

Abstract
Transgenic mice expressing the Notch 4 intracellular domain (ICD) (Int3) in the mammary gland have two phenotypes: arrest of mammary alveolar/lobular development and mammary tumorigenesis. Notch4 signaling is mediated primarily through the interaction of Int3 with the transcription repressor/activator Rbpj. We have conditionally ablated the Rbpj gene in the mammary glands of mice expressing whey acidic protein (Wap)-Int3. Interestingly, Rbpj knockout mice (Wap-Cre+/Rbpj−/−/ Wap-Int3) have normal mammary gland development, suggesting that the effect of endogenous Notch signaling on mammary gland development is complete by day 15 of pregnancy. RBP-J heterozygous (Wap-Cre+/Rbpj−/+/ Wap-Int3) and Rbpj control (Rbpjflox/flox/Wap-Int3) mice are phenotypically the same as Wap-Int3 mice with respect to mammary gland development and tumorigenesis. In addition, the Wap-Cre+/Rbpj−/−/Wap-Int3-knockout mice also developed mammary tumors at a frequency similar to Rbpj heterozygous and Wap-Int3 control mice but with a slightly longer latency. Thus, the effect on mammary gland development is dependent on the interaction of the Notch ICD with the transcription repressor/activator Rbpj, and Notch-induced mammary tumor development is independent of this interaction.
Keywords: RBP-J, Int3, tumorigenesis
Introduction
The Notch signaling pathway is an evolutionarily conserved intercellular signaling mechanism (reviewed by Callahan and Egan, 2004). Genes of the Notch family encode transmembrane receptors that interact with membrane-bound ligands encoded by the Delta/Serrate/ Jagged gene families. The signal induced by ligand binding is transmitted by a process involving proteolytic cleavage of the receptor by γ-secretase followed by nuclear translocation of the Notch intracellular domain (ICD). The Notch ICD translocates to the nucleus and serves as a transcription activator. The Notch ICD does not possess DNA binding activity; rather it associates with the transcription factor Rbpj, the primary transcriptional mediator of canonical Notch signaling. The Notch-ICD–Rbpj complex transactivates promoters containing Rbpj-binding sites (Kato et al., 1996).
Evidence for a link between Notch signaling and mammary tumorigenesis came from observations that integration of the mouse mammary tumor virus (MMTV) into the Notch4 gene leads to the formation of mammary tumors (Gallahan and Callahan, 1987). MMTV integration into Notch4 results in the transcription of a truncated Notch4 mRNA species (designated as Int3) that represents a gain-of-function mutation (Robbins et al., 1992; Kordon et al., 1995; Gallahan and Callahan, 1997; Raafat et al., 2004). Expression of Int3 under control of mammary-specific regulatory elements in transgenic mice confirmed that activation of Notch signaling leads to the establishment of mammary tumors in 100% of female mice (Jhappan et al., 1992; Gallahan et al., 1996). Expression of Int3 as a transgene from the whey acidic protein (Wap) promoter or the MMTV-Long terminal repeat (LTR) in transgenic mice blocks normal mammary lobular development and the ability of these females to lactate (Jhappan et al., 1992; Smith et al., 1995; Gallahan et al., 1996). Wap- and MMTV-Int3 mice develop mammary tumors with 100% penetrance in breeding and nulliparous females (Jhappan et al., 1992; Smith et al., 1995; Gallahan et al., 1996).
A variant Int3 RNA species was detected in certain human tumor cell lines and was designated as h-Int3sh (Imatani and Callahan, 2000). Int3sh is missing sequences encoding the Rbpj-binding region (RAM23) of the Notch4 ICD. We have shown that h-Int3sh can still activate transcription from the Hes1 promoter, although at a reduced efficiency (Raafat et al., 2004). Interestingly, mammary gland development in transgenic mice overexpressing h-Int3sh under the control of Wap promoter (Wap-h-Int3sh) appears normal and the females lactate (Raafat et al., 2004). However, multiparous Wap-h-Int3sh females develop mammary tumors. These results are compatible with the concept of a gradient in Notch4 signaling affecting mammary gland development and tumorigenesis. In this model, impairment of mammary gland development requires high levels of Notch signaling, whereas induction of mammary tumors by activated Notch4 would require lower levels of Notch signaling. The aim of this study was to investigate, in vivo, the biological role of Int3-Rbpj-dependent and Int3-Rbpj-independent signaling pathways in mammary gland development and tumorigenesis.
Results
Inactivation of Rbpj and mammary gland development
The MMTV LTR-Int3 and Wap-Int3 transgenic mice exhibit two phenotypes with 100% penetrance: lack of mammary alveolar/lobular development and mammary tumor development, respectively (Jhappan et al., 1992; Gallahan et al., 1996). To determine whether these phenotypes are a consequence of a canonical Int3/Rbpj signaling pathway, we conditionally deleted exons 6 and 7 of the Rbpj gene in the presence of the mammary gland-specific Wap-Cre transgene (Figure 1a). Exons 6 and 7 encode the DNA-binding and Notch-binding domains, and loss of these exons results in the complete loss of Rbpj-mediated Notch signaling (Han et al., 2002). In addition, RT–PCR analysis of total RNA extracted from three independent tumors of Rbpj knockout and Rbpj control mice showed that only the Rbpj knockout tumor RNAs are negative for neomycin phoshotransferase (Neo) RNA (Figure 1b). Through a series of genetic crosses between Wap-Cre, Rbpjflox/flox and Wap- Int3 mice, female mice with the following genotypes were obtained: Wap-Cre+/Rbpj−/−/Wap-Int3 (Wap-Int3/ Rbpj knockout); Wap-Cre+/Rbpj −/+/Wap-Int3 (Wap- Int3/Rbpj heterozygous); and Rbpjflox/flox/ Wap-Int3 (Wap-Int3/Rbpj control) (see Materials and Methods). Interestingly, the Wap-Cre+/Rbpj−/− females that were used in the generation of the Rbpj knockout mice had no detectable phenotype with regard to mammary gland development. This suggests that the contribution of endogenous Notch/Rbpj signaling to mammary alveolar/ lobular development is completed before the expression of the Wap-Cre transgene.
Figure 1.

Genotyping and analysis of mice. Wap-Cre/Rbpjflox/flox mice were genetically crossed with Wap-Cre/ Rbpj+/flox/Wap-Int3 mice, to generate Wap-Cre+/Rbpjflox/flox/Wap-Int3 (Wap-Int3/Rbpj knockout), Wap-Cre+/Rbpjflox/+/Wap-Int3 (Wap-Int3/Rbpj heterozygous) and Rbpjflox/flox/Wap-Int3 (Wap-Int3/Rbpj control) mice, as described in the Materials and methods. (a) PCR tail DNA analysis of Wap-Cre/Rbpj −/−/Wap-Int3 (lane 1), Wap-Cre/Rbpj−/+/Wap-Int3 (lane 2) and Rbpjflox/flox/Wap-Int3 (lane 3, note that because of the large size of the neomycin phosphotransferase cassette inserted in Rbpj intron 7, Wap-Int3/Rbpj control mice test negative for Rbpj in the tail DNA analysis). (b) Neo RT–PCR analysis of total RNA extracted from three each of Wap-Int3/Rbpj knockout and Wap-Int3/Rbpj control tumors. Only the Wap-Int3/Rbpj knockout tumor RNAs are negative for Neo. RT–PCR, reverse transcriptase PCR.
The functional unit in the mammary gland during lactation is the alveolus, which produces and secretes milk. In the normal mammary gland, the lobules and their alveoli start to expand at day 5 of pregnancy (Oakes et al., 2006) and continue their growth and differentiation during gestation to form functionally active glandular structures that secrete milk proteins. However, as shown in Figure 2 and earlier (Gallahan et al., 1996), by day 1 of lactation, expression of Wap-Int3 in Wap-Int3/Rbpj control mice exhibits an impaired ability to form functionally competent alveolar/lobular structures as observed in whole mounts and histological sections of mammary glands (panels a and e, respectively) as compared with normal lactating wild-type FVB/N mammary glands (panels d and h, respectively). Similarly, alveolar/lobular development in mammary glands from day 1 lactating Wap-Int3/Rbpj heterozygous mice are inhibited to a similar degree, as these females are unable to nurse their pups (Figures 2b and f, respectively). To determine whether the lack of alveolar/ lobular development in the mammary glands of Wap-Int3/Rbpj heterozygous and Wap-Int3/Rbpj control mice was also reflected in milk composition, β-casein levels were measured by immunohistochemistry (IHC) (Figure 2). β-Casein production was high in the FVB/N mammary gland (Figure 2l) and not detectable in the mammary glands of Wap-Int3/Rbpj heterozygous (panel j) and Wap-Int3/Rbpj control mice (panel i).
Figure 2.

Morphology, histology and immunohistochemical analysis of Wap-Int3/RBP-J knockout mammary glands. Photomicrographs of mammary gland wholemounts (a–d); histological sections (e–h); and β-casein immunohistochemistry (i–l) in the Wap-Int3/ Rbpj control (panels a, e, and i), Wap-Int3/Rbpj heterozygous (panels b, f and j), Wap-Int3/Rbpj knockout (panels c, g and k) and FVB (d, h and l) mice collected from the number 4 inguinal mammary gland from day 1 lactating mice. Of the experimental mice, only the Wap-Int3/Rbpj knockout mice were able to lactate, showed normal alveolar development (c, g) and were positive for the milk protein, β-casein (k), confirming the morphological and histological observations. Panels a–h are at ×10 original magnification and panels i–l are at ×40 original magnification. Each treatment group contained at least 10 mice.
The effects of Wap-Int3 expression on mammary gland development are not manifested in Wap-Int3/Rbpj knockout females. The morphology of the mammary gland (in the whole mount and histology section Figure 2c and g, respectfully) from day1 lactating females is very similar to the normal FVB/N mammary gland, and β-casein can be detected in the alveolar/lobular structures (Figure 2k). In addition, the Wap-Int3/Rbpj knockout females can successfully nurse their pups.
Mammary tumorigenesis is independent of Rbpj function
Morphological analysis of mammary glands revealed the presence of focal hyperproliferative lesions arising within the mammary ducts of Wap-Int3/Rbpj knockout, Wap-Int3/Rbpj heterozygous and Wap-Int3/Rbpj control females (Figure 3a). Irrespective of the genotype, the number of the lesions averaged 24 per gland (Figure 3b). By the second pregnancy, 80% of the Wap-Int3/Rbpj heterozygous and Wap-Int3/Rbpj control mice developed mammary tumors (Figure 3c). However, tumor-free survival was longer in the Wap-Int3/Rbpj knockout females: 80% of them developed tumors after three or four pregnancies. Thus, although the loss of Rbpj function seems to have no effect on Int3-induced mammary tumorigenesis, it may affect the rate of tumor cell proliferation.
Figure 3.

Mammary gland lesions, overall tumor-free survival and tumor histological analysis. (a) A representative wholemount from Wap-Int3/Rbpj knockout mouse showing several hyperproliferative lesions in the mammary gland; (b) the frequency of hyperproliferative lesions in Wap-Int3/Rbpj knockout, Wap-Int3/Rbpj heterozygous and Wap-Int3/Rbpj control mice in the fourth inguinal mammary gland, after the second parity; (c) the overall tumor-free survival of Wap-Int3/Rbpj control, Wap-Int3/Rbpj heterozygous and Wap-Int3/Rbpj knockout mice; histopathology of solid mammary adenocarcinomas from (d) Wap-Int3/Rbpj knockout tumor, (e) Wap-Int3/Rbpj heterozygous tumor, and (f) Wap-Int3/Rbpj control tumor; and (g) a papillary adenocarcinoma from Wap-Int3/Rbpj knockout mouse, (h) a Wap-Int3/Rbpj heterozygous tumor and (i) a Wap-Int3/Rbpj control tumor. All figures were hematoxylin-and-eosin-stained and are at ×40 original magnification.
Histological analysis of the primary mammary tumors showed that they are primarily solid (Figures 3d–f) or papillary mammary adenocarcinomas (Figures 3g–i). The solid tumors from Wap-Int3/Rbpj knockout (panel 3d), Wap-Int3/Rbpj heterozygous (panel 3e) and Wap-Int3/Rbpj control (panel 3f) mice consist mostly of tightly packed, eosinophilic epithelial tumor cells. Rare mitotic figures can be identified. In the papillary adenocarcinoma from a Wap-Int3/Rbpj-knockout (panel 3g) mouse, papillary structures project toward a lumen containing eosinophilic material and numerous apoptotic bodies and consist predominantly of a monolayer of tumor epithelial cells lining an identifiable stromal axis (arrows). The papillary structures shown in Figure 3h (arrows) in a Wap-Int3/Rbpj heterozygous tumor are irregular and consist of hyperchromic cells with areas of more florid proliferation. The stromal axis is not as evident within the papillary formations. Streams of dense connective cells can be identified surrounding the papillary lesion. In the Wap-Int3/Rbpj control tumor shown in Figure 3i, the papillary formations are smaller and less numerous (arrows) and are mixed with microglandular structures. Epithelial tumor cells are more tightly packed and regular in shape and size and the stromal component is less evident.
To confirm the malignant nature of the Wap-Int3/ Rbpj knockout tumors, fragments of nine independent tumors were transplanted bilaterally into the mammary glands of nine female nude mice. These were compared with three independent Wap-Int3/Rbpj control tumors transplanted into six mammary glands in nude mice. Transplantation of both Wap-Int3/Rbpj knockout and control tumors was 100% successful (data not shown). The transplanted Wap-Int3/Rbpj knockout tumors had a latency of 8 weeks, whereas the control tumors were detectable at 5 weeks of transplantation.
Molecular consequences of Rbpj inactivation in Wap-Int3/Rbpj knockout mammary tumors
The conditional knockout of Rbpjflox/flox has two molecular consequences: the loss of the Neo gene inserted between the lox sites located in intron 5 and intron 7 and the creation of a frame shift in the open reading frame of Rbpj exon 8 leading to the expression of a truncated Rbpj protein (Han et al., 2002). To ascertain the extent to which Rbpj was knocked out in mammary hyperplasia and primary and transplanted Wap-Int3/Rbpj knockout tumors, sections of each were analysed by IHC for Neo (Figures 4A, a–f) and Rbpj (Figures 4B, a and b). There were virtually no cells expressing Neo in the Wap-Int3/Rbpj knockout hyperplasia (panel 4Aa), primary (panel 4Ac) or transplanted (panel 4Ae) tumors. In contrast, each of the corresponding tissues from Wap-Int3/Rbpj control mice stained positive for Neo (panels 4Ab, d and f, respectively). These results were quantified as shown in Figures 4Ag–i and demonstrate that Neo was virtually undetectable in the Wap-Int3/Rbpj knockout tissue. This conclusion was confirmed using an antibody that reacts with the C′ terminus of Rbpj and therefore should only react with wild type Rbpj. As shown in Figure 4Ba, the vast majority of Wap-Int3/Rbpj knockout tumor cells were not expressing full-length or wild-type Rbpj whereas in the Wap-Int3/Rbpj control tumor (Figure 4Bb), the tumor cells were stained and easily quantified (Figure 4Bc).
Figure 4.
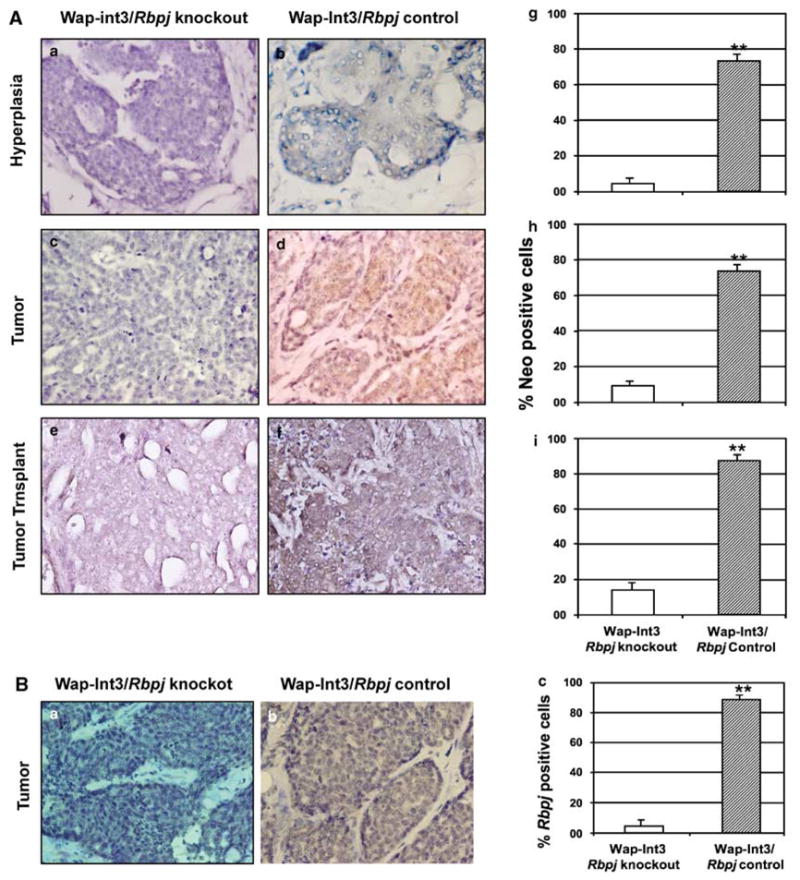
Photomicrographs of immunohistochemical staining of Neo and Rbpj in Wap-Int3/Rbpj knockout hyperplasia, primary mammary tumor and transplanted mammary tumor. (A) IHC analysis of Neo in Wap-Int3/Rbpj knockout (a, c and e) and Wap-Int3/ Rbpj control (b, d and f) hyperplasia (a, b), primary tumor (c, d) and tumor transplants (e, f). Only Wap-Int3/Rbpj control tissue was positive for Neo. Positive cells were scored in the hyperplasia (g), tumor (h) and mammary transplants (i) and labeling index was expressed as a percentage of positive nuclei of 3000 counted cells. In all tissues, neomycin phosphotransferse was significantly lower in Rbpj−/−/Wap-Int3 than Rbpj−/+/Wap-Int3. (B) IHC analysis of Rbpj using an antibody that recognizes the C′-terminal end of the protein (that is, wild-type protein) in a Wap-Int3/Rbpj knockout tumor (a) and Wap-Int3/Rbpj control tumor (b). Positive cells were scored in each and labeling index expressed as a percentage of positive nuclei of 3000 counted cells (c). A total of at least 5–6 mice were used for each experiment. *P<0.05. Original magnification was at ×40. IHC, immunohistochemistry.
Int3 does not activate the expression of Hes1 or Hey2 in the absence of Rbpj
At a molecular level, the loss of active Rbpj should result in a loss of expression of the canonical Rbpj target genes such as Hes1 or Hey2. Therefore, we have stained sections of Wap-Int3/Rbpj knockout andWap-Int3/Rbpj control mammary hyperplasias and primary and transplanted mammary tumors with Hes1 (Figure 5A, compare a, c and e with b, d and f, respectively) or Hey2 antibodies (compare Figure 5Ba and b). The number of cells staining positive for Hes1 (Figures 5Ag–i) and Hey2 (Figures 5Bc) was quantified. In the hyperplasia and primary tumors, there was virtually no Hes1- or Hey2-staining cells in the knockout tissue. The cells that were stained for Hes1 in the transplanted tumor may correspond to infiltrating host vasculature and epithelium. Taken together, these results are consistent with the concept that the growth of Int3-induced tumors is independent of Notch/Rbpj signaling, but does not formally exclude the possibility that Rbpj-dependent signaling is necessary for the initiation of tumorigenesis.
Figure 5.
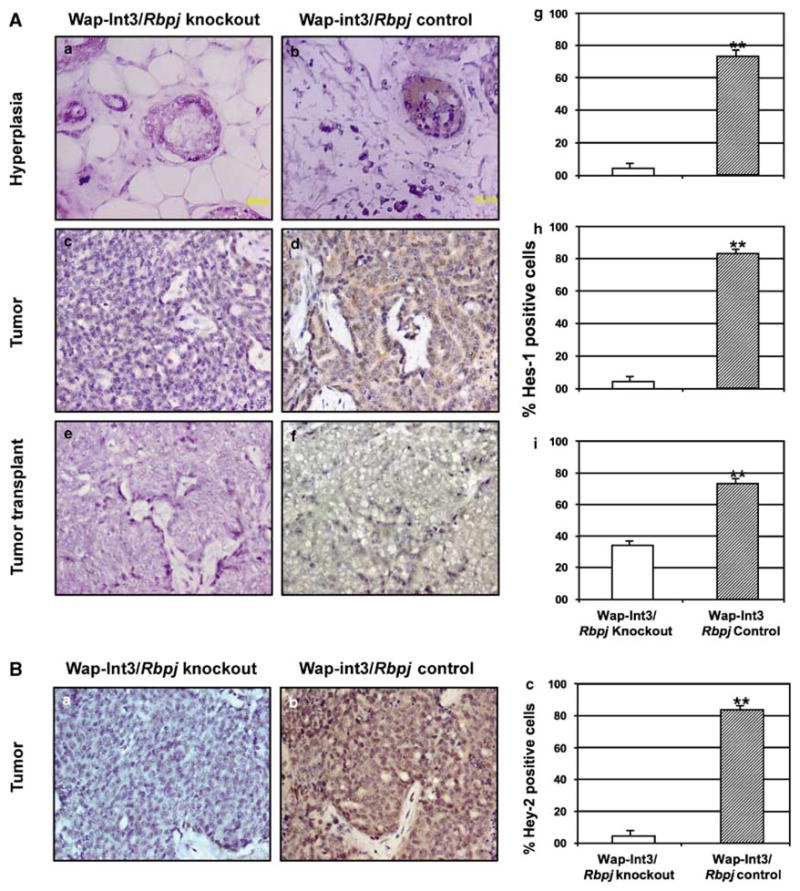
Photomicrographs of immunohistochemical staining of Hes1 and Hey2 in Wap-Int3/Rbpj knockout hyperplasia, primary mammary tumor and transplanted mammary tumor. (A) IHC analysis of Hes-1 in Wap-Int3/Rbpj knockout (a, c and e) and Wap-Int3/ Rbpj control (b, d and f) hyperplasia (a, b), primary tumor (c, d) and transplanted tumor (e, f). Only Wap-Int3/Rbpj control tissue was positive for Hes-1. Positive cells were scored in the hyperplasia (g), tumor (h) and transplanted tumor (i) and labeling index was expressed as a percentage of positive nuclei of at least 3000 counted cells. In all tissues, Hes-1 was significantly lower in Wap-Int3/Rbpj knockout than Wap-Int3/Rbpj control. (B) IHC analysis of Hey2 in a Wap-Int3/Rbpj knockout tumor (a) and Wap-Int3/Rbpj control tumor (b). Positive cells were scored in each, and labeling index was expressed as a percentage of positive nuclei of 3000 counted cells (c). Hey2 was detected only in the Wap-Int3/Rbpj control tumor. A total of 5–6 mice were used for each experiment. *P<0.05. Original magnification was at ×40. IHC, immunohistochemistry.
Cell proliferation and apoptosis in WAP-Int3/Rbpj control and knockout tumors
We examined the effects of Rbpj deletion on cell proliferation (Figure 6a–c) and apoptosis (Figure 6d–f) of the Wap-Int3 tumors. Rbpj deletion did not result in a significant difference in the level of cell proliferation between control and knockout mice (Figure 6c). However, the level of apoptosis is significantly higher in the Rbpj control tumors than in the Rbpj knockout tumors (Figure 6f).
Figure 6.

In vivo effect of Rbpj deletion on proliferation and apoptosis of WAP-Int3/Rbpj knockout and control mammary tumors. WAP-Int3/Rbpj knockout (a and d) and control (b and e) tumor-bearing mice were euthanized and mammary tumor tissue was collected and processed for proliferation (a and b) and apoptosis (d and e) as described in the Materials and methods. Deletion of Rbpj in the WAP-Int3 mice did not affect the tumor proliferation; however, apoptosis was significantly higher in the WAP-Int3/Rbpj knockout mammary tumors than in the WAP-Int3/Rbpj control tumors. The proliferating and apoptotic cells were scored, and labeling index was expressed as a percentage of positive nuclei of at least 3000 counted cells. A total of at least 5–6 mice were used for each experiment. Arrows point to apoptotic cells. *P<0.05. Original magnification was at ×40.
The effect of Rbpj knockout on anchorage-independent growth in soft agar by HC11 and HC11-Int3 mammary epithelial cells
It is possible that Notch ICD may function in two ways as a regulator of transcription. Notch ICD can displace the corepressors from Rbpj and this by itself may be sufficient to indirectly activate the expression of target genes that are independent of a Notch/Rbpj interaction. This has been referred to as a NotchICD permissive signaling pathway (Furriols and Bray, 2001). Alternatively, the Notch ICD may be required not only to alleviate repression by Rbpj, but also to activate the expression of Notch target genes (Furriols and Bray, 2000, 2001; Bray and Furriols, 2001). We therefore have developed HC11 and HC11-Int3 cell lines that were stably infected with Rbpj short hairpin RNA (shRNA) expression vectors. A reverse transcriptase PCR assay of Rbpj RNA expression in these cells is shown in Figure 7a. Compared with HC11 cells (lane1) or HC11 cells with a scrambled vector (lane 2), Rbpj RNA is virtually undetectable in HC11 cells expressing either H5 Rbpj shRNA (lane 3) or H6 Rbpj shRNA (lane 4). Similarly, compared with HC11-Int3 (lane 5) or HC11- Int3 with the scrambled vector, HC11-Int3-H5 Rbpj shRNA (lane 7) or HC11-Int3-H6 Rbpj shRNA (lane 8) exhibited a loss or a significant decrease in the levels of Rbpj RNA. To validate the biological effect of Rbpj shRNA on Notch-Rbpj-dependent signaling, we quantified Hey2 mRNA in HC-11-Int3 (Figure 7b, lane 4) and HC11-Int3- Rbpj H6 shRNA (Figure 7b, lane 5). Clearly, Rbpj H6shRNA inhibits Notch canonical signaling. This inhibition did not affect cell proliferation (Figure 7c), suggesting that cell proliferation is Rbpj independent.
Figure 7.
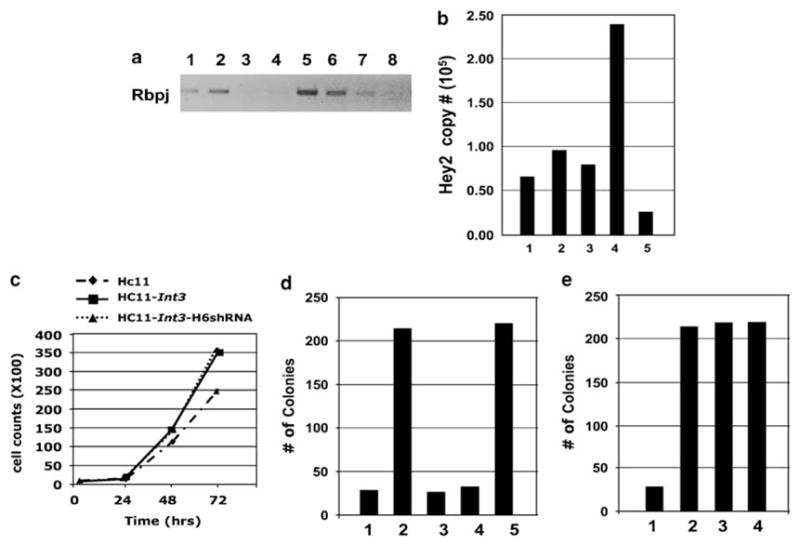
Is Rbpj necessary for anchorage-independent growth of HC11 and HC11-Int3 cells in soft agar? HC11 and HC11-Int3 cells stably expressing Rbpj shRNA (HC11-H5shRNA, HC11-H6shRNA, HC11-Int3-H5shRNA, and HC11-Int3-H6shRNA) were tested for the expression of Rbpj RNA by RT–PCR (a), effect of Rbpj shRNA on Notch signaling (b) and proliferation (c) and for their ability to grow in soft agar (d and e). (a) HC11 (lane 1), HC11+scrambled vector (lane 2), HC11+RbpjH5shRNA (lane 3), HC11+RbpjH6shRNA (lane 4), HC11-int3 (lane 5), HC11-Int3+scrambled vector (lane 6), HC11-Int3+RbpjH5shRNA (lane 7) and HC11-Int3+RbpjH6shRNA (lane 8). RbpjH5 and H6shRNA both blocked Rbpj expression (Lanes 3, 4, 7 and 8), but RbpjH6shRNA was more efficient. (b) Quantitative RT–PCR analysis of Hey2 mRNA in the HC-11 (lane 1), HC11+ scrambled vector (lane 2), HC11+scrambled vector+ Rbpj H6shRNA (lane 3), Hc11+Int3 (lane 4), Hc11+Int3+RbpjH6shRNA (lane 5). Rbpj H6shRNA blocked Notch signaling (lane 4 vs lane 5). (c) Growth curves for HC11, HC11-Int3 and HC11-Int3-H6shRNA mammary epithelial cells. The rate of proliferation of HC11-Int3 and HC11-Int3 cells stably expressing Rbpj H6shRNA was not significantly different. (d) 15 000 cells were seeded in soft agar in the presence and absence of Rbpj H6shRNA. HC11 (lane 1), HC11- Int3 (lane 2), HC11+scrambled vector (lane 3), HC11+Rbpj H6shRNA (lane 4) and HC11-Int3+scrambled vector (lane 5). HC11 cells did not acquire the ability to grow in soft agar in the absence of Rbpj (lanes 4 and 5). (e) Soft agar growth of 15 000 HC11-Int3 cells in the presence and absence of Rbpj H6shRNA. HC11 (lane 1), HC11-Int3 (lane 2), HC11-Int3+scrambled vector (lane 3), HC11- Int3+Rbpj H6shRNA (lane 4). HC11-Int3 cells did not lose the ability to grow in soft agar in the absence of Rbpj (Lane 4). RT–PCR, reverse transcriptase PCR.
The capability for anchorage-independent growth by tissue culture cells in soft agar is accepted as a measure of their tumor-inducing potential. We have shown previously (Robbins et al., 1992) and in Figure 7d (lane 1) that the HC11 mouse mammary epithelial cells cannot grow in soft agar, whereas HC11-Int3 cells do have this capability (Figure 7d, lane2). If NotchICD permissive signaling confers on HC11 cells the ability to grow in soft agar, then HC11 cells that are stably expressing Rbpj shRNA should exhibit this phenotype. To test this possibility, we compared HC11 cells expressing the Rbpj H6shRNA (lane 4) seeded at 15 000 cells per plate for their capacity to grow in soft agar with HC11 (lane 3) or HC11-Int3 (lane 5) cells with the scrambled vector. HC11 cells that were stably expressing Rbpj shRNA do not grow in soft agar (lane 4). Alternatively, if the Int3/Rbpj complex is required to confer the capability for the anchorage-independent growth of HC11-Int3 cells, then the stable expression of Rbpj shRNA should block growth in soft agar of these cells. However, as shown in Figure 7e, expression of Rbpj H6shRNA does not diminish the capability of HC11-Int3 cells (lane 4) for anchorage-independent growth in soft agar compared with HC11- Int3 (lane 2) or HC11-Int3 with the scrambled vector (lane 3). These results are compatible with the conclusion that the ability of HC11-Int3 cells for anchorage-independent growth in soft agar is independent of an Int3/Rbpj signaling pathway.
Discussion
Effect of Rbpj knockout on mammary gland development
Buono et al. (2006) have previously shown that the default developmental pathway in the mammary gland upon deletion of Rbpj in MMTV LTR-Cre/Rbpjfl/fl females results in the formation of basal cell populations and an absence of alveolar structures. Transplantation of mammary tissue from these mice demonstrated that development of the ductal tree was normal, suggesting that Rbpj is not necessary for the establishment of this structure. In contrast, mammary gland development occurs normally in Wap-Cre/Rbpjfl/fl females and they can nurse their pups. Expression from the MMTV LTR begins early during mammary gland development, whereas expression from the Wap promoter peaks around day 15 of pregnancy. This is consistent with there being a window of time during which endogenous Notch/Rbpj signaling is required to promote the development of alveolar/lobular structures. Thus, by the time the Wap promoter is active, endogenous Notch/Rbpj signaling has ceased.
In Wap-Int3 female mammary glands, ductal development is normal but alveolar development does not occur (Gallahan et al., 1996). We interpret this to mean that target cells affected by WAP-Int3/Rbpj signaling must be alveolar committed progenitor cells downstream of those affected by endogenous Notch/Rbpj signaling. Thus, in the Wap-Cre/Rbpjfl/fl/Wap-Int3 females, loss of Rbpj allows mammary gland development to proceed normally. The effect of Int3/Rbpj signaling on mammary gland development is not unique to Notch4/Int3. Transgenic mice containing either mouse MMTV-LTR-Notch1-ICD or mouse Notch3-ICD have mammary gland phenotypes that are very similar to that observed in MMTV LTR-Int3 mice, namely mammary alveolar/lobular development is suppressed and mammary tumor development occurs (Hu et al., 2006). How does inappropriate Notch/Rbpj signaling block normal mammary gland development? One possibility is suggested by the work of Leong et al. (2007), who have shown that the transcriptional E-box binding repressor Slug is a direct target gene in the canonical Notch/Rbpj signaling pathway and that Slug expression leads to epithelial-to-mesenchymal transition by repressing the expression of E-cadherin. Mutations in the E-cadherin gene (CDH1) in breast cancer have been well documented (reviewed in Yoder et al., 2007). E-cadherin is involved in many cellular processes including morphogenesis, adhesion, recognition, communication and oncogenesis. Inactivation of its adhesive properties is often a key step in tumor progression and metastasis (reviewed by Bashyam, 2002).
The effect of Rbpj knockout on mammary tumorigenesis
Although deletion of Rbpj in the mammary gland suppresses the negative effect of Int3 signaling on mammary alveolar development, it has little effect on the frequency of hyperproliferative lesions within individual mammary glands (average of 24 per gland), and also on the frequency of Int3-induced mammary tumors. In fact, 100% of the breeding Wap-Int3/Rbpj knockout females develop mammary tumors that could be transplanted in nude mice, although in each case, respectively, with a longer latency than the Wap-Int3/ Rbpj control tumors. The longer latency of primary and secondary tumor development in the Wap-Int3/Rbpj knockout mice could be due to the fact that these glands fully develop and are more differentiated than the Wap-Int3/Rbpj control mammary glands. Similarly, Notch/Rbpj-induced Slug expression in MDA MB 231 tumor cells leads to epithelial-to-mesenchymal transition and confers on them the ability to form tumors in immunodeficient mice (Leong et al., 2007). Expression of a dominant negative mutant of Rbpj delayed but did not completely block tumor growth of the MDA-MB- 231 cells. Therefore, Notch/Rbpj signaling may be involved in the initiation of tumor development by sequestering or interacting with different transcriptional corepressors or activators or by regulating the expression of a different subset of target genes.
Immunohistochemical analysis of the primary and transplanted Rbpj knockout tumors for Neo, wild-type RBP-J and the targets of Notch signaling, Hes1 and Hey2, demonstrated that these tumors were negative for each of these proteins. This suggests that a minimum level of Int3/Rbpj signaling is not required for the sustained growth of the primary or transplanted mammary tumors. However, our results cannot exclude the possibility that during the first pregnancy of the Wap-Int3/Rbpj knockout mice, there is a heterogeneity of cells in the mammary gland expressing Wap- Cre and WAP-Int3, and that in those cells not expressing Wap-Cre, as noted above, Int3/Rbpj signaling could be involved in the initiation of mammary tumorigenesis.
We have also examined the requirement for Rbpj expression on the ability of HC11 and HC11-Int3 mammary epithelial cells to grow in soft agar. It is possible that the function of Int3 is simply to derepress RBP-J repressed genes and does not affect the activation of their transcription as suggested by Furriols and Bray (2001). Although the expression of several genes was derepressed in HC11 cells that were stably infected with vectors expressing Rbpj shRNA (unpublished data), the expression of these cellular genes did not confer on HC11 cells the capability to grow in soft agar. Furthermore, the stable expression of Rbpj shRNA in HC11-Int3 cells did not affect their ability to grow in soft agar. In a related study, Stylianou et al. (2006) presented evidence that constitutively activated Rbpj- VP16 confers on normal human mammary MCF10A epithelial cells the ability to grow in soft agar, resistance to the induction of apoptosis and a reduction in E-cadherin expression. This is in contrast to our findings in which knockdown of Rbpj expression in HC11-Int3 cells did not negatively affect their ability to grow in soft agar. In addition, the level of apoptosis in the Rbpj knockout tumors was significantly lower than in the Rbpj control tumors. There are several aspects of these experiments that are difficult to evaluate. For instance, it is not known whether the chimeric activated Rbpj- VP16 protein possesses other novel properties that are not inherent to those exhibited by a wild-type Notch ICD/Rbpj interaction. It is also possible that the mechanism by which Notch1 ICD and Int3 induce mammary tumorigenesis is different, as the Notch1 ICD has a trans-activating domain C′ terminal to the ankyrin repeats that is missing in Int3.
Taken together, our data is most consistent with the concept that constitutive Int3 signaling blocks mammary alveolar development through an Rbpj-dependent pathway and that Int3-induced mammary tumorigenesis or mammary tumor growth occurs as a consequence of Int3 signaling that is not dependent on a canonical Rbpj-dependent pathway. Earlier reports have suggested that there are components of Notch signaling that occur independently of Rbpj and, in the case of Notch1, may be involved in neoplastic transformation (Shawber et al., 1996; Dumont et al., 2000; Jeffries and Capobianco, 2000; Levy et al., 2002; MacKenzie et al., 2004). In the literature, at least three additional binding partners for the Notch ICD have been reported: Deltex (Dtx) (luo et al., 2005), Hypoxia-induced factor-1α (Hif1α) (Zheng et al., 2008) and nuclear factor of κ light polypeptide gene enhancer in B-cells (Shin et al., 2006). Whether any one of these candidate binding partners or some as yet unknown binding partner collaborates with the Notch4 ICD to contribute to mammary tumorigenesis will be the subject of future studies.
Materials and methods
Mouse breeding and genotyping
We employed a mouse strain carrying a LoxP-flanked (floxed) Rbpj gene (Rbpjflox/flox) provided by Dr Tasuku Honjo (Kyoto University, Kyoto, Japan), which has been described elsewhere (Han et al., 2002; Buono et al., 2006). To delete Rbpj in mammary epithelial cells in vivo, Rbpj flox/flox mice (Han et al., 2002) were bred with transgenic mice, provided by Dr Lothar Hennighausen (Laboratory of Genetics and Physiology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA), expressing Cre-recombinase under the control of the Wap promoter (Wagner et al., 1997) to generate mice having the genotype Wap-Cre+/Rbpjflox/flox mice. Male Wap-Cre+/Rbpjflox/flox mice were genetically crossed with female Wap-Int3 mice (note, the Wap-Int3 transgene is located on the X chromosome). Among the offspring were male mice having the genotype Wap-Cre+/Rbpjflox/+/WAP-Int3. These mice were genetically crossed with female Wap-Cre+/Rbpjflox/flox mice. The offspring had the following genotypes: Wap-Cre+/Rbpjflox/flox/ Wap-Int3 (designated as Wap-Int3/Rbpj knockout) and Wap- Cre+/Rbpjflox/+/Wap-Int3 (designated as Wap-Int3/Rbpj heterozygous) mice. The Rbpjflox/flox/Wap-Int3 mice, lacking the Wap-Cre transgene, were used as a control group and are referred to as Wap-Int3/Rbpj control mice. All the experimental mice had a mixed (129/C57BL/6/FVB) genetic background. The mice were treated according to the animal protocols approved by the Animal Care and Use Committee at National Institutes of Health.
Screening tail DNA for inheritance of the floxed Rbpj and Wap-Int3 genes was performed by PCR as reported previously (Buono et al., 2006; Gallahan et al., 1996). Wap-Cre transgene was determined by PCR using the following primers: forward, 5′-CATCACTCGTTGCATCGACCGG-3′; and reverse, 5′-TA GAGCTGTGCCAGCCT CTTC-3′. DNAs were amplified for 34 cycles (94 °Cfor 30 s, 56 °Cfor 45 s and 72 °C for 1min).
Preparation of mammary tissue for analysis and transplantation
Mice were euthanized and mammary glands were examined grossly under a dissecting microscope. Mammary whole mounts and histology sections were prepared from the fourth abdominal gland as described previously (Raafat et al., 2004). Tumor tissue transplants were prepared as described previously (Raafat et al., 2007); briefly viable tissue from Rbpj control and knockout mammary tumors was transplanted into the inguinal mammary glands of 10-week-old recipients (homozygous athymic NCR-nu females). All the recipients were kept as nulliparous mice and were palpated twice weekly. Experiments were repeated at least twice, and at least five mice were used in each experiment.
Immunohistochemistry
Immunohistochemical analysis was carried out as described previously (Raafat et al., 2007). Primary antibodies were diluted: 500×; β-casein (SC-30042), 200X; Hes-1 (SC-25392), 200X; Hey-2 (Protein Tech Group, Inc-10597-1-AP), 200X; Neo (USBiological-N2008-05), 500X; and Rbpj (SC-8213), 500×, PCNA (sc-9857), 100×, in phosphate-buffered saline–1% bovine serum albumin. Appropriate biotinylated secondary anti-goat (PK-6105, Vector Laboratories Inc., Burlingame, CA, USA) or anti-rabbit (PK-6101, Vector Laboratories Inc., Burlingame, CA, USA) antibodies were diluted according to the manufacturer’s recommendations. For apoptosis, the Roche in situ cell detection POD kit (1684817, Roche, Indianapolis, IN, USA) was used according to the manufacturer’s recommendations. Labeling index was determined in at least a total of 3000 cells in each experimental condition.
Lentivirus production and transduction, proliferation and colony growth in soft agar
HEK293T/17 cells were purchased from the ATCC (Mannassas, VA, USA) and grown in DMEM, supplemented with 10% fetal bovine serum and Pen/Strep (Invitrogen; Carlsbad, CA, USA). A shRNA knockdown vector for murine Rbpj, derived from the RNAi consortium, was purchased from Open Biosystems Inc. (Huntsville, AL, USA). Purchased Clone ID numbers included TRCN0000097284–88 and were designated as H2 through H6, respectively. A control vector expressing the nontarget shRNA sequence (CCGGCAACAAGATGAA GAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTG TTTTT) was purchased from Sigma-Aldrich (St Louis, MO, USA). The lentiviral helper plasmids, psPax2 and pMD2.G, were purchased from Addgene (Cambridge, MA, USA). All viral work conformed to accepted Biosafety Level 2+ guidelines as described by the National Institutes of Health (http://bmbl.od.nih.gov/contents.htm). Briefly, knockdown vectors (H2–H6) were cotransfected with helper plasmids (psPax2 and pMD2.G) in a ratio of 10:7.5:3, respectively, in HEK293T/17, using FugeneHD (Roche Applied Science; Indianapolis, IN, USA). HC11 mouse mammary epithelial cells were transduced with lentiviral particles at approximately 40% confluence for 8–12 h followed by replacement with fresh media. Cells were selected for puromycin (4.5 mg/ml, Sigma- Aldrich, St Louis, MO, USA) resistance 36 h post-infection. For proliferation, cells were plated at a density of 1×103 per well in 24-well plates in regular growing media (day 0). The number of cells was determined every day until confluence using a Coulter counter.
The proliferation ratios were calculated from the initial start point (day 0) and the data presented are the mean of three independent experiments. Soft agar colony growth was conducted as described previously (Raafat et al., 2007). Colonies measuring 0.2 μm or larger were counted using the AccuCount 1000 colony counter (Biologics Inc., Manassas, VA, USA).
Statistics
Quantitative values are represented as the mean of at least three experiments. All in vivo experiments were repeated at least three times, and at least five mice were used in each experiment. The statistical significance of the difference between groups was determined by the Wilcoxon rank sum test. Comparisons resulting in P-values less than 0.05 were considered as statistically significant and identified in the figures with an asterisk (*).
Acknowledgments
The Rbpjflox/flox mice were kindly provided by Dr Tasuku Honjo, Department of Medical Chemistry, Graduate School of Medicine, Kyoto University, Kyoto, Japan, and the WAP-Cre mice were kindly provided by Dr Lothar Hennighausen, Laboratory of Genetics and Physiology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- Bashyam MD. Understanding cancer metastasis: an urgent need for using differential gene expression analysis. Cancer. 2002;94:1821–1829. doi: 10.1002/cncr.10362. [DOI] [PubMed] [Google Scholar]
- Bray S, Furriols M. Notch pathway: making sense of suppressor of hairless. Curr Biol. 2001;11:R217–R221. doi: 10.1016/s0960-9822(01)00109-9. [DOI] [PubMed] [Google Scholar]
- Buono KD, Robinson GW, Martin C, Shi S, Stanley P, Tanigaki K, et al. The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev Biol. 2006;293:565–580. doi: 10.1016/j.ydbio.2006.02.043. [DOI] [PubMed] [Google Scholar]
- Callahan R, Egan SE. Notch signaling in mammary development and oncogenesis. J Mammary Gland Biol Neoplasia. 2004;9:145–163. doi: 10.1023/B:JOMG.0000037159.63644.81. [DOI] [PubMed] [Google Scholar]
- Dumont E, Fuchs KP, Bommer G, Christoph B, Kremmer E, Kempkes B. Neoplastic transformation by Notch is independent of transcriptional activation by RBP-J signalling. Oncogene. 2000;19:556–561. doi: 10.1038/sj.onc.1203352. [DOI] [PubMed] [Google Scholar]
- Furriols M, Bray S. Dissecting the mechanisms of suppressor of hairless function. Dev Biol. 2000;227:520–532. doi: 10.1006/dbio.2000.9923. [DOI] [PubMed] [Google Scholar]
- Furriols M, Bray S. A model Notch response element detects Suppressor of Hairless-dependent molecular switch. Curr Biol. 2001;11:60–64. doi: 10.1016/s0960-9822(00)00044-0. [DOI] [PubMed] [Google Scholar]
- Gallahan D, Callahan R. Mammary tumorigenesis in feral mice: identification of a new int locus in mouse mammary tumor virus (Czech II)-induced mammary tumors. J Virol. 1987;61:66–74. doi: 10.1128/jvi.61.1.66-74.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallahan D, Callahan R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4) Oncogene. 1997;14:1883–1890. doi: 10.1038/sj.onc.1201035. [DOI] [PubMed] [Google Scholar]
- Gallahan D, Jhappan C, Robinson G, Hennighausen L, Sharp R, Kordon E, et al. Expression of a truncated Int3 gene in developing secretory mammary epithelium specifically retards lobular differentiation resulting in tumorigenesis. Cancer Res. 1996;56:1775–1785. [PubMed] [Google Scholar]
- Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14:637–645. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- Hu C, Dievart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of activated murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am J Pathol. 2006;168:973–990. doi: 10.2353/ajpath.2006.050416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imatani A, Callahan R. Identification of a novel NOTCH-4/ INT-3 RNA species encoding an activated gene product in certain human tumor cell lines. Oncogene. 2000;19:223–231. doi: 10.1038/sj.onc.1203295. [DOI] [PubMed] [Google Scholar]
- Jeffries S, Capobianco AJ. Neoplastic transformation by Notch requires nuclear localization. Mol Cell Biol. 2000;20:3928–3941. doi: 10.1128/mcb.20.11.3928-3941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhappan C, Gallahan D, Stahle C, Chu E, Smith GH, Merlino G, et al. Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev. 1992;6:345–355. doi: 10.1101/gad.6.3.345. [DOI] [PubMed] [Google Scholar]
- Kato H, Sakai T, Tamura K, Minoguchi S, Shirayoshi Y, Hamada Y, et al. Functional conservation of mouse Notch receptor family members. FEBS Lett. 1996;395:221–224. doi: 10.1016/0014-5793(96)01046-0. [DOI] [PubMed] [Google Scholar]
- Kordon EC, Smith GH, Callahan R, Gallahan D. A novel non-mouse mammary tumor virus activation of the Int-3 gene in a spontaneous mouse mammary tumor. J Virol. 1995;69:8066–8069. doi: 10.1128/jvi.69.12.8066-8069.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204:2935–2948. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy OA, Lah JJ, Levey AI. Notch signaling inhibits PC12 cell neurite outgrowth via RBP-J-dependent and -independent mechanisms. Dev Neurosci. 2002;24:79–88. doi: 10.1159/000064948. [DOI] [PubMed] [Google Scholar]
- Luo D, Renault VM, Rando TA. The regulation of Notch signaling in muscle stem cell activation and postnatal myogenesis. Semin Cell Dev Biol. 2005;16:612–622. doi: 10.1016/j.semcdb.2005.07.002. [DOI] [PubMed] [Google Scholar]
- MacKenzie F, Duriez P, Wong F, Noseda M, Karsan A. Notch4 inhibits endothelial apoptosis via RBP-J kappa-dependent and -independent pathways. J Biol Chem. 2004;279:11657–11663. doi: 10.1074/jbc.M312102200. [DOI] [PubMed] [Google Scholar]
- Oakes SR, Hilton HN, Ormandy CJ. The alveolar switch: coordinating the proliferative cues and cell fate decisions that drive the formation of lobuloalveoli from ductal epithelium. Breast Cancer Res. 2006;8:207. doi: 10.1186/bcr1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raafat A, Bargo S, Anver MR, Callahan R. Mammary development and tumorigenesis in mice expressing a truncated human Notch4/Int3 intracellular domain (h-Int3sh) Oncogene. 2004;23:9401–9407. doi: 10.1038/sj.onc.1208187. [DOI] [PubMed] [Google Scholar]
- Raafat A, Zoltan-Jones A, Strizzi L, Bargo S, Kimura K, Salomon D, et al. Kit and PDGFR-alpha activities are necessary for Notch4/Int3-induced tumorigenesis. Oncogene. 2007;26:662–672. doi: 10.1038/sj.onc.1209823. [DOI] [PubMed] [Google Scholar]
- Robbins J, Blondel BJ, Gallahan D, Callahan R. Mouse mammary tumor gene int-3: a member of the notch gene family transforms mammary epithelial cells. J Virol. 1992;66:2594–2599. doi: 10.1128/jvi.66.4.2594-2599.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawber C, Nofziger D, Hsieh JJ, Lindsell C, Bögler O, Hayward D, et al. Notch signaling inhibits muscle cell differentiation through a CBF1-independent pathway. Development. 1996;122:3765–3773. doi: 10.1242/dev.122.12.3765. [DOI] [PubMed] [Google Scholar]
- Shin HM, Minter LM, Cho OH, Gottipati S, Fauq AH, Golde TE, et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006;25:129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GH, Gallahan D, Diella F, Jhappan C, Merlino G, Callahan R. Constitutive expression of a truncated INT3 gene in mouse mammary epithelium impairs differentiation and functional development. Cell Growth Differ. 1995;6:563–577. [PubMed] [Google Scholar]
- Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–1525. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder BJ, Wilkinson EJ, Massoll NA. Molecular and morphologic distinctions between infiltrating ductal and lobular carcinoma of the breast. Breast J. 2007;13:172–179. doi: 10.1111/j.1524-4741.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- Zheng X, Linke S, Dias JM, Gradin K, Wallis TP, Hamilton BR, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci USA. 2008;105:3368–3373. doi: 10.1073/pnas.0711591105. [DOI] [PMC free article] [PubMed] [Google Scholar]