H., Zhang Q.H., Chen S.J., Huang Q.H., Chen Z. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem. 2005;280:35261–35271. doi: 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- 36.Rayasam G.V., Wendling O., Angrand P.O., Mark M., Niederreither K., Song L., Lerouge T., Hager G.L., Chambon P., Losson R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. Embo J. 2003;22:3153–3163. doi: 10.1093/emboj/cdg288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edmunds J.W., Mahadevan L.C., Clayton A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. Embo J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bell O., Wirbelauer C., Hild M., Scharf A.N., Schwaiger M., MacAlpine D.M., Zilbermann F., van Leeuwen F., Bell S.P., Imhof A., Garza D., Peters A.H., Schubeler D. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. Embo J. 2007;26:4974–4984. doi: 10.1038/sj.emboj.7601926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shahbazian M.D., Zhang K., Grunstein M. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol. Cell. 2005;19:271–277. doi: 10.1016/j.molcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 40.Laribee R.N., Krogan N.J., Xiao T., Shibata Y., Hughes T.R., Greenblatt J.F., Strahl B.D. BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr. Biol. 2005;15:1487–1493. doi: 10.1016/j.cub.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 41.Lee J.S., Shukla A., Schneider J., Swanson S.K., Washburn M.P., Florens L., Bhaumik S.R., Shilatifard A. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 42.Kirmizis A., Santos-Rosa H., Penkett C.J., Singer M.A., Vermeulen M., Mann M., Bahler J., Green R.D., Kouzarides T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007;449:928–932. doi: 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dodge J.E., Kang Y.K., Beppu H., Lei H., Li E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 2004;24:2478–2486. doi: 10.1128/MCB.24.6.2478-2486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayashi K., Yoshida K., Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–378. doi: 10.1038/nature04112. [DOI] [PubMed] [Google Scholar]
- 45.Kurash J.K., Lei H., Shen Q., Marston W.L., Granda B.W., Fan H., Wall D., Li E., Gaudet F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 46.O’Carroll D., Erhardt S., Pagani M., Barton S.C., Surani M.A., Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 2001;21:4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters A.H., O’Carroll D., Scherthan H., Mechtler K., Sauer S., Schofer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A., Opravil S., Doyle M., Sibilia M., Jenuwein T. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 48.Steele-Perkins G., Fang W., Yang X.H., Van Gele M., Carling T., Gu J., Buyse I.M., Fletcher J.A., Liu J., Bronson R., Chadwick R.B., de la Chapelle A., Zhang X., Speleman F., Huang S. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes. Dev. 2001;15:2250–2262. doi: 10.1101/gad.870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu B.D., Hess J.L., Horning S.E., Brown G.A., Korsmeyer S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 50.Jaju R.J., Fidler C., Haas O.A., Strickson A.J., Watkins F., Clark K., Cross N.C., Cheng J.F., Aplan P.D., Kearney L., Boultwood J., Wainscoat J.S. Anovel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001;98:1264–1267. doi: 10.1182/blood.v98.4.1264. [DOI] [PubMed] [Google Scholar]
- 51.Ayton P.M., Cleary M.L. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 52.Greiner D., Bonaldi T., Eskeland R., Roemer E., Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat. Chem. Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 53.Kubicek S., O’Sullivan R.J., August E.M., Hickey E.R., Zhang Q., Teodoro M.L., Rea S., Mechtler K., Kowalski J.A., Homon C.A., Kelly T.A., Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 54.Wysocka J., Swigut T., Xiao H., Milne T.A., Kwon S.Y., Landry J., Kauer M., Tackett A.J., Chait B.T., Badenhorst P., Wu C., Allis C.D. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 55.Li H., Ilin S., Wang W., Duncan E.M., Wysocka J., Allis C.D., Patel D.J. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bannister A.J., Zegerman P., Partridge J.F., Miska E.A., Thomas J.O., Allshire R.C., Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 57.Lachner M., O’Carroll D., Rea S., Mechtler K., Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 58.Nakayama J., Rice J.C., Strahl B.D., Allis C.D., Grewal S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 59.Lan F., Collins R.E., De Cegli R., Alpatov R., Horton J.R., Shi X., Gozani O., Cheng X., Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ooi S.K., Qiu C., Bernstein E., Li K., Jia D., Yang Z., Erdjument-Bromage H., Tempst P., Lin S.P., Allis C.D., Cheng X., Bestor T.H. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruthenburg A.J., Li H., Patel D.J., Allis C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell. Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Y., Fang J., Bedford M.T., Zhang Y., Xu R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 63.Lee J., Thompson J.R., Botuyan M.V., Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 2008;15:109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matthews A.G., Kuo A.J., Ramon-Maiques S., Han S., Champagne K.S., Ivanov D., Gallardo M., Carney D., Cheung P., Ciccone D.N., Walter K.L., Utz P.J., Shi Y., Kutateladze T.G., Yang W., Gozani O., Oettinger M.A. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramon-Maiques S., Kuo A.J., Carney D., Matthews A.G., Oettinger M.A., Gozani O., Yang W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc. Natl. Acad. Sci. USA. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collins R.E., Northrop J.P., Horton J.R., Lee D.Y., Zhang X., Stallcup M.R., Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008;15:245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Botuyan M.V., Lee J., Ward I.M., Kim J.E., Thompson J.R., Chen J., Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trojer P., Li G., Sims R.J., 3rd, Vaquero A., Kalakonda N., Boccuni P., Lee D., Erdjument-Bromage H., Tempst P., Nimer S.D., Wang Y.H., Reinberg D. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 69.Min J., Allali-Hassani A., Nady N., Qi C., Ouyang H., Liu Y., MacKenzie F., Vedadi M., Arrowsmith C.H. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007;14:1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 70.Couture J.F., Collazo E., Trievel R.C. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat. Struct. Mol. Biol. 2006;13:698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 71.Han Z., Guo L., Wang H., Shen Y., Deng X.W., Chai J. Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol. Cell. 2006;22:137–144. doi: 10.1016/j.molcel.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 72.Ruthenburg A.J., Wang W., Graybosch D.M., Li H., Allis C.D., Patel D.J., Verdine G.L. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 2006;13:704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schuetz A., Allali-Hassani A., Martin F., Loppnau P., Vedadi M., Bochkarev A., Plotnikov A.N., Arrowsmith C.H., Min J. Structural basis for molecular recognition and presentation of histone H3 by WDR5. Embo J. 2006;25:4245–4252. doi: 10.1038/sj.emboj.7601316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Byvoet P., Shepherd G.R., Hardin J.M., Noland B.J. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 1972;148:558–567. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- 75.Duerre J.A., Lee C.T. In vivo methylation and turnover of rat brain histones. J. Neurochem. 1974;23:541–547. doi: 10.1111/j.1471-4159.1974.tb06057.x. [DOI] [PubMed] [Google Scholar]
- 76.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.A., Casero R.A., Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 77.Tsukada Y., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 78.Stavropoulos P., Blobel G., Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat Struct Mol Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 79.Chen Y., Yang Y., Wang F., Wan K., Yamane K., Zhang Y., Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc. Natl. Acad. Sci. USA. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang M., Gocke C.B., Luo X., Borek D., Tomchick D.R., Machius M., Otwinowski Z., Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 81.Ng S.S., Kavanagh K.L., McDonough M.A., Butler D., Pilka E.S., Lienard B.M., Bray J.E., Savitsky P., Gileadi O., von Delft F., Rose N.R., Offer J., Scheinost J.C., Borowski T., Sundstrom M., Schofield C.J., Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 82.Couture J.F., Collazo E., Ortiz-Tello P.A., Brunzelle J.S., Trievel R.C. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat.Struct.Mol.Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 83.Chen Z., Zang J., Kappler J., Hong X., Crawford F., Wang Q., Lan F., Jiang C., Whetstine J., Dai S., Hansen K., Shi Y., Zhang G. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc.Natl.Acad.Sci.USA. 2007;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen Z., Zang J., Whetstine J., Hong X., Davrazou F., Kutateladze T.G., Simpson M., Mao Q., Pan C.H., Dai S., Hagman J., Hansen K., Shi Y., Zhang G. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 85.Yang M., Culhane J.C., Szewczuk L.M., Gocke C.B., Brautigam C.A., Tomchick D.R., Machius M., Cole P.A., Yu H. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat.Struct.Mol.Biol. 2007;14:535–539. doi: 10.1038/nsmb1255. [DOI] [PubMed] [Google Scholar]
- 86.Swigut T., Wysocka J. H3K27 demethylases, at long last. Cell. 2007;131:29–32. doi: 10.1016/j.cell.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 87.Klose R.J., Kallin E.M., Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat.Rev.Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 88.Shi Y., Whetstine J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol.Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 89.Cloos P.A., Christensen J., Agger K., Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang B., Chen Y., Zhao Y., Bruick R.K. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 91.Wang Y., Wysocka J., Sayegh J., Lee Y.H., Perlin J.R., Leonelli L., Sonbuchner L.S., McDonald C.H., Cook R.G., Dou Y., Roeder R.G., Clarke S., Stallcup M.R., Allis C.D., Coonrod S.A. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 92.Cuthbert G.L., Daujat S., Snowden A.W., Erdjument-Bromage H., Hagiwara T., Yamada M., Schneider R., Gregory P.D., Tempst P., Bannister A.J., Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 93.Perillo B., Ombra M.N., Bertoni A., Cuozzo C., Sacchetti S., Sasso A., Chiariotti L., Malorni A., Abbondanza C., Avvedimento E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 94.Metivier R., Gallais R., Tiffoche C., Le Peron C., Jurkowska R.Z., Carmouche R.P., Ibberson D., Barath P., Demay F., Reid G., Benes V., Jeltsch A., Gannon F., Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 95.Kangaspeska S., Stride B., Metivier R., Polycarpou-Schwarz M., Ibberson D., Carmouche R.P., Benes V., Gannon F., Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 96.Holmquist B., Vallee B.L. Human liver class III alcohol and glutathione dependent formaldehyde dehydrogenase are the same enzyme. Biochem. Biophys. Res. Commun. 1991;178:1371–1377. doi: 10.1016/0006-291x(91)91045-e. [DOI] [PubMed] [Google Scholar]
- 97.Koivusalo M., Baumann M., Uotila L. Evidence for the identity of glutathione-dependent formaldehyde dehydrogenase and class III alcohol dehydrogenase. FEBS Lett. 1989;257:105–109. doi: 10.1016/0014-5793(89)81797-1. [DOI] [PubMed] [Google Scholar]
- 98.Iborra F.J., Renau-Piqueras J., Portoles M., Boleda M.D., Guerri C., Pares X. Immunocytochemical and biochemical demonstration of formaldhyde dehydrogenase (class III alcohol dehydrogenase) in the nucleus. J. Histochem. Cytochem. 1992;40:1865–1878. doi: 10.1177/40.12.1453005. [DOI] [PubMed] [Google Scholar]
- 99.Yamane K., Tateishi K., Klose R.J., Fang J., Fabrizio L.A., Erdjument-Bromage H., Taylor-Papadimitriou J., Tempst P., Zhang Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell. 2007;25:801–812. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 100.Klose R.J., Yan Q., Tothova Z., Yamane K., Erdjument-Bromage H., Tempst P., Gilliland D.G., Zhang Y., Kaelin W.G., Jr. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128:889–900. doi: 10.1016/j.cell.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 101.Klose R.J., Yamane K., Bae Y., Zhang D., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 102.Frescas D., Guardavaccaro D., Bassermann F., Koyama-Nasu R., Pagano M. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature. 2007;450:309–313. doi: 10.1038/nature06255. [DOI] [PubMed] [Google Scholar]
- 103.Lan F., Bayliss P.E., Rinn J.L., Whetstine J.R., Wang J.K., Chen S., Iwase S., Alpatov R., Issaeva I., Canaani E., Roberts T.M., Chang H.Y., Shi Y. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 104.De Santa F., Totaro M.G., Prosperini E., Notarbartolo S., Testa G., Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 105.Agger K., Cloos P.A., Christensen J., Pasini D., Rose S., Rappsilber J., Issaeva I., Canaani E., Salcini A.E., Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 106.Iwase S., Lan F., Bayliss P., de la Torre-Ubieta L., Huarte M., Qi H.H., Whetstine J.R., Bonni A., Roberts T.M., Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 107.Wissmann M., Yin N., Muller J.M., Greschik H., Fodor B.D., Jenuwein T., Vogler C., Schneider R., Gunther T., Buettner R., Metzger E., Schule R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell. Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 108.Whetstine J.R., Nottke A., Lan F., Huarte M., Smolikov S., Chen Z., Spooner E., Li E., Zhang G., Colaiacovo M., Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 109.Yamane K., Toumazou C., Tsukada Y., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 110.Lee M.G., Villa R., Trojer P., Norman J., Yan K.P., Reinberg D., Di Croce L., Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 111.Cloos P.A., Christensen J., Agger K., Maiolica A., Rappsilber J., Antal T., Hansen K.H., Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 112.Tahiliani M., Mei P., Fang R., Leonor T., Rutenberg M., Shimizu F., Li J., Rao A., Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- 113.Loh Y.H., Zhang W., Chen X., George J., Ng H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Okada Y., Scott G., Ray M.K., Mishina Y., Zhang Y. Histone demethylaseJHDM2Ais critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature. 2007;450:119–123. doi: 10.1038/nature06236. [DOI] [PubMed] [Google Scholar]
- 115.Huang, J. and Berger, S.L. (2008) The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. [DOI] [PubMed]
- 116.Zhang K., Lin W., Latham J.A., Riefler G.M., Schumacher J.M., Chan C., Tatchell K., Hawke D.H., Kobayashi R., Dent S.Y. The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell. 2005;122:723–734. doi: 10.1016/j.cell.2005.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huq M.D., Tsai N.P., Khan S.A., Wei L.N. Lysine trimethylation of retinoic acid receptor-alpha: a novel means to regulate receptor function. Mol. Cell. Proteomics. 2007;6:677–688. doi: 10.1074/mcp.M600223-MCP200. [DOI] [PubMed] [Google Scholar]
- 118.Kouskouti A., Scheer E., Staub A., Tora L., Talianidis I. Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol. Cell. 2004;14:175–182. doi: 10.1016/s1097-2765(04)00182-0. [DOI] [PubMed] [Google Scholar]
- 119.Houtz R.L., Stults J.T., Mulligan R.M., Tolbert N.E. Post-translational modifications in the large subunit of ribulose bisphosphate carboxylase/oxygenase. Proc. Natl. Acad. Sci. USA. 1989;86:1855–1859. doi: 10.1073/pnas.86.6.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thompson E.W., Laycock M.V., Ramshaw J.A., Boulter D. The amino acid sequence of Phaseolus aureua L. (mung-bean) cytochrome c. Biochem. J. 1970;117:183–192. doi: 10.1042/bj1170183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang J., Perez-Burgos L., Placek B.J., Sengupta R., Richter M., Dorsey J.A., Kubicek S., Opravil S., Jenuwein T., Berger S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 122.Shi X., Kachirskaia I., Yamaguchi H., West L.E., Wen H., Wang E.W., Dutta S., Appella E., Gozani O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell. 2007;27:636–646. doi: 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Feng L., Lin T., Uranishi H., Gu W., Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chuikov S., Kurash J.K., Wilson J.R., Xiao B., Justin N., Ivanov G.S., McKinney K., Tempst P., Prives C., Gamblin S.J., Barlev N.A., Reinberg D. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 125.Huang J., Sengupta R., Espejo A.B., Lee M.G., Dorsey J.A., Richter M., Opravil S., Shiekhattar R., Bedford M.T., Jenuwein T., Berger S.L. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 126.Olsson A., Manzl C., Strasser A., Villunger A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 2007;14:1561–1575. doi: 10.1038/sj.cdd.4402196. [DOI] [PubMed] [Google Scholar]
- 127.Rao B., Shibata Y., Strahl B.D., Lieb J.D. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol. Cell. Biol. 2005;25:9447–9459. doi: 10.1128/MCB.25.21.9447-9459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pokholok D.K., Harbison C.T., Levine S., Cole M., Hannett N.M., Lee T.I., Bell G.W., Walker K., Rolfe P.A., Herbolsheimer E., Zeitlinger J., Lewitter F., Gifford D.K., Young R.A. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 129.Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., Lee W., Mendenhall E., O’Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E.S., Bernstein B.E. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 131.Guenther M.G., Levine S.S., Boyer L.A., Jaenisch R., Young R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xiao T., Hall H., Kizer K.O., Shibata Y., Hall M.C., Borchers C.H., Strahl B.D. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao X.D., Han X., Chew J.L., Liu J., Chiu K.P., Choo A., Orlov Y.L., Sung W.K., Shahab A., Kuznetsov V.A., Bourque G., Oh S., Ruan Y., Ng H.H., Wei C.L. Whole-genome mapping of histone h3 lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell. Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 134.Pan G., Tian S., Nie J., Yang C., Ruotti V., Wei H., Jonsdottir G.A., Stewart R., Thomson J.A. Whole-genome analysis of histone h3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell. Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 135.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Jaenisch R., Wagschal A., Feil R., Schreiber S.L., Lander E.S. Abivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 136.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 137.Muller J., Hart C.M., Francis N.J., Vargas M.L., Sengupta A., Wild B., Miller E.L., O’Connor M.B., Kingston R.E., Simon J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 138.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Issaeva I., Zonis Y., Rozovskaia T., Orlovsky K., Croce C.M., Nakamura T., Mazo A., Eisenbach L., Canaani E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell. Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jepsen K., Solum D., Zhou T., McEvilly R.J., Kim H.J., Glass C.K., Hermanson O., Rosenfeld M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature. 2007;450:415–419. doi: 10.1038/nature06270. [DOI] [PubMed] [Google Scholar]
- 141.Guccione E., Bassi C., Casadio F., Martinato F., Cesaroni M., Schuchlautz H., Luscher B., Amati B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–937. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- 142.Hyllus D., Stein C., Schnabel K., Schiltz E., Imhof A., Dou Y., Hsieh J., Bauer U.M. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007;21:3369–3380. doi: 10.1101/gad.447007. [DOI] [PMC free article] [PubMed] [Google Scholar]