

Abstract
The prion (infectious protein) concept has evolved with the discovery of new self-propagating protein states in organisms as diverse as mammals and fungi. The infectious agent of the mammalian transmissible spongiform encephalopathies (TSE) has long been considered to be the prototypical prion, and recent cell-free propagation and biophysical analyses of TSE infectivity have now firmly established its prion credentials. Other disease-associated protein aggregates, such as some amyloids, can also have prion-like characteristics under certain experimental conditions. However, most amyloids appear to lack the natural transmissibility of TSE prions. One feature that distinguishes the latter from the former is the glycophosphatidylinositol membrane anchor on prion protein, the molecule that is corrupted in TSE diseases. The presence of this anchor profoundly affects TSE pathogenesis, which involves major membrane distortions in the brain, and may be a key reason for the greater neurovirulence of TSE prions relative to many other autocatalytic protein aggregates.
Keywords: protein misfolding diseases, glycophosphatidylinositol anchor, exosomes, tunneling nanotubes, membranes
INTRODUCTION
In the 1960s, the notion that corrupted host proteins could act as infectious pathogens was proposed by JS Griffith as an explanation for the mysterious sheep disease called scrapie (1). In 1982, Stanley Prusiner coined the term prion for such proteinaceous infectious agents that apparently lack their own specific nucleic acid genome (2). Shortly thereafter, Prusiner discovered a host protein, prion protein (PrP), whose alteration is critical in the pathogenesis of the transmissible spongiform encephalopathies (TSEs) of mammals. However, it was exceptionally difficult to show unequivocally that prions are composed solely of modified PrP without any prion-specific nucleic acid. Thus, for a long time it remained uncertain whether prions exist as infectious pathogenic proteins in mammals. Only recently has there been a report of the propagation of robust mammalian prions in vitro using semi-purified molecular constituents. This directly implicates PrP conformational change in prion propagation and seems to rule out the need for an agent-specific protein-encoding nucleic acid (3, 4). Meanwhile, in 1994, Reed Wickner argued compellingly that mysterious epigenetic elements of yeast could also be explained by the transfer of aggregated proteins from one cell to another (5). In doing so, Wickner proposed that the prion concept be extended to include these proteinaceous entities that could act as infectious agents and/or convey heritable changes in phenotype without mediation by nucleic acids. Many diverse biological phenomena have now been described as being prions or prion-like. There are several basic requirements for a prion (6). First, there must be a self-propagating state of a protein (the prion) that is biologically accessible but rarely formed spontaneously. Second, prions must replicate themselves by acting on their non-prion substrate protein. And third, prions must spread to naive hosts and find new substrate pools for replication. Typically, prions also cause phenotypic changes in the host.
In this review, we will first describe the challenging hunt for molecularly defined mammalian TSE prions. We will then consider recent insights into their three-dimensional structure, biogenesis, and neuropathogenic effects. Emphasis will be given to a unique aspect of mammalian TSE diseases, that is, the anchoring of the protein of interest (PrP) to membranes by a glycophosphatidylinositol (GPI) moiety. This feature strongly influences TSE prion pathogenicity. Moreover, neuronal membrane abnormalities figure prominently in the pathognomonic lesions of TSE diseases, prompting us to also consider the extent to which membrane tethering plays a role in distinguishing TSE prion biology from the biology of other types of prions or prion-like phenomena in mammals. A number of different proteins of mammals and fungi can be found in altered states with at least some properties of prions. These examples extend the prion concept and help to define its fundamental features and useful limits.
IN VITRO PROPAGATION OF PRIONS
PrP conversion
The most compelling way to demonstrate the composition of prions is to propagate them in vitro using defined biochemical constituents. Mammalian prion propagation is usually associated with the post-translational conversion of the host’s normal, soluble, protease-sensitive PrP (PrPC or PrP-sen) to a less soluble and more proteinase-K resistant state (PrPRES or PrPSc) (7). Initial attempts to propagate prions in extensively purified cell-free systems showed that PrPRES can cause PrPC to convert to a PrPRES-like protease-resistant state in a highly species- and sequence-dependent manner consistent with known prion transmission barriers in vivo (8–11). PrPRES with PrPC converting activity is oligomeric, and, newly converted PrP molecules become associated with PrPRES oligomers (12–14). These and other features of the conversion reaction are consistent with either non-catalytic or autocatalytic (templated) seeded polymerization mechanisms initially outlined by Carleton Gadjusek and Peter Lansbury (Figure 1) (12, 15, 16). PrPRES oligomers of different prion strain-associated conformations are capable of imposing those distinct conformations on PrPC molecules during conversion (8). This provides a potential molecular basis for the propagation of prion strains without the need for a prion-specific nucleic acid. Although the presence of cell-free PrPC converting activity correlates with the presence of prion infectivity (17), the yields of the initial cell-free PrPC conversion reactions were usually substoichiometric relative to the PrPRES seed, and the generation of prion infectivity was not demonstrable (18).
Figure 1.
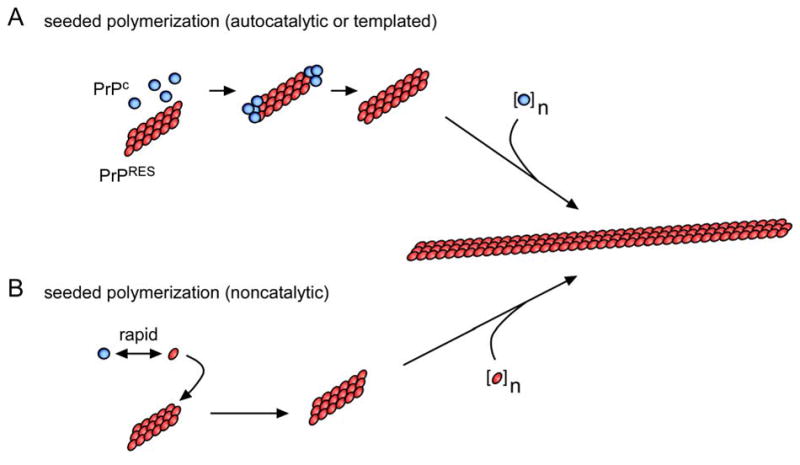
Seeded PrP polymerization mechanisms. In the non-catalytic model, the conformational interchange between the PrPC and PrPRES conformations is rapid, but the PrPRES conformer is poorly populated unless stabilized by binding to an existing PrPRES multimer. In the autocatalytic model, the conformational conversion of PrPC to PrPRES is rare unless catalyzed by contact with an existing PrPRES multimer.
Synthetic prions
More recently, synthetic amyloid fibrils composed of a truncated fragment of recombinant PrPC (PrP residues 90-231) were shown to accelerate clinical disease when inoculated into transgenic mice that overexpress PrP90-231 by ~16-fold (19, 20). Furthermore, brain tissue from these sick mice contained prions that caused TSE disease when inoculated into wild-type mice. These striking results suggested that amyloid preparations of recombinant PrP alone instigated transmissible prion disease, albeit in an unnatural host that, by virtue of PrP overexpression, was likely to be strongly primed for PrP aggregation and susceptibility to both induced and spontaneous prion disease. Importantly, the initial synthetic fibrils had no activity in wild-type mice, implying that these “synthetic prions” were many orders of magnitude less infectious than bona fide scrapie prions. There are several possible explanations for these results: 1) recombinant PrP90-231 amyloid may be infectious, but with extremely poor efficiency; 2) the infectious entity might not be the amyloid itself, but a different form of PrP-containing particle that is present at only trace quantities in the synthetic amyloid preparation; 3) robust recombinant PrP prions might require another molecular constituent [e.g. polyanions such as glycosaminoglycans (GAGs) or RNA (4)]; or 4) the amyloid preparations might not be infectious at all but may accelerate a neurodegenerative process that occurs spontaneously in transgenic mice overexpressing truncated PrP90-231. In this latter case, the infectivity for wild-type mice could have arisen spontaneously in the transgenic mice, as has been reported in other transgenic mice that overexpress PrP mutants (21). Further studies will be required to discriminate between these possibilities.
PMCA
In a major breakthrough, brain-homogenate-based reactions were developed that allowed unlimited amplification of both PrPRES and robust prions that were infectious for wild-type rodents (3, 22, 23). This amplification protocol, called PMCA for protein misfolding cyclic amplification, involves introducing minute amounts of an infectious PrPRES-containing seed into a homogenate of brain from an uninfected animal which provides PrPC and any cofactors that might be needed. The suspension is subjected to cycles of incubation, which allows for seeded polymerization of the PrPC, and sonication, which fragments the polymers to generate more seeds (Figure 2). Two striking features of the PMCA reaction is its sensitivity and amplification power (24). As little as ~1 attogram (10−18 g) or the equivalent of ~20 PrPRES molecules can be detected by this method, providing the basis for extraordinarily sensitive prion assays and potentially, diagnostic tests for prion diseases.
Figure 2.
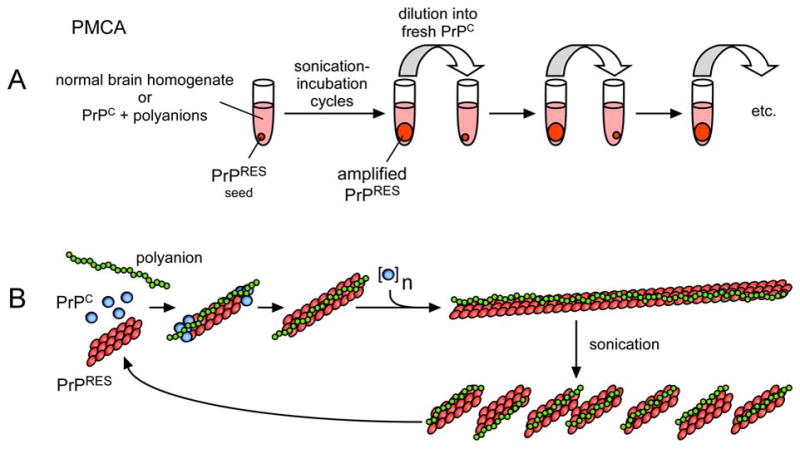
Models of TSE prion amplification by PMCA. A. PMCA flow chart based on (3, 4). B. Seeded polymerization model for PrPRES formation with the aid of a polyanion (e.g. polyA RNA) (4). Adapted from (156).
Prion amplification has been reported in either prion-seeded or spontaneous conversion reactions containing largely purified, brain-derived PrPC and synthetic polyA RNA (Figure 2) (4). Lipids and perhaps other molecules copurify with the PrPC in these preparations and therefore might also be important in prion formation. This seminal report not only provided long-awaited direct support for the PrP-based prion hypothesis, but suggested that other non-protein molecules, such as RNA molecules, are critical either as prion components or accessories in the PrP conformational conversion (25). The spontaneous instigation of prion formation in these experiments may represent an in vitro equivalent of sporadic Creutzfeldt-Jakob disease, which, for epidemiological reasons, is widely assumed to be an example of spontaneous prion disease in humans.
Recombinant PrP-PMCA and related amplification reactions
In attempts to further refine the understanding of prion composition, PMCA-like amplification reactions (rPrP-PMCA and QuIC) have been performed using purified bacterially expressed recombinant PrPC as a substitute for brain-derived PrPC (26, 27). PrPRES-seeded rPrP-PMCA and QuIC reactions produce a protease-resistant, largely beta-sheet, fibrillar PrP recombinant conversion product that serves as a highly amplified marker of sub-femptogram amounts of PrPRES. Furthermore, these and related reactions can be much faster than conventional PMCA reactions, improving prospects for the development of practical prion assays and diagnostic tests (26–28). However, inoculations of hamster rPrP-PMCA product into hamsters have not yet revealed clear evidence of prion infectivity (R. Atarashi, G. Raymond and B. Caughey, unpublished data). Thus, like the truncated recombinant PrP amyloid fibrils called “synthetic prions” (19), the infectivity of the rPrP-PMCA product appears to be much lower than either PrPRES itself or the product of PrPRES amplification in PMCA reactions using brain-derived PrPC, if it proves to be infectious at all. It should be revealing to determine whether the apparently vast difference in infectivity between these various PrP aggregates is a matter of molecular composition, conformation, protease-resistance, ultrastructure, cofactors and/or aggregation conditions. Two notable features of natural PrPC that are lacking on the recombinant PrPC are N-linked glycans and the GPI anchor. Interestingly, these post-translational modifications do not appear to be essential for the generation of infectious prions as indicated by studies of scrapie-infected transgenic mice that express PrPC molecules lacking these modifications (29–31). These observations suggest that conformational details and/or non-PrP molecular cofactors are more critical determinants of prion infectivity.
STRUCTURAL STUDIES OF PrP OLIGOMERS AND FIBRILS
One of the greatest hurdles in understanding PrPRES is the lack of biophysical techniques suitable for determining the high-resolution structures of non-crystalline fibrillar protein assemblies. The preparations of PrPRES that are of the highest purity have most often contained amyloid-like fibrils which have been characterized in terms of fibril dimensions (32–34) and secondary structure composition (35–38). Visualization of the protein core of wild-type prion fibrils can be obscured by heavy glycosylation and GPI anchors (Figure 3A). However, plaques and fibrils containing mostly unglycosylated and anchorless PrPRES have been isolated from scrapie-infected transgenic mice expressing only anchorless PrPC (34). These fibrils are composed of protofilaments 3.0–3.5 nm in width as measured by negative stain transmission electron microscopy (Figure 3A). The protofilament widths and twist periodicities can vary significantly with scrapie strain, indicating strain-dependent fibril ultrastructures. This expands the list of strain-dependent features of PrPRES which includes secondary structures (37, 38), proteolytic sensitivities (39), glycoform patterns (40–43), stabilities (44) and conformational templating activities (8).
Figure 3.
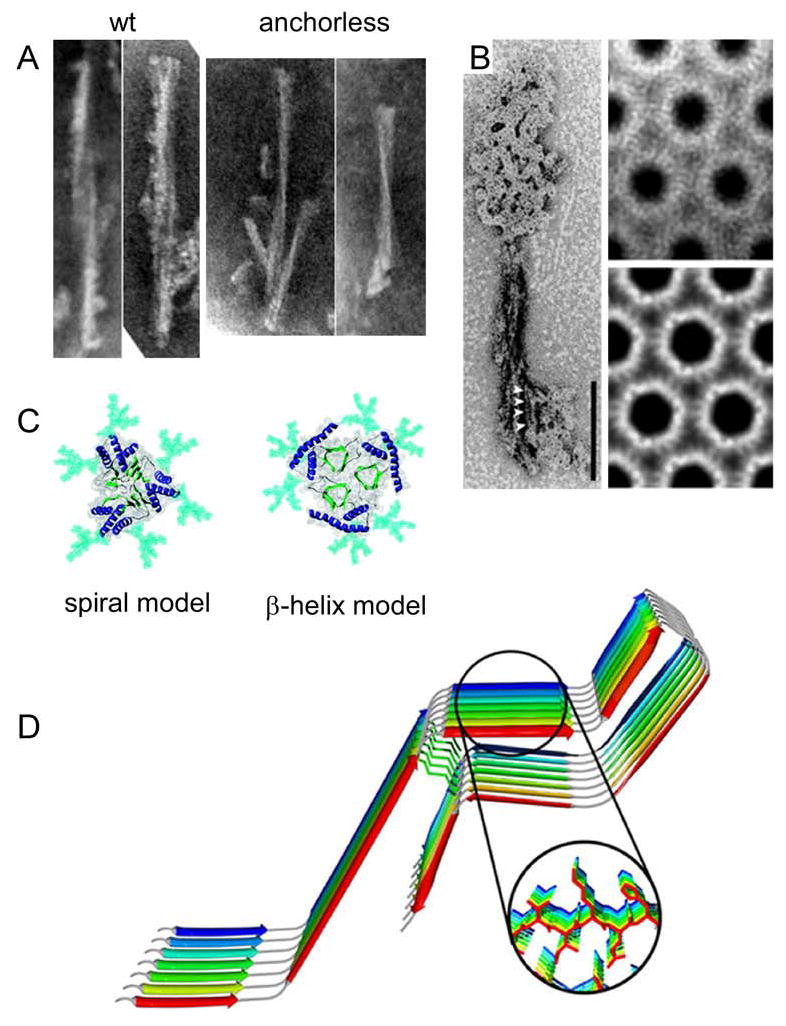
Ultrastructures and models of PrPRES fibrils. A. Negative-stained transmission electron micrographs of proteinase K-treated wild-type or GPI-anchorless PrPRES amyloid fibrils (34). B. Electron micrographs and crystallographically refined images of hexagonal 2D crystals and of proteinase K-treated PrPRES. On the left, the 2D crystals are shown together with fibrils. Adapted from (45). C. Top-down view of PrPRES trimers according to the spiral (47, 48) and β-helix (46) models. Glycans (aqua), α-helices (blue) and β-strands (green) are highlighted. Adapted from (48). D. Parallel, in-register β-sheet model of synthetic fibrils of human PrP residues 120-231. Adapted from (50).
Electron crystallography and modelling
So far, the highest resolution information derived from infectious PrPRES preparations has come from analyses of 2-dimensional crystals that appeared along with fibrils in certain PrPRES preparations (45) (Figure 3B). Image processing and electron crystallography of these structures revealed that they were likely composed of PrP trimers. Based on molecular modelling and mutational analyses, a β-helix model was proposed (46) (Figure 3C). In this model a flexibly disordered domain and helix 1 of PrPC are coiled into a β-helix, which then aligns with the β-helices of two other PrPRES monomers to form a trimer. N-linked glycans are displayed on the outside of the trimer. An alternative model has been developed on the basis of molecular dynamics simulations and docking procedures (47, 48) (Figure 3C). In the resulting spiral model, monomers are joined together via intermolecular β-sheets and can be readily assembled into continuous twisting filaments. Both the β-helix and spiral models are close to being consistent with the protofilament dimensions measured in the anchorless PrPRES fibrils (34); however, the spiral model is more consistent with a variety of other biochemical characteristics of PrPRES (48).
Other probing of fibril structure
Recent studies of synthetic recombinant PrP 90–231 amyloids using hydrogen/deuterium exchange and electron spin resonance provided evidence that residues ~160–220 could form parallel, in-register β-sheet structures that were unlike either the spiral or β-helix models (49, 50) (Figure 3D). In contrast, solid state NMR studies of fibrils of recombinant human PrP residues 23-144, which corresponds to a Gerstmann-Straussler-Scheinker syndrome-associated mutant PrP (Y145stop) in humans, showed that residues 112-144 could form a compact, highly ordered core, while the remainder of the residues toward the N-terminus were largely unordered (51). Earlier studies of disease-inducing fibrils of murine PrP residues 89–143 showed that residues 112–124 formed an extended β-sheet conformation without the β-strands being in a parallel, in-register alignment (52). These and other recent developments in the analysis of a variety of other prion proteins and peptide amyloids (53–64) are laying the foundation for more detailed investigation of the PrPRES structures. Particularly noteworthy are the first high-resolution X-ray diffraction-based structures of amyloid fibrils of short peptides determined by the Eisenberg group (65, 66). One important theme that emerges from these structures is that amyloids can be stabilized by short stretches of amino acid residues that align to form β-sheets which, in turn, are welded to adjacent sheets by the close interdigitation of side chains, forming a dehydrated “steric zipper”.
The most infectious particle
Assessments of the relative infectivity of PrPRES aggregates of various sizes indicated that the most infectious particle per unit protein is ~300–600 kDa, or the mass equivalent of 14–24 PrP monomers, and the smallest oligomer with cell-free PrPC converting activity is larger than a PrP pentamer in the presence of a low concentration of SDS (13). These studies provide evidence that as long as the infectious particles are above a certain size, presumably related to a stability threshold of some sort, it is the concentration of particles rather than the concentration of PrPRES molecules per se that correlates best with infectivity titer. Still, the multimeric nature and heterogeneity of infectious PrP oligomers, as well as uncertainties about the role of other potential molecular constituents, greatly complicates more detailed analyses of their structures by conventional techniques of structural biology.
BIOGENESIS OF PrPRES
One of the difficulties in reconstituting prion propagation with defined molecular constituents may be the recapitulation of the membrane microenvironment that supports PrPRES formation in vivo. The PrPC polypeptide is first synthesized as a nascent GPI-anchored glycoprotein in the endoplasmic reticulum (ER), subjected to glycan modifications in the Golgi apparatus, and then transported to the cell surface (Figure 4) where it is associated primarily with cholesterol-rich raft membrane subdomains. In the case of familial TSE-associated mutant PrPC molecules, spontaneous folding abnormalities can occur in the endoplasmic reticulum and/or Golgi apparatus [reviewed in (67)]. In scrapie-infected cells, the conversion of mature PrPC to PrPRES occurs on the cell surface and/or in endosomal vesicles that are internalized from the cell surface (68–70). In contrast to PrPC, PrPRES is resistant to detergent solubilization, proteinase K, and phosphatidylinositol-specific phospholipase C (which cleaves the GPI anchor). PrPRES, with the apparent assistance of cofactors, directly induces PrPC conversion via an ill-defined templating or seeding mechanism which results in PrPC joining the PrPRES oligomer/polymer. These apparent cofactors include sulfated glycosaminoglycan (GAG)-containing proteoglycans (71–73) and laminin receptor and its precursor (LRP/LR) (74). Although the mechanistic role of these accessory molecules is unclear, there is evidence that each can interact directly with PrP molecules and might therefore i) prime PrPC conformationally for conversion, ii) pre-organize or orient PrPC and PrPRES molecules for efficient conversion and/or iii) help sequester PrPC and PrPRES together in the membrane microdomain or subcellular compartment where efficient conversion occurs. Another possible “cofactor” in conversion could be membrane surfaces, which like GAGs, can be polyanionic and induce conformational changes in PrP molecules (see below). The importance of membrane domain structure is indicated by the influence of alterations in raft membrane components such as cholesterol (75), cholesterol esters (76) and sphingolipids (77) on PrPRES formation, but the mechanism of such effects are not clear.
Figure 4.
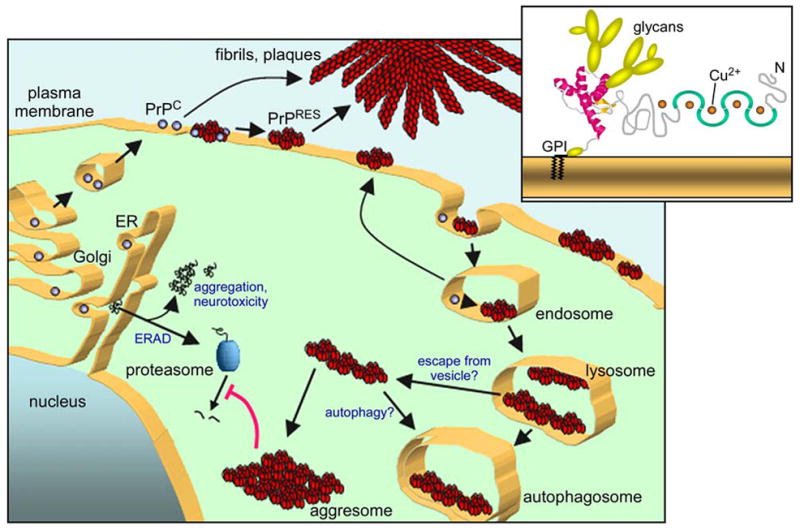
Model of biogenesis and accumulation of PrPRES in scrapie-infected cells. As a GPI-anchored plasma membrane glycoprotein (upper right), PrPC is first synthesized in the endoplasmic reticulum (ER), processed in the Golgi apparatus, and transported to the cell surface (bottom). PrPRES, together with apparent cofactors, directly induces the conversion of GPI-anchored PrPC on the cell surface and/or in endosomes. PrPC that is released from the cell may be converted on extracellular deposits such as amyloid fibrils. Once PrPRES is made, it can accumulate on the cell surface, in intracellular vesicles (e.g. lysosomes) and aggresomes, or in extracellular deposits. Under conditions of mild proteasome inhibition, cytotoxic cytoplasmic PrP aggregates (e.g., aggresomes) can be found (157, 158). Scrapie infection alone can inhibit proteasomes, apparently due to the presence of cytoplasmic PrP oligomers (140).
FIBRILLAR VS NONFIBRILLAR PrPRES DEPOSITS: INFLUENCE OF MEMBRANE ASSOCIATION
Once PrPRES is made, it can accumulate on the cell surface, in intracellular vesicles (e.g. lysosomes), or in extracellular deposits such as amyloid plaques (Figure 4). Amyloid plaques are comprised of bundles of fibrillar protein aggregates with a core β-sheet secondary structure that typically gives a cross-beta X-ray diffraction pattern and characteristic staining by Congo red and thioflavins S and T. In most types of TSE diseases in natural hosts expressing GPI-anchored PrPC, amyloid plaques are uncommon or absent altogether. More typical PrPRES deposits such as those described at the light microscopy level as granular or diffuse punctate patterns (Figure 5) are usually associated with membranous ultrastructures that lack visible fibrils (78). It is unclear whether these PrPRES deposits are structurally distinct from amyloids or simply amyloid fibrils whose visualization is impaired by their small size, irregular length, or association with membranes and/or other structures. It has been reported that amyloid fibrils of PrPRES (scrapie-associated fibrils or prion rods) that are recovered in purified preparations typically arise only when PrPRES is extracted from membranes with detergent (79). Since the evidence that PrPRES can exist in a stable, biologically relevant monomeric state is tenuous, such a result could be consistent with PrPRES existing as a small oligomer or aggregate in its membrane-bound state and assembling into fibrils upon extraction from membranes. A radiation inactivation target size of ~55 kDa suggests that a dimeric form of PrPRES might be the key infectious unit in liposomes (80). On the other hand, detergent-extracted PrPRES preparations can have regular non-fibrillar two-dimensional arrays of PrPRES trimers (45, 46) while size-based fractionations of PrPRES particles in detergents indicate that PrPC converting activity is associated with oligomers larger than PrP pentamers (13). It is clear that PrPRES can also exist in much larger highly stable ordered aggregates under a wide variety of conditions both in vitro and in vivo, with amyloid fibrils and plaques being the most obvious examples in brain tissue (Figure 5). Further study will be required to determine the nature of membrane-bound states of PrPRES in vivo and whether they include individual PrPRES monomers or small oligomers.
Figure 5.
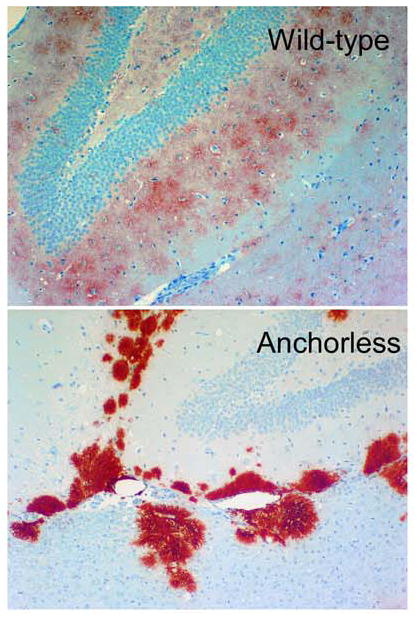
Different patterns of PrPRES deposition in brain tissue of scrapie-infected mice expressing wildtype (top) versus anchorless PrPC (bottom) (29). The sections shown are from the hippocampus in the vicinity of the dentate gyrus. The photos are courtesy of Dr. Bruce Chesebro, Rocky Mountain Laboratories.
Although rare in many TSE diseases, amyloid plaques are especially prominent in humans with familial Gerstmann-Straussler-Scheinker syndrome (GSS) and kuru, and in scrapie-infected transgenic mice expressing only mutated PrPC that lacks a GPI-anchor and is secreted from cells (29). In GSS patients, the abnormal PrP plaque amyloid that accumulates (PrPGSS) is composed primarily of truncated internal PrP fragments that lack the GPI anchor. In scrapie-infected anchorless PrP transgenic mice, PrPRES accumulates almost exclusively in prominent extracellular amyloid plaques that assemble around brain blood vessels (Figure 5). These perivascular plaques have close morphologic similarity to the cerebrovascular plaques of Alzheimer’s disease, including initial fibrilization within capillary basement membranes. This suggests that both the concentration of PrPRES multimers and extracellular matrix components within the endothelial basement membranes are important for the conversion of diffusible forms of PrP into fibrillar amyloid (71, 73, 81, 82). As plaques are formed in several tissues of the scrapie-infected anchorless PrP transgenic mice the anchorless PrPC substrate must be capable of being secreted by a number of different cell types and diffusing to the amyloid plaques for conversion at the growing tips of fibrils.
TSE PRION TRANSMISSION, UPTAKE AND TRANSPORT MECHANISMS
TSE prions can be naturally or experimentally transmitted to new individuals via inoculation into several peripheral sites with subsequent and critical spread to the central nervous system (CNS). After oral challenge, infectivity can be taken up by gut mucosa and Peyer’s patches and then be transferred somehow to intestinal nerve endings (83–85). However, orally acquired prion diseases do not invariably affect lymphoid tissues or the peripheral nervous system. In such situations haematogenous neuroinvasion, most probably involving specialized capillaries within the circumventricular organs of the brain, is a likely alternative route (86). Haematogenous neuroinvasion can also occur following blood transfusion from infected donors (87). With intra-tongue inoculations, transport to the CNS occurs efficiently via cranial nerves (88). Clearly, the spread of prion infections to and within the CNS requires the transfer of prions within and between cells.
PrPRES uptake and acute induction of PrPC conversion
A variety of cultured cell types are capable of sustaining prion infections. Neuronal cells can endocytose PrPRES aggregates and transport them (see Supplemental Material link in the online version of this article or at http://www.annualreviews.org/) via acidic transport vesicles throughout the cytoplasm and along neuritic projections to points of contact with other cells (Figure 6) (89). Concurrently, new PrPRES formation and chronic scrapie infections can be established (89, 90). The uptake mechanism does not appear to be specific to GPI-anchored PrPRES because similar uptake and vesicular transport can occur with other types of amyloid fibrils such as those composed of Alzheimer’s beta peptide or anchorless recombinant PrP. Uptake of exogenous PrPRES can depend upon heparan sulfate-containing proteoglycans [(72, 91), but also consider (92)] and 37/67 kDa laminin receptor (93), but not PrPC (89, 90, 92). However, cell surface PrPC expression is obviously required for new PrPRES formation and the maintenance of scrapie infections. Presumably, contacts between incoming PrPRES and the endogenous PrPC can begin on the cell surface, but the subcellular sites at which the PrPC is converted to new PrPRES remain unclear. Virtually all of the PrPRES produced in cultured N2a neuroblastoma cells is internalized and rapidly truncated at the N-terminus by endolysosomal proteases (69). Evidence for similar events can be found in scrapie brain and lymphoid tissues (94). However, much of the PrPRES is only visualized in vivo at the cell membrane where it usually remains full-length, suggesting that it may be formed and initially accumulated on the cell surface without being endocytosed and exposed to intracellular proteases.
Figure 6.
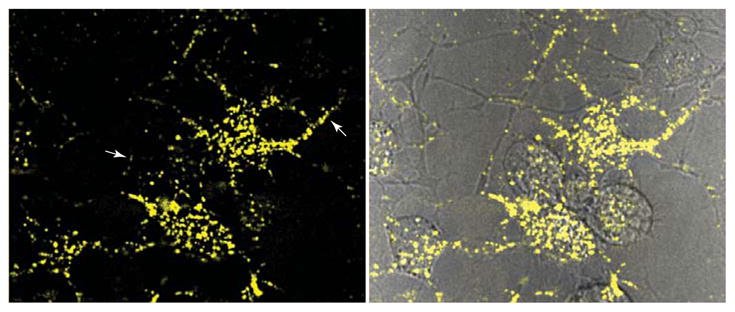
Neuritic transport of PrPRES during acute infection. SN56 mouse neuroblastoma cells were treated with Alexa Fluor 568-labeled mouse PrPRES and imaged at 4 d post-infection. Arrows (left panel) indicate examples of fluorescent PrPRES particles transported within neuritic processes that contact other neurites and cells (89). Left panel shows Alexa Fluor 568 fluorescence. Right panel shows Alexa Fluor 568 fluorescence superimposed on a differential interference contrast image.
Although isolated detergent-extracted PrPRES can instigate PrPC conversion and chronic infection in neuroblastoma cells, PrPRES that is still associated with membranous vesicles is much more efficient at doing so per unit of PrPRES (90, 95). Cell-free conversion reactions have shown that when PrPC is GPI-anchored to membranes, PrPRES can much more readily induce PrPC conversion when the two isoforms are attached to the same membrane (96, 97). On the other hand, if the GPI-anchor of PrPC is cleaved by a phospholipase, then it is free to be converted by PrPRES in a separate membrane. Interestingly, PrPC need not be fully dissociated from its membrane to be converted by PrPRES on a separate membrane. For example, GPI-anchorless PrPC can bind to certain membranes with the help of residues 34-94 in the flexible N-terminal domain and, in this configuration, can be converted by PrPRES on separate membrane vesicles (96). Altogether, these findings support the view that the GPI-anchoring of PrPC to membranes constrains its interactions with PrPRES such that conversion is only efficient when PrPRES is anchored in cis to the same membrane. However, in apparent contradiction to this conclusion are studies showing that TSE infectivity tightly bound to steel surfaces can readily induce infections in vivo (98). At present, it is not clear how to reconcile these observations unless cells can extract PrPRES/infectivity from the wires. Another possibility is that a distinct orientation of steel-bound PrPRES, perhaps with exposed GPI anchors, allows it to induce the conversion of membrane-bound PrPC without dissociating from the steel.
CELL-TO-CELL SPREAD OF TSE PRIONS
Once an individual cell is propagating prions, a central unresolved issue is how the infection then spreads to other cells. Cell proximity appears to be important for efficient spread in cell culture (99). The greater apparent specific infectivity of infectivity associated with membrane-bound PrPRES (95), as well as the greater efficiency of conversion when GPI-anchored PrPC and PrPRES are inserted into the same membranes (96, 97), led us to propose that the cell-to-cell spread of infection might occur primarily via transfer of PrPRES-containing membrane microparticles (Figure 7) (97). Fusion of such particles into recipient cells could insert PrPRES into the plasma membrane in a topology compatible with conversion of GPI anchored PrPC molecules.
Figure 7.

Potential mechanisms of intercellular spread of prions. A) Exosomes/membrane microparticles. Prion-infected cells release PrPRES-containing membrane vesicles that deliver PrPRES (red squares) to membranes of uninfected cells to initiate new PrPRES formation (blue squares) in recipient cells (95–97). These include exosomes, vesicles generated by invagination of the limiting membranes of multivesicular bodies (MVBs) and released by MVB fusion with the plasma membrane (100). Delivery of membrane vesicles could occur at the cell surface (not shown) or in an endocytic compartment. B) Tunneling nanotubes (TNTs). TNTs are visualized as thin intercellular projections (Top view, Merged, arrow) through space (see Side view, Merged) in a co-culture of two cell lines expressing either a GPI-anchored protein tagged with GFP (green) or one tagged with mCherry (magenta) fluorescent protein. The arrowhead (Top view, Merged, white area) shows a TNT that has facilitated the transfer of GFP-tagged protein onto the surface of the mCherry-expressing cell. Side view (Merged) corresponds to a side view of a 3D volume rendering of the area indicated by the dotted rectangle in Top view (Merged). DIC, differential interference contrast image.
Exosomes
Consistent with this idea are observations that PrPC, PrPRES, and scrapie infectivity can be released from cultured scrapie-infected cells in association with small (50–90 nm) vesicles called exosomes (100). Exosomes are assembled in cytoplasmic organelles known as multivesicular bodies and secreted via exocytosis (Figure 7). Released exosomes can then fuse with other cells and deliver their cargos. Appropriately, the topology of exosomes is such that exofacial membrane proteins such as PrPRES would maintain an exofacial topology upon fusion with the plasmalemma of a new cell. Both non-neuronal and neuronal prion-infected cell lines can release exosomes that are infectious for mice, supporting the concept that exosomes may be a significant mechanism for the spread of these infections in vivo (100, 101). However, extensive efforts have failed so far to visualize exosomes in brain tissue (78). Exosomes can be found in germinal centers of lymphoid tissues, where they derive from B cells and not follicular dendritic cells, the cell type which putatively amplifies infectivity. In scrapie infected follicles, B cell-derived exosomes lack demonstrable abnormal PrP (102).
Tunneling nanotubes
Another potential spreading mechanism is the transfer of membrane-associated PrPRES between cells via recently-described entities called tunneling nanotubes (89, 103, 104). Tunneling nanotubes are thin membranous bridges that can form between cells and mediate the transfer of organelles, plasma membrane components (including GPI-anchored proteins), cytoplasmic molecules, and pathogens (Figure 7) (105). Interestingly, tunneling nanotubes can transport vesicles that, like those responsible for neuritic transport of PrPRES (89), are small, acidic and of endosomal/lysosomal origin. Although their existence remains to be documented in vivo, tunneling nanotubes have been observed in a variety of cultured cell types (105). For example, murine retroviruses are known to exploit tunneling nanotube-like cell bridges to spread between cells. These viruses can induce the outgrowth of filapodia from uninfected cells to virus-infected cells and then migrate along the bridge back to the target cell. It is notable that murine retroviruses are also known to enhance the release of scrapie prions into the medium of cultured cells (106). In the latter study, the prions were found in exosome-like structures; however, it is possible that vesicular structures can also be derived from the shearing of tunneling nanotubes.
PrP-MEMBRANE ASSOCIATION AND PATHOGENESIS
The usual proximity between PrP molecules and cellular membranes raises the issue of the influence of these interactions in the neurodegenerative pathology of TSE diseases. The anchorless PrP transgenic mouse model provides striking evidence that, although the lack of GPI-anchored PrP does not prevent PrPRES and prion accumulation, it profoundly alters the manifestations of scrapie infections (29). Indeed, most of the typical clinical and neuropathogical characteristics of scrapie are either absent or greatly reduced in these mice despite the accumulation of brain PrPRES to levels comparable to those in scrapie-infected wild-type mice. This reduced brain damage could be due either to a need for anchored PrPC on brain cells for toxicity induced by PrPRES and/or to a lower pathogenicity of PrPRES amyloid plaques compared to the more dispersed, amorphous, and membrane-associated PrPRES deposits seen in most other TSE/prion diseases. This latter situation would be consistent with the emerging view that in other protein misfolding diseases such as Alzheimer’s disease, it is not amyloid plaques, but smaller misfolded protein oligomers that are the most neuropathologic (107, 108). It is likely that potentially toxic protein aggregates are relatively benign when deposited in the extracellular space as plaques with relatively low surface area-to-mass ratios. Moreover, microglia tend to surround amyloid plaques and may seal them off from exposure to the rest of the brain tissue while attempting to degrade them [eg., (109, 110)]. This is not to say that PrPRES amyloid deposits are innocuous, because they clearly can be associated with severe pathogenic lesions and clinical disease. However, these pathogenic effects are different and appear to be slower to result in clinical deficits than those associated with diffuse types of PrPRES deposits [(29) and B. Chesebro, personal communication].
In TSE-infected wild-type hosts expressing GPI-anchored PrPC, a number of ultrastructural neuropathological lesions of neuronal and/or astrocyte membranes are considered pathognomonic. These include membrane proliferation and microfolding, highly abnormal coated pits, tubulovesicular bodies, and spiral membrane inclusions of axons (111). Other findings that are typical, but not specific for TSEs, include vacuolation, gliosis, degenerate processes, dystrophic axons, autophagic vacuoles, clumped synaptic vesicles, and neuronal death. TSE-associated pathology often involves membrane distortions in proximity to the accumulation of what has been aptly described as disease-associated PrP (PrPd) because it can be labeled specifically in histological sections of infected hosts without determination of the various biochemical properties that are usually used to identify PrPRES. PrPd accumulation corresponds ultrastructurally with abnormal endocytosis, increased endo-lysosomes, and both finger-like extensions and invaginations (i.e., microfolding) of plasma membranes (Figure 8) (78). Extracellular accumulations of PrPd are also observed, as well as suggestions of inter-cellular transfer of PrPd between neurons and/or glia. PrPd can appear to fill the narrow gap (~10 nm) between the closely opposed plasmalemmas (Figure 8A). Some aberrant membrane structures, notably tubulovesicular structures, appear to lack PrPd (78). Tubulovesicular bodies are small (20–35 nm), irregular spherical or short tubular structures that are packed into dendrites and occasionally axon terminals (112, 113). Tubovesicular body clusters have some resemblance to multivesicular bodies but differ in their smaller size, lack of a limiting membrane, and absence of PrPd which suggests that they are not the same as the PrPRES-containing exosomes that may mediate transfer of infectivity between cultured cells.
Figure 8.

Abnormal PrP (PrPd) accumulation and membrane distortions on the surface of neurons and astrocytes in scrapie-infected sheep brain tissue. PrPd is labeled with immunogold particles. A. PrPd on the plasma membrane of a neuron without morphological changes. B. Complex dendrite membrane disturbances (microfolding-asterisks) associated with the formation of abnormal pits (not immunolabeled). C. PrPd accumulation associated with the formation of excess coated pits (arrows) and with the transfer to membranes of adjacent processes (arrow heads). D. coated membrane invaginations, one of which shows a spiral, twisted neck (not immunolabeled). E. PrPd on the plasma membrane of astrocytic processes containing abundant glial filaments. Parts of the plasmalemma of some processes have linear segments (arrowheads) suggesting increased membrane rigidity and incipient fibril formation. Adapted from (78).
How might membrane-associated PrPRES be neurotoxic?
The importance of GPI-anchoring of PrP molecules in TSE pathogenesis is likely connected mechanistically to the distortions of cellular membranes and membrane trafficking associated with TSE disease. When PrPC is anchored to membranes by a GPI (or GPI-like) moiety, it typically assumes the α-helical conformation of native soluble PrPC (114, 115), unless its concentration is raised above a threshold level (116). In the latter situation, fully glycosylated, GPI-anchored PrPC can oligomerize into a form with higher β-sheet content in raft-like membranes, but it is not clear whether the polypeptides penetrate the lipid bilayer. In the absence of GPI-anchors, full-length PrPC and PrP90-231 can bind to lipid membranes and undergo major changes in conformation, protease-resistance and aggregation state (96, 117–122). In doing so, such PrP molecules can disrupt the lipid bilayer (120, 122) and, in the case of the GSS-associated mutant PrP fragment 82–146, form channels (118). Moreover, oligomers of full length anchorless PrP can be neurotoxic when added to culture media or inoculated into the brain (123). When anchorless PrP is artificially expressed in the cytoplasm rather than in the secretory pathway, its interactions with membranes correlates with cytotoxicity (119). Despite these multiple observations of seemingly unhealthy interactions between anchorless PrP molecules and membranes, the fact remains that transgenic mice that express anchorless PrP are normally healthy and, when infected with scrapie, have neuropathological manifestations that are quite different from those of wild-type mice. This again highlights the GPI anchor as a key arbiter in TSE pathogenesis in vivo.
Given that PrPRES tends to be oligomeric, membrane-associated, and difficult for cells to degrade, it is possible that the mere accumulation of PrPRES aggregates on membranes compromises fundamental membrane functions. Because most PrPRES molecules have GPI moieties (124), the GPI anchoring of PrPRES oligomers to membranes should be multivalent. Multivalent membrane interactions should limit the quaternary interactions between PrPRES molecules within the plane of the membrane and may promote sheet-like lateral structures (125) compatible with the 2-D PrPRES crystals visualized by Wille et al. (45). On the other hand, whole-cell atomic force microscopy suggests that fibrillar PrPRES aggregates might be present on the surface of scrapie-infected cells in culture (126).
Whatever the preferred PrPRES quaternary structure(s), it is likely that the accumulation of any relatively large PrPRES aggregate that is multiply anchored to membranes could distort its local structure, composition, flexibility, fluidity, dynamics, integrity and, hence, functionality. The presence of PrPd in abnormal spiral clefts at the cell surface and in endosome-derived subcellular structures points to aberrant attempts by cells to internalize membranes containing PrPd aggregates (Figure 8). Membranes containing intense PrPd accumulation can also be unusually linear and close to short extracellular fibrils (Figure 8E). This suggests that PrPd accumulation might impart an increased rigidity to membranes which could affect PrPd internalization and degradation as well as normal endocytic processes. PrPd often appears to be associated with ubiquitin and clathrin on opposite sides of lipid bilayers of spiral clefts and internalized coated membranous structures (78). Thus ubiquitin tagging and the membrane-inverting forces of clathrin could play roles in the generation of these distorted structures. The packing of PrPd between closely opposed membranes of adjacent cells (Figure 8A) and into narrow spiral clefts (78) also suggests that PrPd, alone or in conjunction with other molecules, may cause abnormal adhesion between membrane surfaces.
A pathological role for GPI-anchored PrPC on neurons?
Following infection, transgenic PrP knockout mice that lack PrP altogether (127) or turn off PrPC expression in adulthood (128) do not exhibit the symptoms of TSE disease. Thus, although other recent data suggest that some aspects of neurological disease associated with inherited PrP mutations might be due to deficits in the apparent neuroprotective activities of PrPC (129), TSE pathogenesis does not appear to be due simply to a loss of PrPC function. On the contrary, there is evidence that clinical disease may require the presence of PrPC as well as PrPRES. For instance, wild-type brain tissue grafts in scrapie-infected PrP knockout mice produce PrPRES that does not cause neuropathology in surrounding tissue that lacks PrPC (130). Moreover, Mallucci and colleagues have shown that cessation of PrPC expression in the neurons of adult scrapie-infected transgenic mice reverses spongiosis and delays clinical disease long term without stopping further accumulation of PrPRES produced in other cell types (131). In addition, transgenic mice expressing PrPC exclusively in neurons are susceptible to scrapie (132). These results suggest that neuronal PrPC expression is needed to mediate most PrPRES-induced neuropathogenesis. However, this is not always true because Raeber and colleagues have shown that TSE disease can develop in scrapie-infected transgenic mice that express PrPC only in astrocytes and not to any detectable extent in neurons (133). The reason for the apparent discrepancy between the Mallucci and Raeber results is not clear but could relate to differences in the strain and species of prion, the sequence of expressed PrPC, and/or the levels of PrPC expression in the respective transgenic mouse models.
Direct and indirect mechanisms of PrPRES neurotoxicity
Collectively, these studies leave open the possibilities of both direct and indirect mechanisms of PrPRES neurotoxicity, which may vary in importance between various TSE disease models or neuroanatomical sites within hosts. Direct PrPRES toxicity might be mediated by the membrane disturbances described above, which could have profound effects on processes such as neuronal homeostasis, intercellular contacts, synaptogenesis, synaptic functions, and axonal transport. Indirect effects of PrPRES could be mediated by perturbations of glial functions that in turn cause neuronal lesions (112, 134, 135). Alternatively PrPRES could corrupt, rather than knock out, PrPC function (136). Transgenic mice expressing various mutant PrPC molecules have revealed that perturbations of PrPC structure, expression level or functions alone can cause neurological disease, even without TSE infection [reviewed in (136)]. It follows then that the accumulation of PrPRES might cause disease by altering the metabolism or activities of PrPC. To decipher these possibilities it would be helpful to understand the physiological function of PrPC, but this remains elusive (104, 137). Nonetheless, a number of studies have provided evidence that PrPC has cytoprotective, anti-apoptotic activities against various cellular stresses [reviewed in (104, 136, 137)]. At the same time, injections of PrP antibodies into the brain can lead to neuronal apoptosis providing evidence that PrPC crosslinking or interference with PrPC-ligand binding can have adverse consequences (138). Thus, it is possible that contact between PrPRES and PrPC could have similar effects in TSE-infected individuals. It has been proposed that PrPC interacts with a physiological ligand in two sites and that disruption of one of those interactions can elicit cell death (139). Still another possibility is that an intermediate or byproduct of PrPRES formation is more neurotoxic than mature PrPRES itself. This scenario could account for the enhanced neuropathogenesis observed in scrapie-infected hosts expressing GPI-anchored PrPC in neurons.
Interestingly, small amounts of PrPRES have been detected in the cytosol and can inhibit the ubiquitin-proteosome system, with oligomeric aggregation intermediates being the most inhibitory in this system (140). At this point, it is unclear which of these direct and indirect mechanisms of PrPRES-associated neuropathogenesis, or some combination thereof, is most important in TSE diseases. It seems unlikely that a single discrete mechanism is at play in all TSE diseases, given their phenotypic diversity.
STRETCHING THE REALM OF PRIONS
Since Prusiner’s coining of the term prion and Wickner’s expansion of the prion concept to fungal epigenetics, a number of biological phenomena have been described as prions or prion-like. These include both functional [reviewed in (141, 142)] and disease-associated amyloids in a variety of organisms. The prions of Saccharomyces cerevisiae and other fungi have become the best characterized both genetically and biochemically [reviewed in (143)]. The [URE3], [PSI+], [PIN+] and [Het-s] prions are amyloid forms of normally soluble proteins, Ure2p, Sup35p, Rnq1p and HETs, respectively. The prion forms of Ure2p and Sup35p are inactive and result in loss-of-function phenotypes for the cell. Rnq1p amyloid primes yeast for [PSI+] formation (144), but the function of soluble Rnq1p is unknown. The HETs protein function is also unknown but its amyloid (prion) form is active in mediating heterokaryon incompatibility in Podospora anserina (145). Thus, unlike most prions, the [Het-s] prion may play a positive physiological role in the host organism. Wickner and Roberts have also reported evidence for a novel non-amyloid prion of S. cerevisiae, [β], which takes the form of a self-activating vacuolar protease B that is necessary for meiosis and aids survival in the stationary phase of the cell cycle. Even a self-activating protein regulatory network, as opposed to an altered form of an individual protein, has been shown to share many properties with fungal prions. In this case, a self-activating MAP kinase cascade appears to be responsible for a non-Mendelian hereditary unit called [C] which is responsible for inheritance, and possibly the cell-to-cell spread, of the crippled growth phenotype in P. anserina (146).
Whether in fungi or mammals, the ability of amyloids to nucleate or seed their own growth gives them self-propagating activity that is at least partly analogous to that of prions. Indeed, some experimental amyloidoses in mammals have shown evidence of being transmissible in the sense that amyloid taken from an amyloidotic individual can sometimes greatly accelerate amyloidosis in another individual. However, it is important to emphasize that such “transmissions” have only been shown under unnatural, experimental conditions in which the recipient animal is highly primed for susceptibility to amyloidosis and would be expected to develop amyloidosis spontaneously given time.
AA amyloidosis
One prototypic example of an apparently transmissible amyloid disease is AA amyloidosis, a malady of humans and animals involving the widespread accumulation of serum amyloid A (AA) protein amyloid, especially in the kidney, liver and spleen. Most cases of AA amyloidosis in mammals appear to occur spontaneously due to chronic inflammation or genetic peculiarities that elevate blood levels of AA and predispose the organism to AA amyloid formation. However, it has long been known that inoculation of extracts of material from amyloidotic mice (a.k.a., amyloid enhancing factor) into other mice can rapidly induce amyloidosis as long as the recipients are themselves strongly primed for amyloidosis by proinflammatory treatments such as silver nitrate injections (147). Evidence now points to aggregated AA as amyloid enhancing factor (148). As with the synthetic prion experiments discussed above (19), these experiments raise the question of whether the amyloid inoculum initiates disease, as is clearly the case with legitimate TSE prions, or merely enhances an ongoing pathogenic process.
AA amyloidosis is wreaking havoc in the captive cheetah population, complicating efforts to rescue this endangered species from extinction (149, 150). Amyloidotic cheetahs shed AA amyloid in the feces, and this fecal amyloid can accelerate AA amyloidosis when injected intravenously into silver nitrate-primed mice (151). Given that murine AA amyloid can accelerate amyloidosis in mice when administered orally, these results suggest a plausible fecal-oral route for the natural transmission of AA amyloidosis in cheetahs. More study will be required to demonstrate the actuality of such a transmission cycle. If proven to be true, cheetah AA amyloidosis might become the first known naturally transmissible, non-PrP prion in mammals. Even so, the question of whether the transmitted amyloid initiates or enhances amyloidosis in naïve cheetahs will remain. Moreover, one striking characteristic of captive cheetahs relative to those in the wild is their chronic inflammation and high blood AA levels (150). These factors could make them susceptible to spontaneous amyloidosis, as is the case with silver nitrate-treated mice. If so, then the high incidence of amyloidosis in captive cheetahs might be due to spontaneous disease rather than infections with amyloid from other animals.
Apolipoprotein AII (ApoAII) amyloidosis
ApoAII is an amyloidogenic protein that causes mouse senile amyloidosis and a hereditary human disease due to a mutation in the APOA2 gene. Like experimental AA amyloidosis, mouse senile amyloidosis affects visceral organs and can be stimulated by the experimental administration of exogenous amyloid via the gastrointestinal tract, possibly after consumption of ApoAII amyloid in the feces of amyloidotic cagemates (152). ApoAII amyloid accumulation also occurs spontaneously in aged mice, raising the possibility that inoculated ApoAII amyloid merely accelerates systemic amyloidosis.
Aβ amyloidoses
In Alzheimer’s disease (AD), cerebral Aβ angiopathy, and, to some extent, in many aged humans who are cognitively normal, Aβ, a small proteolytic fragment of the Aβ precursor protein (βAPP), accumulates in amyloid deposits in the brain. Whether the amyloid form of Aβ is the primary pathogenic entity in AD remains a matter of debate, with recent evidence leaning toward smaller sub-amyloid Aβ oligomers being more neurotoxic (107, 108). Although there is no evidence that AD per se is transmissible, studies in transgenic mouse models of AD have suggested that Aβ amyloidosis can be stimulated by intracerebral injections of brain extracts from AD patients or amyloidotic βAPP transgenic mice (153). Once again, as was the case with the AA and ApoAII amyloidosis inoculation studies noted above, it is unclear whether the Aβ amyloid caused amyloidosis or merely accelerated an ongoing pathogenic process because the transgenic recipients ultimately develop deposits spontaneously without inoculation exogenous of the Aβ amyloid extracts. Another major unresolved issue, when comparing this amyloid-inducing activity with a TSE prion disease, is the extent to which the induced Aβ-amyloidotic lesions can spread from localized sites of seeding by the inoculum to other sites within the host.
CPEB
Another protein that has been described as having prion-like properties is cytoplasmic polyadenylation element binding protein (CPEB) (154). The behavior of ectopically expressed neuronal CPEB or its N-terminal domain in yeast led Si and coworkers to propose that distinct CPEB states in neurons might serve as a mechanism for stabilizing plastic changes in synapses. In their model, a self-perpetuating aggregated state of CPEB is induced in activated synapses, altering local mRNA translation and helping to sustain long term facilitation. Although this is an intriguing potential mechanism for synaptic regulation, it would be difficult to consider it a prion-like phenomenon in neurons because of the lack of CPEB-mediated transmission of the phenotypic change to other cells and organisms.
Amyloid spreading mechanisms?
Consideration of the above examples of prion or prion-like entities raises the key issue of spreading mechanisms. It is now commonplace to find proteins that can exist in two or more states with at least one of those states being able to perpetuate itself via self-seeding or autocatalytic activity. Indeed, most proteins are predicted to be capable of forming amyloids under certain conditions (142). As noted above, the ability of a self-perpetuating protein state to spread between cells and organisms is a hallmark of prions. In yeast and at least some other fungi, the spread of cytoplasmic prions can occur through cytoplasmic exchange or transfer during cell division, mating (in which cells fuse), or cytoduction (a largely experimental abortive mating procedure in which cytoplasmic mixing occurs while nuclear fusion is blocked). In mammals, horizontal prion spreading mechanisms would tend to be much more complex, involving host-to-host transfers through the environment and tissue barriers, and cell-to-cell transfers without overt cytoplasmic mixing. With most mammalian cells, mechanisms for the horizontal exchange of cytoplasmic molecules larger than 1 kDa are not readily apparent, except perhaps in some cases for exosomes and tunneling nanotubes as described above. The apparent lack of such mechanisms would seem to reduce the likelihood of finding purely cytoplasmic prions in mammals.
As noted above, many mammalian diseases lead to the accumulation of extracellular, rather than cytoplasmic, amyloid which might more readily migrate between cells to spread within tissues and hosts. With inducible systemic amyloidoses, such as AA and ApoAII amyloidoses, hematogenous spread appears to occur. Blood-borne amyloid is particularly capable of penetrating some tissues via fenestrated capillaries, which allow passage of large molecules such as proteins (155). On the other hand, access of such blood-borne amyloids to the central nervous system is severely limited. Furthermore, it is not yet clear whether extracellular amyloids, such as Aβ amyloid, that are produced within the brain are capable of spreading effectively from their sites of initial deposition to other regions. If it turns out to be true that various brain amyloids have little capacity to propagate throughout the central nervous system, or to invade the CNS from peripheral tissues, then the hazards of localized amyloid inoculations will likely be small. However, if amyloids can propagate throughout the CNS like TSE prions, then there might be real reason to fear the possibility of amyloid infections. For the time being it appears that, relative to other self-propagating protein aggregates, TSE prions are unusually adept at spreading between and within mammalian hosts by practical routes. TSEs are also the only prion diseases known to involve a protein that is normally GPI anchored, a feature that accounts at least in part for the unique biology and neurovirulence of these devastating transmissible diseases.
KEY TERMS/DEFINITIONS
prion: an infectious protein or self-propagating protein-based element of epigenetic inheritance with the capacity to spread between hosts
protein misfolding disease: disease associated with the deposition of a misfolded host protein
amyloid: protein fibrils exhibiting a cross β-core structure and specific staining characteristics with diagnostic dyes (thioflavins S and T, Congo red)
protofilament: fibrillar protein polymers that form amyloid fibrils by lateral association
SUMMARY LIST
Cell-free propagation of TSE prions has effectively ruled out the need for an exogenous, pathogen-encoding nucleic acid, but polyanions such as poly A RNA, can play an important role in prion replication.
The 3D structure of TSE prions remains largely a mystery, but recent technical advances have greatly enhanced the understanding of the molecular architecture of yeast prions and other amyloid fibrils.
The ability to propagate within and between hosts is a key characteristic of prions which distinguishes them from other autocatalytic protein states.
The GPI anchor distinguishes prion protein from other amyloidogenic proteins and has strong influences on TSE pathogenesis.
TSE pathogenesis involves major membrane disturbances in the brain, which may in part be mediated by the accumulation of abnormal GPI-anchored PrP molecules.
Several non-PrP-based mammalian amyloidoses appear to be transmissible under experimental conditions. Further studies will be required to determine if transmission of such amyloids between hosts under practical circumstances poses a risk to humans or animal health.
FUTURE ISSUES
Complete molecular composition of TSE prions and basis for poor infectivity of amyloid fibrils derived from recombinant PrPC
3D structures of prions and their different strains
Neurotoxic entities in TSE diseases and other protein misfolding diseases
Critical neuropathogenic pathways in TSE diseases
In vivo mechanism(s) of cell-to-cell spread of TSE prions
Natural spreading capacities of disease-associated non-PrP amyloids and amyloid-like protein aggregates: Do they pose risks of infection?
Effective diagnostic tests and therapies for TSE diseases
How widespread and diverse are prions in biology?
Supplementary Material
Supplemental Video. Real-time neuritic transport of fluorescent PrPRES particles. The video shows the trafficking of small fluorescent PrPRES (scrapie prion protein) particles derived from a large cell surface PrPRES aggregate. The particles undergo transport within the cell body and a neuritic projection that makes contact with an adjacent cell (Magalhães et al., 2005).
Acknowledgments
We thank Drs. Valerie Sim, Kelly Barton, Winslow Caughey, Leah Christensen and Kim Hasenkrug for helpful comments on this manuscript. Gary Hettrick provided excellent graphics assistance. This work was supported by the Intramural Research Program of the NIAID, NIH.
ABBREVIATIONS/ACRONYMS
- PrPC
normal cellular isoform of the prion protein (PrP)
- PrPRES
abnormal, partially protease-resistant, prion disease-associated isoform of PrP; largely synonymous with PrPSc
- PMCA
protein misfolding cyclic amplification
- rPrP-PMCA
recombinant PrP PMCA
- QuIC
quaking-induced conversion; an rPrP-PMCA-like reaction that is shaken rather than sonicated
- PrPd
disease-specific isoform of PrP detected by immunological staining of tissues without implying any other biochemical or infectious properties
- TSE
transmissible spongiform encephalopathy
- GPI
glycophosphatidylinositol
- PK
proteinase K
LITERATURE CITED
- 1.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–4. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–6. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae [see comments] Science. 1994;264:566–9. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 6.Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem. 2004;73:617–56. doi: 10.1146/annurev.biochem.72.121801.161837. [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural studies of a major scrapie prion protein. Cell. 1984;38:127–34. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- 8.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Jr, Caughey B. Nongenetic propagation of strain-specific phenotypes of scrapie prion protein. Nature. 1995;375:698–700. doi: 10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- 9.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, et al. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–4. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 10.Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT, Jr, Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci USA. 1995;92:3923–7. doi: 10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raymond GJ, Hope J, Kocisko DA, Priola SA, Raymond LD, et al. Molecular assessment of the transmissibilities of BSE and scrapie to humans. Nature. 1997;388:285–8. doi: 10.1038/40876. [DOI] [PubMed] [Google Scholar]
- 12.Caughey B, Kocisko DA, Raymond GJ, Lansbury PT. Aggregates of scrapie associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem & Biol. 1995;2:807–17. doi: 10.1016/1074-5521(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 13.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, et al. The most infectious prion protein particles. Nature. 2005;437:257–61. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callahan MA, Xiong L, Caughey B. Reversibility of scrapie-associated prion protein aggregation. J Biol Chem. 2001;276:28022–8. doi: 10.1074/jbc.M103629200. [DOI] [PubMed] [Google Scholar]
- 15.Gadjusek DC. Transmissible and nontransmissible amyloidoses: Autocatalytic post-translational conversion of host precursor proteins to beta-pleated configurations. J Neuroimmunol. 1988;20:95–110. doi: 10.1016/0165-5728(88)90140-3. [DOI] [PubMed] [Google Scholar]
- 16.Jarrett JT, Lansbury PT., Jr Seeding “One-Dimensional Crystallization” of Amyloid: A Pathogenic Mechanism in Alzheimer’s Disease and Scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 17.Caughey B, Raymond GJ, Kocisko DA, Lansbury PT., Jr Scrapie infectivity correlates with converting activity, protease resistance, and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J Virol. 1997;71:4107–10. doi: 10.1128/jvi.71.5.4107-4110.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill AF, Antoniou M, Collinge J. Protease-resistant prion protein produced in vitro lacks detectable infectivity. J Gen Virol. 1999;80:11–4. doi: 10.1099/0022-1317-80-1-11. [DOI] [PubMed] [Google Scholar]
- 19.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, et al. Synthetic mammalian prions. Science. 2004;305:673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 20.Legname G, Nguyen HO, Baskakov IV, Cohen FE, DeArmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A. 2005;102:2168–73. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsiao KK, Groth D, Scott M, Yang SL, Serban H, et al. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci U S A. 1994;91:9126–30. doi: 10.1073/pnas.91.19.9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–3. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 23.Weber P, Giese A, Piening N, Mitteregger G, Thomzig A, et al. Cell-free formation of misfolded prion protein with authentic prion infectivity. Proc Natl Acad Sci U S A. 2006;103:15818–23. doi: 10.1073/pnas.0605608103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saa P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem. 2006;281:35245–52. doi: 10.1074/jbc.M603964200. [DOI] [PubMed] [Google Scholar]
- 25.Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, et al. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem. 2007;282:36341–53. doi: 10.1074/jbc.M704447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, et al. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5:211–2. doi: 10.1038/nmeth0308-211. [DOI] [PubMed] [Google Scholar]
- 27.Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods. 2007;4:645–50. doi: 10.1038/nmeth1066. [DOI] [PubMed] [Google Scholar]
- 28.Colby DW, Zhang Q, Wang S, Groth D, Legname G, et al. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci U S A. 2007;104:20914–9. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–9. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 30.Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, et al. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol. 2008;6:e100. doi: 10.1371/journal.pbio.0060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neuendorf E, Weber A, Saalmueller A, Schatzl H, Reifenberg K, et al. Glycosylation deficiency at either one of the two glycan attachment sites of cellular prion protein preserves susceptibility to bovine spongiform encephalopathy and scrapie infections. J Biol Chem. 2004;279:53306–16. doi: 10.1074/jbc.M410796200. [DOI] [PubMed] [Google Scholar]
- 32.Merz PA, Somerville RA, Wisniewski HM, Iqbal K. Abnormal fibrils from scrapie-infected brain. Acta Neuropathol. 1981;54:63–74. doi: 10.1007/BF00691333. [DOI] [PubMed] [Google Scholar]
- 33.Prusiner SB, McKinley MP, Bowman KA, Bendheim PE, Bolton DC, et al. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–58. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 34.Sim VL, Caughey B. Ultrastructures and strain comparison of under-glycosylated scrapie prion fibrils. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 35.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–80. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 36.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion protein. Proc Natl Acad Sci USA. 1993;90:10962–6. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem. 1998;273:32230–5. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 38.Spassov S, Beekes M, Naumann D. Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim Biophys Acta. 2006;1760:1138–49. doi: 10.1016/j.bbagen.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 39.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–68. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Somerville RA, Hamilton S, Fernie K. Transmissible spongiform encephalopathy strain, PrP genotype and brain region all affect the degree of glycosylation of PrPSc. J Gen Virol. 2005;86:241–6. doi: 10.1099/vir.0.80251-0. [DOI] [PubMed] [Google Scholar]
- 41.Kascsak RJ, Rubenstein R, Carp RI. Evidence for Biological and Structural Diversity Among Scrapie Strains. In: Chesebro B, editor. Transmissible Spongiform Encephalopathies: Scrapie, BSE and Related Human Disorders. Berlin-Heidelberg: Springer-Verlag; 1991. pp. 139–152. [DOI] [PubMed] [Google Scholar]
- 42.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–78. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 43.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature. 1996;383:685–90. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 44.Safar J, Wille H, Itri V, Groth D, Serban H, et al. Eight prion strains have PrP(Sc) molecules with different conformations [see comments] Nat Med. 1998;4:1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 45.Wille H, Michelitsch MD, Guenebaut V, Supattapone S, Serban A, et al. Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci U S A. 2002;99:3563–8. doi: 10.1073/pnas.052703499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci U S A. 2004;101:8342–7. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demarco ML, Daggett V. From conversion to aggregation: protofibril formation of the prion protein. Proc Natl Acad Sci U S A. 2004;101:2293–8. doi: 10.1073/pnas.0307178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demarco ML, Silveira J, Caughey B, Daggett V. Structural Properties of Prion Protein Protofibrils and Fibrils: An Experimental Assessment of Atomic Models. Biochemistry. 2006;45:15573–82. doi: 10.1021/bi0612723. [DOI] [PubMed] [Google Scholar]
- 49.Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci U S A. 2007;104:1510–5. doi: 10.1073/pnas.0608447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in-register beta-structure. Proc Natl Acad Sci U S A. 2007;104:18946–51. doi: 10.1073/pnas.0706522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP. Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105:6284–9. doi: 10.1073/pnas.0711716105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim KH, Nguyen TN, Damo SM, Mazur T, Ball HL, et al. Solid-state NMR structural studies of the fibril form of a mutant mouse prion peptide PrP89-143(P101L) Solid State Nucl Magn Reson. 2006;29:183–90. doi: 10.1016/j.ssnmr.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 53.Baxa U, Wickner RB, Steven AC, Anderson DE, Marekov LN, et al. Characterization of beta-sheet structure in Ure2p1-89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry. 2007;46:13149–62. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 54.Shewmaker F, Ross ED, Tycko R, Wickner RB. Amyloids of shuffled prion domains that form prions have a parallel in-register beta-sheet structure. Biochemistry. 2008;47:4000–7. doi: 10.1021/bi7024589. [DOI] [PubMed] [Google Scholar]
- 55.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc Natl Acad Sci U S A. 2006;103:19754–9. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wickner RB, Dyda F, Tycko R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register beta-sheet structure. Proc Natl Acad Sci U S A. 2008;105:2403–8. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–72. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, et al. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, et al. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–8. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wasmer C, Lange A, Van MH, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–6. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 61.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–7. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–9. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 63.Sun Y, Breydo L, Makarava N, Yang Q, Bocharova OV, Baskakov IV. Site-specific conformational studies of prion protein (PrP) amyloid fibrils revealed two cooperative folding domains within amyloid structure. J Biol Chem. 2007;282:9090–7. doi: 10.1074/jbc.M608623200. [DOI] [PubMed] [Google Scholar]
- 64.Anderson M, Bocharova OV, Makarava N, Breydo L, Salnikov VV, Baskakov IV. Polymorphism and ultrastructural organization of prion protein amyloid fibrils: an insight from high resolution atomic force microscopy. J Mol Biol. 2006;358:580–96. doi: 10.1016/j.jmb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 65.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 67.Chiesa R, Harris DA. Prion diseases: what is the neurotoxic molecule? Neurobiol Dis. 2001;8:743–63. doi: 10.1006/nbdi.2001.0433. [DOI] [PubMed] [Google Scholar]
- 68.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem. 1991;266:18217–23. [PubMed] [Google Scholar]
- 69.Caughey B, Raymond GJ, Ernst D, Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol. 1991;65:6597–603. doi: 10.1128/jvi.65.12.6597-6603.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion protein in the endocytic pathway. J Biol Chem. 1992;267:16188–99. [PubMed] [Google Scholar]
- 71.Wong C, Xiong L-W, Horiuchi M, Raymond LD, Wehrly K, et al. Sulfated glycans and elevated temperature stimulate PrPSc dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001;20:377–86. doi: 10.1093/emboj/20.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Horonchik L, Tzaban S, Ben Zaken O, Yedidia Y, Rouvinski A, et al. Heparan sulfate is a cellular receptor for purified infectious prions. J Biol Chem. 2005;280:17062–7. doi: 10.1074/jbc.M500122200. [DOI] [PubMed] [Google Scholar]
- 73.Shaked GM, Meiner Z, Avraham I, Taraboulos A, Gabizon R. Reconstitution of prion infectivity from solubilized protease-resistant PrP and nonprotein components of prion rods. J Biol Chem. 2001;276:14324–8. doi: 10.1074/jbc.M007815200. [DOI] [PubMed] [Google Scholar]
- 74.Leucht C, Simoneau S, Rey C, Vana K, Rieger R, et al. The 37 kDa/67 kDa laminin receptor is required for PrP(Sc) propagation in scrapie-infected neuronal cells. EMBO Rep. 2003;4:290–5. doi: 10.1038/sj.embor.embor768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taraboulos A, Scott M, Semenov A, Avraham D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129:121–32. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pani A, Norfo C, Abete C, Mulas C, Putzolu M, et al. Antiprion activity of cholesterol esterification modulators: a comparative study using ex vivo sheep fibroblasts and lymphocytes and mouse neuroblastoma cell lines. Antimicrob Agents Chemother. 2007;51:4141–7. doi: 10.1128/AAC.00524-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Naslavsky N, Shmeeda H, Friedlander G, Yanai A, Futerman AH, et al. Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J Biol Chem. 1999;274:20763–71. doi: 10.1074/jbc.274.30.20763. [DOI] [PubMed] [Google Scholar]
- 78.Jeffrey M, McGovern G, Goodsir CM, Gonzalez L. Strain-Associated Variations in Abnormal PrP Trafficking of Sheep Scrapie. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00150.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKinley MP, Meyer RK, Kenaga L, Rahbar F, Cotter R, et al. Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J Virol. 1991;65:1340–51. doi: 10.1128/jvi.65.3.1340-1351.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bellinger-Kawahara CG, Kempner E, Groth D, Gabizon R, Prusiner SB. Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology. 1988;164:537–41. doi: 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- 81.Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem. 2005;280:26873–9. doi: 10.1074/jbc.M503973200. [DOI] [PubMed] [Google Scholar]
- 82.Ben-Zaken O, Tzaban S, Tal Y, Horonchik L, Esko JD, et al. Cellular heparan sulfate participates in the metabolism of prions. J Biol Chem. 2003;278:40041–9. doi: 10.1074/jbc.M301152200. [DOI] [PubMed] [Google Scholar]
- 83.Mabbott NA, MacPherson GG. Prions and their lethal journey to the brain. Nat Rev Microbiol. 2006;4:201–11. doi: 10.1038/nrmicro1346. [DOI] [PubMed] [Google Scholar]
- 84.Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- 85.Prinz M, Heikenwalder M, Junt T, Schwarz P, Glatzel M, et al. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature. 2003;425:957–62. doi: 10.1038/nature02072. [DOI] [PubMed] [Google Scholar]
- 86.Siso S, Jeffrey M, Gonzalez L. Neuroinvasion in sheep transmissible spongiform encephalopathies: the role of the haematogenous route. Neuropathol Appl Neurobiol. 2008 doi: 10.1111/j.1365-2990.2008.00978.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 87.Houston F, McCutcheon S, Goldmann W, Chong A, Foster J, et al. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood. 2008 doi: 10.1182/blood-2008-04-152520. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 88.Bartz JC, Kincaid AE, Bessen RA. Rapid prion neuroinvasion following tongue infection. J Virol. 2003;77:583–91. doi: 10.1128/JVI.77.1.583-591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Magalhaes AC, Baron GS, Lee KS, Steele-Mortimer O, Dorward D, et al. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J Neurosci. 2005;25:5207–16. doi: 10.1523/JNEUROSCI.0653-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greil CS, Vorberg IM, Ward AE, Meade-White KD, Harris DA, Priola SA. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology. 2008;379:284–93. doi: 10.1016/j.virol.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hijazi N, Kariv-Inbal Z, Gasset M, Gabizon R. PrPSc incorporation to cells requires endogenous glycosaminoglycan expression. J Biol Chem. 2005;280:17057–61. doi: 10.1074/jbc.M411314200. [DOI] [PubMed] [Google Scholar]
- 92.Paquet S, Daude N, Courageot MP, Chapuis J, Laude H, Vilette D. PrPc does not mediate internalization of PrPSc but is required at an early stage for de novo prion infection of Rov cells. J Virol. 2007;81:10786–91. doi: 10.1128/JVI.01137-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gauczynski S, Nikles D, El-Gogo S, Papy-Garcia D, Rey C, et al. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J Infect Dis. 2006;194:702–9. doi: 10.1086/505914. [DOI] [PubMed] [Google Scholar]
- 94.Jeffrey M, Martin S, Gonzalez L. Cell-associated variants of disease-specific prion protein immunolabelling are found in different sources of sheep transmissible spongiform encephalopathy. J Gen Virol. 2003;84:1033–45. doi: 10.1099/vir.0.18825-0. [DOI] [PubMed] [Google Scholar]
- 95.Baron GS, Magalhaes AC, Prado MA, Caughey B. Mouse-adapted scrapie infection of SN56 cells: greater efficiency with microsome-associated versus purified PrP-res. J Virol. 2006;80:2106–17. doi: 10.1128/JVI.80.5.2106-2117.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor-dependent and - independent prion protein association with model raft membranes on conversion to the protease-resistant Isoform. J Biol Chem. 2003;278:14883–92. doi: 10.1074/jbc.M210840200. [DOI] [PubMed] [Google Scholar]
- 97.Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 2002;21:1031–40. doi: 10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Flechsig E, Hegyi I, Enari M, Schwarz P, Collinge J, Weissmann C. Transmission of scrapie by steel-surface-bound prions. Mol Med. 2001;7:679–84. [PMC free article] [PubMed] [Google Scholar]
- 99.Paquet S, Langevin C, Chapuis J, Jackson GS, Laude H, Vilette D. Efficient dissemination of prions through preferential transmission to nearby cells. J Gen Virol. 2007;88:706–13. doi: 10.1099/vir.0.82336-0. [DOI] [PubMed] [Google Scholar]
- 100.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–8. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol. 2007;211:582–90. doi: 10.1002/path.2145. [DOI] [PubMed] [Google Scholar]
- 102.McGovern G, Jeffrey M. Scrapie-specific pathology of sheep lymphoid tissues. PLoS ONE. 2007;2:e1304. doi: 10.1371/journal.pone.0001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–10. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 104.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–10. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 105.Gerdes HH, Carvalho RN. Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol. 2008;20:470–5. doi: 10.1016/j.ceb.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 106.Leblanc P, Alais S, Porto-Carreiro I, Lehmann S, Grassi J, et al. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J. 2006;25:2674–85. doi: 10.1038/sj.emboj.7601162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 108.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 109.Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Farquhar C. Morphogenesis of amyloid plaques in 87V murine scrapie. Neuropath Appl Neurobiol. 1994;20:535–42. doi: 10.1111/j.1365-2990.1994.tb01007.x. [DOI] [PubMed] [Google Scholar]
- 110.Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Belichenko PV, Brown D, Jeffrey M, Fraser JR. Dendritic and synaptic alterations of hippocampal pyramidal neurones in scrapie-infected mice. Neuropathol Appl Neurobiol. 2000;26:143–9. doi: 10.1046/j.1365-2990.2000.026002143.x. [DOI] [PubMed] [Google Scholar]
- 112.Jeffrey M, Goodsir CM, Race RE, Chesebro B. Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Ann Neurol. 2004;55:781–92. doi: 10.1002/ana.20093. [DOI] [PubMed] [Google Scholar]
- 113.Liberski P. Tht tubulovesicular structures-the ultrastructural hallmark for all prion diseases. Acta Neurobiol Exp. 2008;68:113–21. doi: 10.55782/ane-2008-1679. [DOI] [PubMed] [Google Scholar]
- 114.Eberl H, Tittmann P, Glockshuber R. Characterization of recombinant, membrane-attached full-length prion protein. J Biol Chem. 2004;279:25058–65. doi: 10.1074/jbc.M400952200. [DOI] [PubMed] [Google Scholar]
- 115.Hicks MR, Gill AC, Bath IK, Rullay AK, Sylvester ID, et al. Synthesis and structural characterization of a mimetic membrane-anchored prion protein. FEBS J. 2006;273:1285–99. doi: 10.1111/j.1742-4658.2006.05152.x. [DOI] [PubMed] [Google Scholar]
- 116.Elfrink K, Ollesch J, Stohr J, Willbold D, Riesner D, Gerwert K. Structural changes of membrane-anchored native PrP(C) Proc Natl Acad Sci U S A. 2008;105:10815–9. doi: 10.1073/pnas.0804721105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem. 1999;274:36859–65. doi: 10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- 118.Bahadi R, Farrelly PV, Kenna BL, Kourie JI, Tagliavini F, et al. Channels formed with a mutant prion protein PrP(82–146) homologous to a 7-kDa fragment in diseased brain of GSS patients. Am J Physiol Cell Physiol. 2003;285:C862–C872. doi: 10.1152/ajpcell.00077.2003. [DOI] [PubMed] [Google Scholar]
- 119.Wang X, Wang F, Arterburn L, Wollmann R, Ma J. The interaction between cytoplasmic prion protein and the hydrophobic lipid core of membrane correlates with neurotoxicity. J Biol Chem. 2006;281:13559–65. doi: 10.1074/jbc.M512306200. [DOI] [PubMed] [Google Scholar]
- 120.Wang F, Yang F, Hu Y, Wang X, Wang X, et al. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry. 2007;46:7045–53. doi: 10.1021/bi700299h. [DOI] [PubMed] [Google Scholar]
- 121.Kazlauskaite J, Sanghera N, Sylvester I, Venien-Bryan C, Pinheiro TJ. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry. 2003;42:3295–304. doi: 10.1021/bi026872q. [DOI] [PubMed] [Google Scholar]
- 122.Sanghera N, Pinheiro TJ. Binding of prion protein to lipid membranes and implications for prion conversion. J Mol Biol. 2002;315:1241–56. doi: 10.1006/jmbi.2001.5322. [DOI] [PubMed] [Google Scholar]
- 123.Simoneau S, Rezaei H, Sales N, Kaiser-Schulz G, Lefebvre-Roque M, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007;3:e125. doi: 10.1371/journal.ppat.0030125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry. 1990;29:8879–84. doi: 10.1021/bi00490a001. [DOI] [PubMed] [Google Scholar]
- 125.Caughey B. Interactions between prion protein isoforms: the kiss of death? Trends Biochem Sci. 2001;26:235–42. doi: 10.1016/s0968-0004(01)01792-3. [DOI] [PubMed] [Google Scholar]
- 126.Wegmann S, Miesbauer M, Winklhofer KF, Tatzelt J, Muller DJ. Observing fibrillar assemblies on scrapie-infected cells. Pflugers Arch. 2008;456:83–93. doi: 10.1007/s00424-007-0433-x. [DOI] [PubMed] [Google Scholar]
- 127.Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp H-P, et al. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–82. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 128.Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, et al. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–10. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li A, Piccardo P, Barmada SJ, Ghetti B, Harris DA. Prion protein with an octapeptide insertion has impaired neuroprotective activity in transgenic mice. EMBO J. 2007;26:2777–85. doi: 10.1038/sj.emboj.7601726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–43. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 131.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–4. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 132.Race R, Oldstone M, Chesebro B. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: Role of prion protein expression in peripheral nerves and spleen. J Virol. 2000;74:828–33. doi: 10.1128/jvi.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Raeber AJ, Race RE, Brandner S, Priola SA, Sailer A, et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 1997;16:6057–65. doi: 10.1093/emboj/16.20.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marella M, Gaggioli C, Batoz M, Deckert M, Tartare-Deckert S, Chabry J. Pathological prion protein exposure switches on neuronal mitogen-activated protein kinase pathway resulting in microglia recruitment. J Biol Chem. 2005;280:1529–34. doi: 10.1074/jbc.M410966200. [DOI] [PubMed] [Google Scholar]
- 135.Giese A, Brown DR, Groschup MH, Feldmann C, Haist I, Kretzschmar HA. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998;8:449–57. doi: 10.1111/j.1750-3639.1998.tb00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–7. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 137.Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 138.Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–6. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- 139.Watts JC, Westaway D. The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta. 2007;1772:654–72. doi: 10.1016/j.bbadis.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 140.Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26:175–88. doi: 10.1016/j.molcel.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 141.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–24. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 142.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 143.Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol. 2007;5:611–8. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)] Cell. 2001;106:171–82. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 145.Coustou-Linares V, Maddelein ML, Begueret J, Saupe SJ. In vivo aggregation of the HET-s prion protein of the fungus Podospora anserina. Mol Microbiol. 2001;42:1325–35. doi: 10.1046/j.1365-2958.2001.02707.x. [DOI] [PubMed] [Google Scholar]
- 146.Kicka S, Bonnet C, Sobering AK, Ganesan LP, Silar P. A mitotically inheritable unit containing a MAP kinase module. Proc Natl Acad Sci U S A. 2006;103:13445–50. doi: 10.1073/pnas.0603693103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kisilevsky R. Preparation and propagation of amyloid-enhancing factor. Methods Mol Biol. 2005;299:237–41. doi: 10.1385/1-59259-874-9:237. [DOI] [PubMed] [Google Scholar]
- 148.Lundmark K, Westermark GT, Nystrom S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A. 2002;99:6979–84. doi: 10.1073/pnas.092205999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Papendick RE, Munson L, O’Brien TD, Johnson KH. Systemic AA amyloidosis in captive cheetahs (Acinonyx jubatus) Vet Pathol. 1997;34:549–56. doi: 10.1177/030098589703400602. [DOI] [PubMed] [Google Scholar]
- 150.Munson L, Terio KA, Worley M, Jago M, Bagot-Smith A, Marker L. Extrinsic factors significantly affect patterns of disease in free-ranging and captive cheetah (Acinonyx jubatus) populations. J Wildl Dis. 2005;41:542–8. doi: 10.7589/0090-3558-41.3.542. [DOI] [PubMed] [Google Scholar]
- 151.Zhang B, Une Y, Fu X, Yan J, Ge F, et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc Natl Acad Sci USA. 2008 doi: 10.1073/pnas.0800367105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xing Y, Nakamura A, Chiba T, Kogishi K, Matsushita T, et al. Transmission of mouse senile amyloidosis. Lab Invest. 2001;81:493–9. doi: 10.1038/labinvest.3780257. [DOI] [PubMed] [Google Scholar]
- 153.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 154.Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. 2003;115:879–91. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 155.Walker LC, LeVine H, III, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006;29:438–43. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lee KS, Caughey B. A simplified recipe for prions. Proc Natl Acad Sci U S A. 2007;104:9551–2. doi: 10.1073/pnas.0703910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–5. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- 158.Kristiansen M, Messenger MJ, Klohn PC, Brandner S, Wadsworth JD, et al. Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J Biol Chem. 2005;280:38851–61. doi: 10.1074/jbc.M506600200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video. Real-time neuritic transport of fluorescent PrPRES particles. The video shows the trafficking of small fluorescent PrPRES (scrapie prion protein) particles derived from a large cell surface PrPRES aggregate. The particles undergo transport within the cell body and a neuritic projection that makes contact with an adjacent cell (Magalhães et al., 2005).