

Abstract
The main olfactory bulb (MOB) is the first site of synaptic processing in the central nervous system for odor information that is relayed from olfactory receptor neurons in the nasal cavity via the olfactory nerve (ON). Glutamate and ionotropic glutamate receptors (iGluRs) play a dominant role at ON synapses. Similarly, glutamate and iGluRs mediate dendrodendritic transmission between several populations of neurons within the MOB network. Neuroanatomical studies demonstrate that metabotropic glutamate receptors (mGluRs) are densely expressed through the MOB network, and they are particularly abundant at dendrodendritic synapses. Until recently, the physiological roles of mGluRs in the MOB were poorly understood. Over the past several years, mGluRs have been shown to play surprisingly powerful neuromodulatory roles at ON synapses and in dendrodendritic neurotransmission in the MOB. This chapter focuses on recent advances in our understanding of mGluR-mediated signaling components at dendrodendritic synapses.
Keywords: olfaction, glutamate, GABA, mitral cells, granule cells
Dendrodendritic Synapses in Main Olfactory Bulb
The main olfactory bulb (MOB) is the initial site that performs neural computations to decipher patterns of glomerular activity. Within the MOB, dendrodendritic synapses are thought to play fundamentally important roles in transforming the odor information, sharpening odorant signals, and increasing odorant discrimination. Ultimately, these dendrodendritic synapses are thought to sharpen patterns of odorant representation among the output neurons of the MOB, mitral and tufted (M/T) cells. Dendrodendritic synapses are formed between neuronal populations in several discrete sites in the MOB, including the glomerular layer (GL) and external plexiform layer (EPL). We refer readers elsewhere to detailed descriptions of the architecture and neuroanatomical organization of the MOB.1 The GL contains several populations of neurons, including external tufted (ET), short axon (SA), and periglomerular (PG) cells. All extend dendrites into the glomerular neuropil. ET neurons receive direct glutamatergic monosynaptic input from olfactory nerve (ON) terminals, whereas SA cells and most PG cells do not. However, PG and SA cells receive monosynaptic glutamatergic dendrodendritic excitatory input from ET cells, other tufted cell types, and mitral cells.1 PG cells, in turn, form inhibitory GABAergic dendrodendritic synapses with ET as well as M/T cells within the same glomerulus.1 Thus, in response to ON input, excitation of ET and M/T cells will trigger dendrodendritic glutamate release onto PG cells, which will cause dendrodendritic GABA release onto ET and M/T cells. This dendrodendritic inhibition can be directed back onto the initial ET and M/T cell providing the initial excitation of the PG cell—feedback or reciprocal inhibition—or onto other ET and M/T cells that did not contribute to the PG cell excitation—lateral or feedforward inhibition. This dendrodendritic inhibition in the GL is thought to be intraglomerular; that is, it is restricted to those cells that extend their dendrites into the same glomerulus. This M/T–PG cell dendrodendritic inhibition may provide a scaling function to prevent overexcitation (i.e., saturation) of the intraglomerular network by strong odors and/or to reset glomerular excitability to allow synchrony with the respiration cycle.2 Other circuits may provide substrates for interglomerular inhibitory interactions.2,3
In the deeper EPL, similar dendrodendritic interactions occur between the lateral dendrites of M/T cells and the dendrites of GABAergic granule cells (GCs). The somata of deep GCs (dGCs) are densely packed within the GC layer (GCL), whereas the somata of superficial GCs (sGCs) occur more superficially, interspersed within the mitral cell layer (MCL).1 The apical dendrites of both GC subtypes extend radially into the EPL, where they form dendrodendritic synapses with the lateral dendrites of M/T cells. Neurotransmission at dendrodendritic synapses within the EPL has been well studied. Glutamate released by M/T cell lateral dendrites activates AMPA and N-methyl D-aspartate (NMDA) receptors on GCs, which in turn drives GABAergic feedback/feedforward inhibition of the same or neighboring M/T cells.4–7 NMDA receptors play a particularly important role, however.7,8 Like their superficial cousins in the GL, these dendrodendritic synapses provide feedforward and feedback inhibition, but in this case over longer distances that bridge mitral cells associated with spatially distant glomeruli. Mitral cell lateral dendrites can extend up to 1–2 mm in the EPL, and studies in vitro suggest that spikes actively propagate along the entire lateral dendrite.9 Thus, feedforward inhibition along this tier of dendrodendritic processing is assumed to occur over a much larger spatial extent, and notably, among populations of M/T cells associated with different glomeruli. This circuitry is thought to play a key role in sharpening odorant representation patterns in the MOB, providing contrast enhancement that is essential to odor discrimination.9–11 These dendrodendritic synapses are also thought to shape the output of the MOB to higher-order olfactory structures because they are well suited to regulate the mitral cell spike generation.11
mGluRs in the MOB
Of the eight cloned metabotropic glutamate receptors (mGluRs), seven have been found in the MOB. mGluRs are divided into three subgroups: group I (mGluR1 and -5 subtypes), group II (mGluR2 and -3), and group III (mGluR4, -6, -7, and -8).12,13
GL neurons, collectively termed here juxtaglomerular cells, express several types of mGluRs. ET cells appear to have the same mGluR makeup as mitral cells; that is, they express high levels of mGluR1 and lower levels of mGluR7 and mGluR8.14–18 Other, as yet unidentified juxtaglomerular cells have been reported to express low to moderate levels of group I (mGluR5), group II (mGluR2), and group III (mGluR4, -7, and -8) receptors.14,15,17,19
M/T cells express high levels of mGluR1 (group I), which appear to be distributed along their somata, and apical and lateral dendrites.18,20–23 Studies from several laboratories demonstrate that mGluR1 participates in M/T cell synaptic responses to glutamatergic inputs from ON terminals and may mediate recurrent excitation after glutamate release from the apical or lateral dendrites of M/T cells (i.e., glutamate spillover).24–31 M/T cells do not express group II receptors (mGluR2 and -3), which contrasts markedly with the expression of mGluR2 in mitral cells in the accessory olfactory bulb (AOB).14,15 MOB M/T cells express mRNA for mGluR7 and mGluR8,16,17 although immunocytochemical studies indicate that these receptors are present selectively on the axon terminals of M/T cells in the piriform cortex.32,33
GCs express the highest levels of mGluR5 (group I) in the brain.19 At the cellular level, mGluR5 staining is strongest within the deep GCL.19,34 Electron microscopy studies have shown that mGluR5 is localized on portions of GC dendrites in the EPL apposed to presynaptic glutamatergic synapses from M/T cell lateral dendrites.23 This finding suggests that mGluR5 may mediate, at least in part, responses of GCs to glutamatergic inputs from M/T cells. GCs in the GCL do not appear to express mGluR1.18,23 The GCL stains only weakly for mGluR1 immunocytochemistry,20,23,34 and faint mGluR1 expression has been observed in this layer with in situ hybridization.22 GCs may express low to moderate levels of mGluR2 (group II)14,15 and low levels of mGluR4 and mGluR7 (group III).16,17,33 The precise cellular localization of these group II and III mGluRs on GCs and other cell types in the GCL is not known. The expression of mGluRs by other cell types in the GCL is unknown.
mGluR Modulation of GC Excitability
Given the high level of expression of mGluR5 on GCs, we and others hypothesized that mGluRs would modulate GC excitability and responsiveness to glutamatergic synaptic input from M/T cells and, as a result, modulate GC-mediated GABAergic inhibition of M/T cells. To test these hypotheses, we first investigated the consequences of activation or blockade of mGluR5 on dGCs in the GCL. These and other studies reviewed in this article used electrophysiological and optical imaging approaches and were performed in rat and mouse MOB slices.
Activation of mGluR5 Directly Increases dGC Excitability
Current clamp recordings from dGCs demonstrated that these cells were uniformly depolarized 10–20 mV by group I (DHPG) or group I–II (ACPD) agonists (Figs. 1 and 2). mGluR exerted both direct and indirect excitations on GCs. Identical experiments performed in the presence of antagonists of ionotropic glutamate receptor (iGluR) and GABA receptors (APV–CNQX–gabazine, “synaptic blockers”) produced a more modest depolarization of approximately 8 mV. The reduced depolarization in the antagonist condition is a result of blocking the indirect effects of DHPG or ACPD due to mGluR1-mediated excitation of mitral cells,18 which increases glutamate release onto GCs (Fig. 1). Thus, in the presence of synaptic blockers, only the direct ACPD- or DHPG-evoked depolarization of GCs persists.35 The direct action of DHPG on GCs is mediated exclusively via activation of mGluR5 because (1) it is unaffected by the mGluR1 antagonist LY367385 at concentrations that eliminate DHPG-evoked, mGluR1-mediated excitation of mitral cells18; (2) DHPG-evoked depolarization of dGCs was eliminated in mGluR5-, but not mGluR1-, knockout mice36; and (3) neither group II nor group III mGluR agonists elicited currents in GCs in voltage clamp recording, whereas DHPG evoked robust inward currents at the GC resting potential. The DHPG-evoked current (−8 pA, measured at −60 mV in the presence of synaptic blockers) was comparable to the DHPG-evoked depolarization measured under similar conditions in current clamp.35 This current reversed in polarity near the estimated K+ equilibrium potential (approximately −100 mV) and was abolished by K+ channel antagonists35 (Fig. 2). These results indicate that activation of mGluR5 increases GC excitability and is due, at least in part, to closure of K+ channels. Such an action would be expected to increase GABAergic inhibition of mitral cells. Experiments performed to test this hypothesis are detailed further below. However, these experiments led to the discovery of an unexpected and robust mGluR1-mediated increase in dendrodendritic inhibition of mitral cells. This finding in turn suggested the presence of an undetected population of inhibitory neurons whose excitability was increased by activation of mGluR1. This is addressed next.
Figure 1.

Group I and group I/II mGluR agonists directly and indirectly excite deep GCs (dGCs). Upper two traces: bath application of the group I/II mGluR agonist ACPD (50 μM, at lower bars) depolarized and increased spiking in this rat dGC (top trace). Application of blockers of fast synaptic transmission (synaptic blockers; CNQX, 10 μM; APV, 50 μM; gabazine, 5 μM) to the same cell reduced the baseline spontaneous synaptic activity (bottom trace); the depolarizing response to ACPD under these conditions was smaller than that without synaptic blockers and did not reach spike threshold. Lower two traces: the group I mGluR agonist DHPG (50 μM) depolarized and increased the firing rate of this typical mouse dGC in normal ACSF (top trace). In the same cell, blockers of fast synaptic transmission reduced baseline spontaneous synaptic activity and DHPG evoked a more moderate depolarization (bottom trace). Adapted from Ref. 35.
Figure 2.
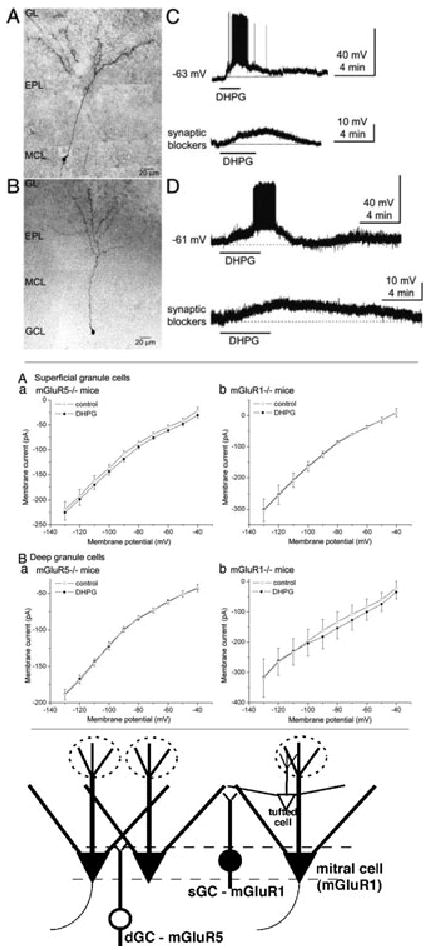
Deep and superficial GCs differentially express mGluR1 and mGluR5. Upper four panels: The group I mGluR agonist DHPG (50 μM) depolarized and increased spike firing in superficial GCs (sGCs) and deep GCs (dGCs) in olfactory bulb slices from wild-type mice. Photomicrograph montage of a biocytin-filled sGC (A) and dGC (B); note soma location in the mitral cell layer (MCL) for the sGC. GCL, granule cell layer; GL, glomerular layer; EPL, external plexiform layer. (C) Bath application of DHPG depolarized and increased the discharge of an sGC (upper trace). Lower trace: DHPG applied in the presence of blockers of fast synaptic transmission (CNQX, APV, gabazine) resulted in smaller membrane depolarization in the same sGC. (D) DHPG also depolarized and increased the firing of dGCs (upper trace). Lower trace: DHPG applied in the presence of blockers of fast synaptic transmission resulted in smaller depolarization of the same dGC. Middle four panels: (A) I–V curves for sGCs in slices from mGluR5−/− (a) and mGluR1−/− mice (b), before (control, open circles) and during bath application of DHPG (50 μM, solid circles). Plots represent group data (mean ± SEM) from four sGCs (a) and five sGCs (b). DHPG evoked a current in mGluR5−/− (a) but not mGluR1−/− (b) mice. (B) Similar experiments for dGCs in slices from mGluR5−/− (a) and mGluR1−/− (b) mice. No DHPG-induced current was recorded from dGCs in slices from mGluR5−/− mice, whereas a robust current was observed in slices from mGluR1−/− mice. The inward current of dGCs in slices from mGluR1−/− mice diminished near the estimated K+ equilibrium potential (−96 mV). DHPG evoked a significant inward current (P < 0.05 compared with control) at −90 mV and more positive voltages. All experiments were performed with blockers of fast synaptic transmission and TTX in the bath. Lower illustration: MOB circuit, depicting mGluR1 expression by mitral cells and sGCs and mGluR5 expression by dGCs. Superficial dendritic extensions of deep and superficial GCs interact with different portions of M/T cell lateral dendrites. Adapted from Ref. 36.
Differential Group I mGluR Expression in Two Distinct Populations of GCs
GCs can be distinguished based on the soma location superficially in the MCL (sGCs) or deeper within the GCL (dGCs). Of the group I mGluRs, only mGluR5 has been reported to be expressed by GCs,19,34 and the preceding electrophysiological results are consistent with these observations. Unexpectedly, our studies also revealed a strong mGluR1-driven component of GABAergic inhibition of mitral cells. This finding suggested another, previously undisclosed, population of GABAergic neurons in the MOB that express mGluR1. One such population is sGCs within the MCL. We compared mGluR1 and mGluR5 responses in deep versus superficial GCs.
Intrinsic properties, resting spontaneous discharge, and responses to DHPG did not differ significantly between sGCs and dGCs in slices harvested from wild-type mice.36 However, responses of the two GC populations differed markedly in mGluR1 and mGluR5 Knockout (−/−) mice (Fig. 2). DHPG depolarized sGCs in slices from mGluR5−/− mice, but it had no effect on sGCs in slices from mGluR1−/− mice. By contrast, DHPG depolarized dGCs in slices from mGluR1−/− mice, but it had no effect on dGCs in slices from mGluR5−/− mice. Also, DHPG had a relatively larger effect on dGCs in mGluR1 Knockout (−/−) mice, indicating that there may be compensatory changes in mGluR5 expression or dynamics of the MOB network in these mice. These findings indicate that the two GC subtypes differentially express group I mGluRs: sGCs express mGluR1, whereas dGCs express mGluR5 (Fig. 2). Mitral cells also express mGluR1, but not mGluR5. Thus, sGCs in the MCL are more similar to mitral cells than dGCs in terms of mGluR expression. Our finding that dGCs express mGluR5 but not mGluR1 is consistent with observations that GCL somata stain for mGluR5, that this staining is strongest within the deep GCL,19,34 and that the GCL stains weakly for mGluR1.20,22,23,34
Activation of Group I mGluRs Increases GABAergic Inhibition of Mitral Cells
Excitatory mGluRs on GCs may function, in part, to facilitate feedback and feedforward inhibition of mitral cells. To address this question we used voltage clamp recordings to determine whether DHPG-evoked activation of GCs alters GABAergic inhibitory input to mitral cells. As shown in Figure 3, DHPG increased the frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) and miniature IPSCs (mIPSCs) in mitral cells.35,37 The increase in IPSC frequency was dose-dependent and occurred in the absence of any significant changes in IPSC amplitude or kinetics. These observations suggest that DHPG acts presynaptically to increase GABA release from inhibitory neurons onto mitral cells. Furthermore, the DHPG-evoked increase in IPSC frequency persisted in the presence of tetrodotoxin (TTX) which blocks spikes in the MOB network but was abolished in the presence of antagonists of voltage-gated Ca2+ channels. This finding indicates that DHPG depolarizes the dendrites of inhibitory interneurons sufficiently to engage voltage-gated Ca2+ currents necessary for GABA release. We next addressed two issues: (1) the mGluR subtypes involved in DHPG-evoked IPSCs and (2) the neuronal populations involved in DHPG-evoked GABA release.
Figure 3.
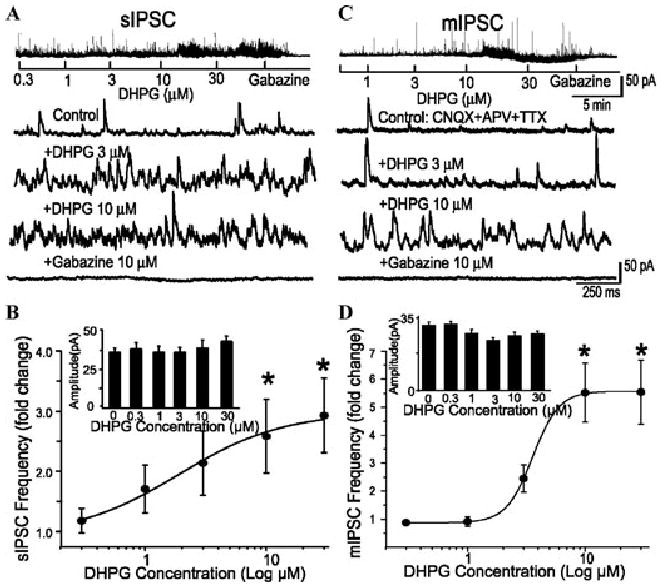
Activation of group I mGluRs increases the frequency of spontaneous GABAergic input to mitral cells. Voltage clamp recordings (HP = 0 mV) of mitral cells show that the group I mGluR agonist, DHPG, dose-dependently increased the frequency of GABAergic sIPSCs (A) and mIPSCs (C); gabazine eliminated all IPSCs, as shown in lower traces. sIPSCs were recorded in normal ACSF (Control) and mIPSCs were recorded in the presence of CNQX (10 μM), APV (50 μM), and TTX (1 μM). (B and D) Group data showing the dose–response relationship for DHPG effects on sIPSC (n = 6) and mIPSC (n = 7) frequency and amplitude (inset bar graphs) recorded in a 1-min epoch. *P < 0.05 compared with control, ANOVA followed by Neuman–Keuls post-hoc comparisons. DHPG did not affect the amplitude of sIPSCs or mIPSCs. Adapted from Ref. 37.
We found that DHPG-evoked increases in IPSCs in mitral cells were mediated by both mGluR1 (LY367385-sensitive component) and mGluR5 (MPEP-sensitive component). Individually, LY367385 or MPEP attenuated some, but not all, of the DHPG-evoked IPSCs. The effects of DHPG were blocked by combined application of both LY367385 and MPEP, and this condition did not differ from that when 100 μM LY341495, which blocks all known mGluRs, was used. Also, LY367385 reduced the basal IPSC frequency,35,37 indicating that endogenous glutamate tonically activates mGluR1 to drive GABA release onto mitral cells; there was an additional reduction in basal IPSC frequency when LY367385 and MPEP were combined. The DHPG-evoked increase in IPSC frequency also occurred, and was in fact enhanced, in mGluR1 KO mice; this finding parallels the enhanced effect of DHPG on dGCs in mGluR1 Knockout (−/−) mice (see earlier) and indicates that activation of mGluR5 alone can drive GABAergic inhibition of mitral cells. However, these overall findings indicate that both mGluR1 and mGluR5 are involved in the DHPG-evoked inhibition, with a predominant role for mGluR1. The findings presented to this point—involvement of mGluR1 and mGluR5 in increasing GC excitability and GABA release onto mitral cells—are entirely consistent with findings in the AOB.38
The second issue we investigated is the location of the DHPG-sensitive GABAergic neurons responsible for the increased inhibition of mitral cells. The DHPG-evoked increase in IPSCs could result from actions on GCs and/or PG cells in the GL. To address this question, we tested the effects of DHPG in MOB slices in which the GL layer was removed (Fig. 4). In such slices, DHPG increased spontaneous, but not miniature (TTX insensitive), IPSCs in mitral cells. Excision of the GL reduced sIPSCs in mitral cells by approximately 50% and abolished IPSC bursts in mitral cells. Interestingly, the frequency of IPSC bursts in mitral cells (0.5 ± 0.2 Hz) in intact slices is nearly identical to that in ET cells in the GL (0.4 ± 0.05 Hz39). These results suggest that IPSC bursts in mitral cells originate in the GL, probably from GABAergic PG cells whose spontaneous excitatory synaptic and spiking activity is strongly driven by ET cell spike bursts.39–41
Figure 4.
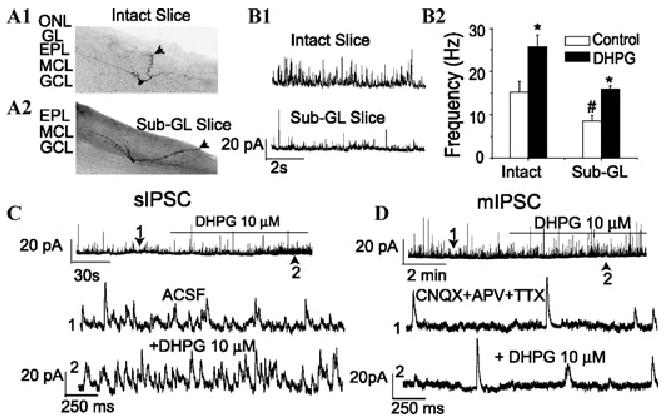
DHPG failed to increase mIPSC frequency in mitral cells after excision of the glomerular layer (GL, Sub-GL slices). Examples of biocytin-filled mitral cells recorded in intact (A1) and Sub-GL (A2) slices; arrows mark the apical dendrites. In panel A2 the GL and ONL layer (ONL) have been excised and the mitral cell apical dendrite is truncated within the EPL. (B1) Voltage clamp recordings in normal ACSF show that the frequency of sIPSCs in mitral cells is higher in intact slices (upper trace) than in Sub-GL slices (lower trace). (B2) Bar graph of group data showing lower basal IPSC frequency in Sub-GL slices and that DHPG readily increases the frequency of sIPSCs recorded in normal ACSF in intact and Sub-GL slices. (C) DHPG readily increased sIPSCs in a mitral cell recorded in a Sub-GL slice. (D) DHPG application did not affect the frequency of mIPSCs (recorded in the presence of TTX) in a mitral cell recorded in a Sub-GL slice. Adapted from Ref. 37.
To further investigate sources of DHPG-driven GABAergic input to mitral cells, we made focal DHPG puffs into individual layers in intact MOB slices. DHPG puffs into the GL, EPL, MCL, or GCL increased sIPSCs in mitral cells (Fig. 5). However, when the same experiment was repeated in the presence of TTX, DHPG puffs into the GL, but not the GCL, increased mIPSCs in mitral cells.37 These findings indicate that DHPG-evoked GABA release from GCs requires action potentials, whereas in the GL DHPG facilitates PG cell GABA release via both spike-dependent and spike-independent presynaptic mechanisms. Functionally, these results suggest that mGluRs amplify spike-driven lateral inhibition through the mitral-cell-to-GC circuit, whereas GL mGluRs may play a more important role in amplifying intraglomerular inhibition after excitatory input to PG cells that is below the threshold for spike generation in these cells. There is an important caveat to this conclusion, however. mIPSC recordings from mitral cells were performed in the presence of APV–CNQX, a condition that hyperpolarizes GCs and lowers their excitability. Thus, it is possible that DHPG may increase GC-mediated mIPSCs if similar experiments are performed in the absence of APV–CNQX.
Figure 5.
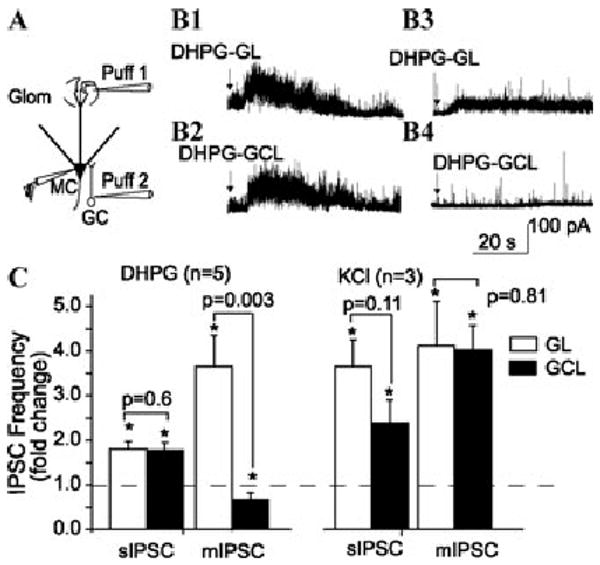
Relative contribution of GCs and PG cells to DHPG evoked-IPSCs in mitral cells. (A) Experimental approach. Two pipettes were positioned to focally puff DHPG (20 nL, 1 mM in ACSF) into the GL and GCL, respectively, while recording IPSCs in a mitral cell. DHPG puffs into the GL (B1) or GCL (B2) increased the frequency of sIPSCs (left column) recorded in normal ACSF; same cell in panels B1 and B2. (B3, B4) Recordings from the same mitral cell in panels B1 and B2 after perfusion with CNQX plus APV plus TTX. DHPG puffs into the GL (B3), but not the GCL (B4), increased the frequency of mIPSCs. (C) Group data showing the effects of DHPG and KCl puffs into the GL or GCL on the frequency of sIPSCs and mIPSCs. *P < 0.05, compared with control, paired t tests. KCl puffs increase IPSCs in all cases. Adapted from Ref. 37.
mGluR Inactivation Reduces M/T Cell–Evoked Excitation of GCs
The findings presented to this point demonstrate that pharmacological activation of group I mGluRs with exogenously applied agonists increases GC excitability and enhances spontaneous release of GABA onto mitral cells. Functionally, however, it is important to determine whether endogenous activation of mGluRs plays a role in GC responses to glutamatergic synaptic input from mitral cells, and if and how these receptors participate in GC-mediated synaptic inhibition of mitral cells. To address these issues, we first assessed the effects of the mGluR inactivation on the operation of the mitral-to-GC synapse (Fig. 6). Current clamp recordings were first used to assess the effects of mGluR blockade on the responses of individual dGCs to M/T cell input. Single shocks applied to the GL directly activated mitral cells, and, in turn, elicited spikes in GCs as previously reported.5,42 LY341495 reversibly reduced the mean response probability to M/T cell input, that is, the occurrence of a spike, from approximately 80% to 50%.35 A cell-by-cell analysis revealed that the reduction of evoked spiking by LY341495 varied with the degree of hyperpolarization. The response probability to M/T cell input decreased with increasing hyperpolarization. Also, LY341495 significantly hyperpolarized those cells that exhibited the largest reduction in evoked spiking. The spike threshold was not significantly affected by LY341495 in these cells. LY341495 produced a small, but nonsignificant, reduction in the amplitude of evoked excitatory postsynaptic potentials (EPSPs), and there was no consistent relationship between the EPSP amplitude and the degree of hyperpolarization. In parallel voltage clamp recordings, LY341495 did not significantly affect the amplitude, onset latency, kinetics, or integral of the single-shock, GL-evoked EPSC in GCs. Taken together, these findings suggest that the reduction of evoked spiking in GCs by LY341495 was not due to presynaptic modulation of glutamate release from M/T cell dendrites but may be due to the degree of GC hyperpolarization. Perhaps tonic excitation of dGCs via mGluR5 provides a favorable climate for NMDA receptor–mediated synaptic activation of GCs (see Discussion). Although we cannot ascribe these effects solely to blockade of mGluR5, these result suggest that activation of mGluRs by endogenously released glutamate tonically modulates the excitability of GCs.
Figure 6.
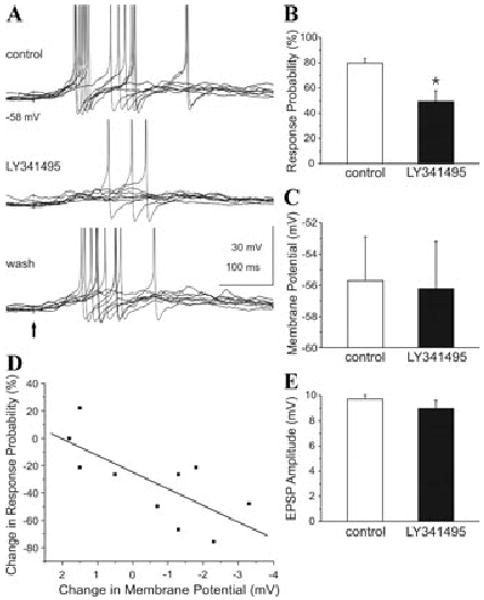
Blockade of mGluRs attenuates M/T cell–evoked excitation of GCs. (A) Upper trace: single shocks (marked by arrow in lower trace) applied to the GL to directly activate M/T cells and evoke excitatory postsynaptic potentials (EPSPs) and spikes in a mouse GC. Middle trace: application of LY341495 (100 μM) markedly reduced evoked excitation of the same GC in a reversible manner (lower trace). (B) Bar graphs of group data (n = 10) show the percentage of GL–evoked spikes before (control) and during application of LY341495. *P < 0.05. (C) LY341495 did not significantly alter the resting membrane potential in the group of 10 cells tested. (D) Regression analysis revealed that the reduction of evoked spikes in individual GCs varied with the degree of hyperpolarization. Data are fitted with a regression line (r = −0.73; P = 0.01). (E) LY341495 did not alter the mean amplitude of the EPSP (n = 10 cells). Adapted from Ref. 35.
We next used optical imaging of voltage-sensitive dye signals (RH-414) to investigate how inactivation of mGluRs influences the operation of the mitral-to-GC synapse at the population level. Such imaging was used to investigate activity patterns evoked by focal stimulation of the lateral olfactory tract (LOT), which antidromically activates M/T cells and drives M/T-to-GC glutamatergic synapses. We recently reported43 that single shocks applied to the LOT elicited optical responses that were first observed in the EPL and then subsequently spread into the superficial GCL. LOT-evoked responses were abolished by removal of Ca2+ from the ACSF, by application of TTX or CNQX–APV, indicating that they were postsynaptically mediated.43 These and other studies demonstrated that the LOT-evoked optical signals predominantly reflected postsynaptic depolarization of the distal dendrites and somata of GCs. We found that the broad-spectrum mGluR antagonist LY341495 significantly suppressed the LOT-evoked responses (Fig. 7).35 LY341495 reduced the peak amplitude, time course, and spatial extent of the LOT-evoked voltage-sensitive dye signals in the EPL by approximately 50% (Fig. 7).35 These findings suggest that mGluR blockade reduces magnitude, duration, and spatial envelope of GC excitability to M/T cell synaptic input across the network of GCs. Taken together, these results show that mGluR inactivation profoundly suppresses M/T cell-to-GC synaptic activation, an effect that would be expected in turn to reduce lateral inhibition of mitral cells.
Figure 7.
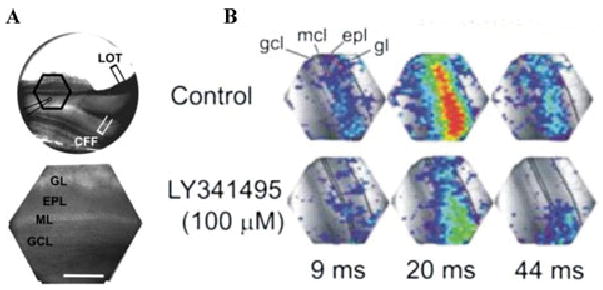
mGluR inactivation reduces the amplitude, duration, and spatial extent of M/T cell–evoked excitation of GCs. (A) Optical recording setup used for voltage-sensitive dye imaging. Upper panel shows a videographic image of a 400-μm-thick forebrain-MOB slice. Hexagonal frame demarcates the border of the 464-element photodiode array. The position of the LOT stimulation electrode is indicated. Lower panel shows a higher-magnification videographic image of the area within the hexagon in upper panel. (B) Upper row shows images of optical activity elicited by one shock to the LOT. Each panel shows the amplitude of optical responses, recorded by each of 464 photodiodes. Signal amplitudes, expressed as a percentage above mean baseline values, are color coded. Intervals beneath the images are relative to the onset of LOT stimulation. Lower row: application of LY341495 reduced the LOT-evoked optical signals. CFF, centrifugal fibers; gl, glomerular layer; epl, external plexiform layer; mcl, mitral cell layer; gcl, granule cell layer; LOT, lateral olfactory tract. Adapted from Ref. 35. (In color in Annals online.)
Blockade of mGluRs Reduces Feedback Inhibition of Mitral Cells
The preceding experiments indicate that inactivation of mGluRs significantly attenuates mitral cell–evoked excitation of GCs. Does blockade of mGluRs decrease GC-mediated inhibition of mitral cells, and specifically, feedback inhibition of mitral cells? To address this question, we determined whether LY341495 alters feedback inhibitory postsynaptic potentials (IPSPs) elicited by direct depolarization of individual mitral cells. As previously reported,4 depolarizing current steps applied to mitral cells in the presence of TTX and tetra ethylammonium (TEA) were followed by a feedback IPSP (Fig. 8). Bath application of LY341495 reduced the peak amplitude and integral of the feedback IPSP.35 These results demonstrated that blockade of mGluRs reduces GC-mediated feedback inhibition of mitral cells. Blockade of mGluR1 similarly reduces feedback inhibition of mitral cells in the AOB.38
Figure 8.
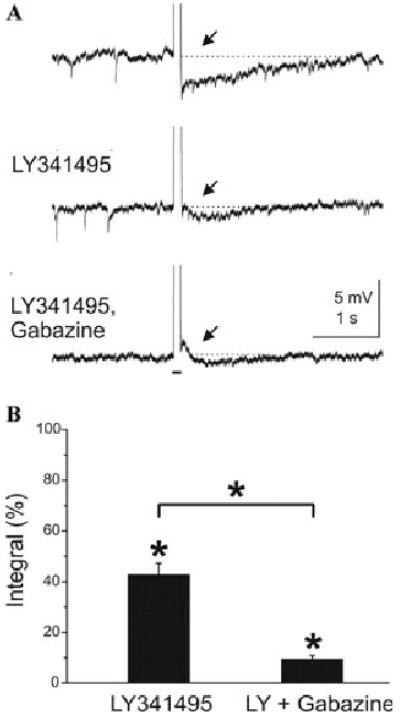
Blockade of mGluRs reduces GC-mediated feedback inhibition of mitral cells. (A) Depolarizing current pulses (100 ms, 300 pA, indicated by bar beneath trace) injected into a mitral cell in the presence of TTX and TEA evoked a Ca2+ spike followed by a GC-mediated feedback IPSPs (at arrow). Middle trace shows that the mGluR antagonist LY341495 reduced the IPSP, and subsequent addition of the GABAa receptor antagonist gabazine abolished any remaining feedback IPSP as well as spontaneous IPSPs (lower trace). (B) Group data from similar experiments. *P < 0.05. Adapted from Ref. 35.
mGluR Actions at Dendrodendritic Synapses in the GL
The role of mGluRs at dendrodendritic synapses in the GL is less well studied than at those in the EPL. As noted earlier, focal DHPG puffs into the GL robustly increase IPSCs in mitral cell. This finding indicates that activation of group I mGluRs excites GABAergic interneurons in the GL, causing them to release GABA onto mitral cells. To confirm this finding, we investigated whether DHPG increases GABAergic inhibition of ET cells in the GL. Because these cells do not extend dendrites into the deeper layers, their GABAergic input is entirely derived from local neurons in the GC, namely, PG neurons. Consistent with this hypothesis, DHPG application significantly increased the frequency of sIPSC in ET cells (Fig. 9).37 This increase persisted in the presence of iGluR antagonists and TTX. The DHPG-evoked increase in IPSCs was abolished by antagonists of voltage-gated Ca2+ channels. These findings suggest that DHPG depolarizes PG cells sufficiently to activate voltage-gated Ca2+ channels, which are necessary for dendritic GABA release.
Figure 9.
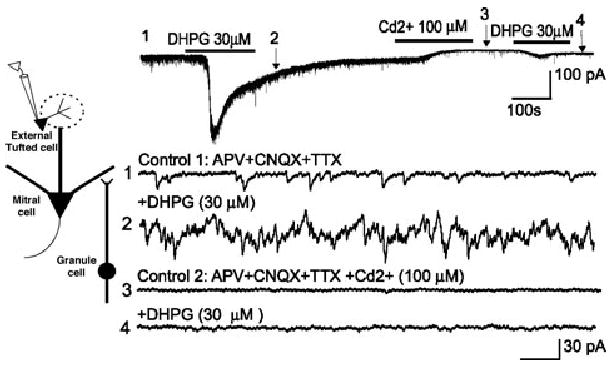
DHPG increases mIPSCs in ET cells. Upper trace: voltage clamp recordings (HP = −60 mV) from an ET cell in the presence of CNQX (10 μM), APV (50 μM), and TTX (1 μM). Because the chloride equilibrium potential in these conditions was near 0 mV, mIPSCs were recorded as inward currents (downward deflections). The four lower traces correspond to recordings from the first trace displayed at faster time scale. DHPG (30 μM) application produces a slow inward current associated with a robust increase in the frequency of mIPSCs. Subsequent application of Cd2+ (100 μM) decreases basal and DHPG-evoked mIPSCs. Inset at left shows diagram of recording from an ET cell in the GL. Adapted from Ref. 37.
Discussion
These overall results indicate that several classes of GABAergic interneurons in the MOB express group I mGluRs. Pharmacological activation of these receptors robustly increases the excitability of these cells, causing them to depolarize to a level that triggers GABA release. This process in turn leads to increased inhibition of mitral cells and ET cells in the GL. Inactivation of mGluRs has several important consequences. First, it decreases the basal or tonic level of GABAergic inhibition of mitral cells, indicating that endogenous glutamate tonically activates mGluRs on GABAergic neurons to facilitate ongoing GABA release. Second, mGluR blockade significantly attenuates GC excitability and excitatory transmission at mitral cell-to-GC synapses. Third, mGluR inactivation exerts a surprisingly robust suppression of feedback inhibition of mitral cells. As noted earlier, most of these results summarized here for mitral cells in the MOB have also been observed in mitral cells in the AOB.38 Thus, mGluRs exert a parallel regulation of dendrodendritic synapses in both the main and accessory olfactory bulbs.
Several unexpected results emerged from these studies. First, superficial and deep GCs differentially express group I mGluRs. The functional significance of this finding is unknown, but it may be related to the morphologically distinct populations of superficial and deep GCs that extend dendrites into different sublamina of the EPL (Fig. 2; see Ref. 36 for review). dGCs tend to extend dendrites into the deep and intermediate EPL and sGCs (and tufted cells) into the superficial EPL. This dendritic stratification presumably allows the two populations of GCs to interact with different populations of M/T cells with similar stratification of lateral dendrites in different EPL sublamina (see Refs. 1 and 36 for review). Thus, sGCs expressing mGluR1 may preferentially interact with M/T cell lateral dendrites in the superficial EPL, and dGCs expressing mGluR5 with mitral cell dendrites located in the deep EPL. Alternatively, a significant percentage of adult-born GCs that enter the MOB via the rostral migratory stream eventually preferentially distribute to the MCL layer and superficial EPL. It is therefore reasonable to speculate that these cells preferentially express mGluR1. In this regard it is interesting that integration of adult-born GCs into the MOB circuitry is enhanced by odor enrichment and that the expression of mGluR1 in the MOB is regulated by sensory input.1,44 Thus, mGluR1 expression by sGCs may play a role in experience-dependent plasticity.
A second surprising result was the marked difference in the ability of DHPG to drive GC versus PG cell-mediated GABAergic inhibition of mitral cells. The ability of DHPG to drive IPSCs in mitral cells in slices with the GL excised was abolished in the presence of TTX, indicating that it was mediated by spike-driven GABA release from GCs. Similarly, focal DHPG puffs into the GCL of intact slices failed to drive GABA release onto mitral cells. However, in intact slices in the presence of TTX, bath-applied DHPG or DHPG puffs into the GL readily triggered GABA release onto mitral cells. This finding leads us to conclude that in the presence of TTX, DHPG-evoked GABAergic inhibition is mediated predominantly by PG cells in the GL. Why DHPG fails to evoke GABA release from GCs in TTX is unclear, but it may be because of their relatively hyperpolarized membrane potential in the presence of TTX (and iGluR antagonists). In these conditions, DHPG-evoked excitation may be insufficient to depolarize GCs to a level necessary to engage NMDA receptors and/or voltage-gated Ca2+ currents, which are necessary for GABA release from dendritic spines.
Perhaps the most striking finding of this and other recent studies was the robust suppression of feedback inhibition of MOB and AOB mitral cell by mGluR blockade. The degree of suppression was nearly equivalent to that produced by GABAa receptor antagonists.35,38 This result was entirely unexpected because of the strong dependence of dendrodendritic feedback inhibition of mitral cells on NMDA receptor activation.4–8,42 Studies from Nathan Urban's laboratory have clarified this finding. When similar experiments are repeated in conditions that favor NMDA receptor activation (e.g., low extracellular Mg2+), the suppressive effect of mGluR blockade on feedback inhibition is reduced.38 This finding indicates that glutamate released from M/T cells, acting at mGluRs to depolarize GCs, produces a favorable condition for activation of NMDA receptors that are critical for GABA release from GC dendritic spines.
We are still at an early stage of exploring mGluR modulation of dendrodendritic inhibition, and much remainto be understood. Under what conditions or operational states of the MOB network are mGluRs engaged? The results presented here indicate, at least in vitro, that single impulses in M/T cells can release sufficient glutamate at dendrodendritic synapses to activate mGluRs on GCs. However, in many circuits group I mGluRs are selectively engaged by high-frequency synaptic activity.9,13 Thus, these receptors may preferentially facilitate or amplify dendrodendritic inhibition during odor-driven spike bursts in M/T cells or when the MOB network processes odor inputs during high-frequency sniffing.
Acknowledgments
This work was supported in part by the Whitehall Foundation; the Howard University New Faculty Research Program; Biomedical Research Foundation of Northwest Louisiana; and United States Public Health Service Grants S06GM08016 (NIGMS/NIH), U54NS039407 (NINDS/NIH), DC008702, DC003195, DC06356, DC07123, DC009049, DC007876, and RR020146.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Ennis M, Hamilton KA, Hayar A. Neurochemistry of the main olfactory system. In: Lajtha A, editor. Handbook of Neurochemistry and Molecular Neurobiology. 3rd. Vol. 20 2007. Ed. in Chief. [Google Scholar]; Johnson DA, editor. Sensory Neurochemistry. Springer; Heidelberg: pp. 137–204. Volume Ed. [Google Scholar]
- 2.Wachowiak M, Shipley MT. Coding and synaptic processing of sensory information in the glomerular layer of the olfactory bulb. Semin Cell Dev Biol. 2006;17:411–423. doi: 10.1016/j.semcdb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Aungst JL, Heyward PM, Puche AC, et al. Center-surround inhibition among olfactory bulb glomeruli. Nature. 2003;426:623–629. doi: 10.1038/nature02185. [DOI] [PubMed] [Google Scholar]
- 4.Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 5.Schoppa NE, Kinzie JM, Sahara Y, et al. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;18:6790–6802. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aroniadou-Anderjaska V, Ennis M, Shipley MT. Current-source density analysis in the rat olfactory bulb: laminar distribution of kainate/AMPA and NMDA receptor-mediated currents. J Neurophysiol. 1999;81:15–28. doi: 10.1152/jn.1999.81.1.15. [DOI] [PubMed] [Google Scholar]
- 7.Chen WR, Xiong W, Shepherd GM. Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron. 2000;25:625–633. doi: 10.1016/s0896-6273(00)81065-x. [DOI] [PubMed] [Google Scholar]
- 8.Halabisky B, Friedman D, Radojicic M, Strowbridge B. Calcium influx through NMDA receptors directly evokes GABA release in olfactory bulb granule cells. J Neurosci. 2000;20:5124–5134. doi: 10.1523/JNEUROSCI.20-13-05124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ennis M, Hayar A. Physiology of the olfactory bulb. In: Basbaum A, Kaneko A, Shepherd G, editors. The Senses: A Comprehensive Reference. Vol. 4 2008. Eds.-In Chief. [Google Scholar]; Firestein S S, Beauchamp G G, editors. Olfaction and Taste. Academic Press; San Diego: pp. 641–686. [Google Scholar]
- 10.Yokoi M, Mori K, Nakanishi S. Refinement of odor molecule tuning by dendrodendritic synaptic inhibition in the olfactory bulb. Proc Natl Acad Sci USA. 1995;92:3371–3375. doi: 10.1073/pnas.92.8.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepherd GW, Chen WR, Greer CA. Olfactory bulb. In: Shepherd GM, editor. The Synaptic Organization of the Brain. Oxford; New York: 2004. pp. 165–216. [Google Scholar]
- 12.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 13.Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 14.Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neurosci. 1993;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- 15.Ohishi H, Neki A, Mizuno N. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res. 1998;30:65–82. doi: 10.1016/s0168-0102(97)00120-x. [DOI] [PubMed] [Google Scholar]
- 16.Kinzie JM, Saugstad JA, Westbrook GL, Segerson TP. Distribution of metabotropic glutamate receptor 7 messenger RNA in the developing and adult rat brain. Neuroscience. 1995;69:167–176. doi: 10.1016/0306-4522(95)00244-d. [DOI] [PubMed] [Google Scholar]
- 17.Saugstad JA, Kinzie JM, Shinohara MM, et al. Cloning and expression of rat metabotropic glutamate receptor 8 reveals a distinct pharmacological profile. Mol Pharmacol. 1997;51:119–125. doi: 10.1124/mol.51.1.119. [DOI] [PubMed] [Google Scholar]
- 18.Heinbockel T, Heyward P, Conquet F, Ennis M. Regulation of main olfactory bulb mitral cell excitability by metabotropic glutamate receptor mGluR1. J Neurophysiol. 2004;92:3085–3096. doi: 10.1152/jn.00349.2004. [DOI] [PubMed] [Google Scholar]
- 19.Romano C, Sesma MA, McDonald CT, et al. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 20.Martin LJ, Blackstone CD, Huganir RL, Price DL. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron. 1992;9:259–270. doi: 10.1016/0896-6273(92)90165-a. [DOI] [PubMed] [Google Scholar]
- 21.Masu M, Tanabe Y, Tsuchida K, et al. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- 22.Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurol. 1992;322:121–135. doi: 10.1002/cne.903220110. [DOI] [PubMed] [Google Scholar]
- 23.Van Den Pol A. Presynaptic metabotropic glutamate receptors in adult and developing neurons: autoexcitation in the olfactory bulb. J Comp Neurol. 1995;359:253–271. doi: 10.1002/cne.903590206. [DOI] [PubMed] [Google Scholar]
- 24.Aroniadou-Anderjaska V, Ennis M, Shipley MT. Dendrodendritic recurrent excitation in mitral cells of the rat olfactory bulb. J Neurophysiol. 1999;82:489–494. doi: 10.1152/jn.1999.82.1.489. [DOI] [PubMed] [Google Scholar]
- 25.Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/s0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 26.Carlson GC, Shipley MT, Keller A. Long-lasting depolarizations in mitral cells of the rat olfactory bulb. J Neurosci. 2000;20:2011–2021. doi: 10.1523/JNEUROSCI.20-05-02011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman D, Strowbridge BW. Functional role of NMDA autoreceptors in olfactory mitral cells. J Neurophysiol. 2000;84:39–50. doi: 10.1152/jn.2000.84.1.39. [DOI] [PubMed] [Google Scholar]
- 28.Salin PA, Lledo PM, Vincent JD, Charpak S. Dendritic glutamate autoreceptors modulate signal processing in rat mitral cells. J Neurophysiol. 2001;8:1275–1282. doi: 10.1152/jn.2001.85.3.1275. [DOI] [PubMed] [Google Scholar]
- 29.Schoppa NE, Westbrook GL. Glomerulus-specific synchronization of mitral cells in the olfactory bulb. Neuron. 2001;31:639–651. doi: 10.1016/s0896-6273(01)00389-0. [DOI] [PubMed] [Google Scholar]
- 30.De Saint Jan D, Westbrook GL. Detecting activity in olfactory bulb glomeruli with astrocyte recording. J Neurosci. 2005;25:2917–2924. doi: 10.1523/JNEUROSCI.5042-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Saint Jan D, Westbrook GL. Disynaptic amplification of metabotropic glutamate receptor 1 responses in the olfactory bulb. J Neurosci. 2007;27:132–140. doi: 10.1523/JNEUROSCI.2439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinoshita A, Shigemoto R, Ohishi H, et al. Immunohistochemical localization of metabotropic glutamate receptors, mGluR7a and mGluR7b, in the central nervous system of the adult rat and mouse: a light and electron microscopic study. J Comp Neurol. 1998;393:332–352. [PubMed] [Google Scholar]
- 33.Wada E, Shigemoto R, Kinoshita A, et al. Metabotropic glutamate receptor subtypes in axon terminals of projection fibers from the main and accessory olfactory bulbs: a light and electron microscopic immunohistochemical study in the rat. J Comp Neurol. 1998;393:493–504. [PubMed] [Google Scholar]
- 34.Sahara Y, Kubota T, Ichikawa M. Cellular localization of metabotropic glutamate receptors mGluR1, 2/3, 5 and 7 in the main and accessory olfactory bulb of the rat. Neurosci Lett. 2001;312:59–62. doi: 10.1016/s0304-3940(01)02184-x. [DOI] [PubMed] [Google Scholar]
- 35.Heinbockel T, Laaris N, Ennis M. Metabotropic glutamate receptors in the main olfactory bulb drive granule cell-mediated inhibition. J Neurophysiol. 2007;97:858–870. doi: 10.1152/jn.00884.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heinbockel T, Hamilton KA, Ennis M. Group I metabotropic glutamate receptors are differentially expressed by two populations of olfactory bulb granule cells. J Neurophysiol. 2007;97:3136–3141. doi: 10.1152/jn.01202.2006. [DOI] [PubMed] [Google Scholar]
- 37.Dong HW, Hayar A, Ennis M. Activation of group I metabotropic glutamate receptors on main olfactory bulb granule cells and periglomerular cells enhances synaptic inhibition of mitral cells. J Neurosci. 2007;27:5654–5663. doi: 10.1523/JNEUROSCI.5495-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castro JB, Hovis KR, Urban NN. Recurrent dendrodendritic inhibition of accessory olfactory bulb mitral cells requires activation of group I metabotropic glutamate receptors. J Neurosci. 2007;27:5664–5671. doi: 10.1523/JNEUROSCI.0613-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayar A, Shipley MT, Ennis M. Olfactory bulb external tufted cells are synchronized by multiple intraglomerular mechanisms. J Neurosci. 2005;25:8197–8208. doi: 10.1523/JNEUROSCI.2374-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayar A, Karnup S, Ennis M, Shipley MT. External tufted cells: a major excitatory element that coordinates glomerular activity. J Neurosci. 2004;24:6676–6685. doi: 10.1523/JNEUROSCI.1367-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy GJ, Darcy DP, Isaacson JS. Intraglomerular inhibition: signaling mechanisms of an olfactory microcircuit. Nat Neurosci. 2005;8:354–364. doi: 10.1038/nn1403. [DOI] [PubMed] [Google Scholar]
- 42.Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- 43.Laaris N, Puche A, Ennis M. Complementary postsynaptic activity patterns elicited in olfactory bulb by stimulation of mitral/tufted and centrifugal fiber inputs to granule cells. J Neurophysiol. 2007;97:296–306. doi: 10.1152/jn.00823.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casabona G, Catania MV, Storto M, et al. Deafferentation up-regulates the expression of the mGlu1a metabotropic glutamate receptor protein in the olfactory bulb. Eur J Neurosci. 1998;10:771–776. doi: 10.1046/j.1460-9568.1998.00076.x. [DOI] [PubMed] [Google Scholar]