

Summary
Reactive nitrogen species (RNS) function as powerful antimicrobials in host defence, but so far little is known about their bacterial targets. In this study, we set out to identify Escherichia coli proteins with RNS-sensitive cysteines. We found that only a very select set of proteins contain cysteines that undergo reversible thiol modifications upon nitric oxide (NO) treatment in vivo. Of the 10 proteins that we identified, six (AtpA, AceF, FabB, GapA, IlvC, TufA) have been shown to harbour functionally important thiol groups and are encoded by genes that are considered essential under our growth conditions. Media supplementation studies suggested that inactivation of AceF and IlvC is, in part, responsible for the observed NO-induced growth inhibition, indicating that RNS-mediated modifications play important physiological roles. Interestingly, the majority of RNS-sensitive E. coli proteins differ from E. coli proteins that harbour H2O2-sensitive thiol groups, implying that reactive oxygen and nitrogen species affect distinct physiological processes in bacteria. We confirmed this specificity by analysing the activity of one of our target proteins, the small subunit of glutamate synthase. In vivo and in vitro activity studies confirmed that glutamate synthase rapidly inactivates upon NO treatment but is resistant towards other oxidative stressors.
Introduction
Nitric oxide (NO) is a lipophilic free radical gas with complex and diverse biological functions (Moncada et al., 1991). In bacteria, low concentrations of NO are produced during anaerobic growth when nitrate and nitrite are used as terminal electron acceptors (Ji and Hollocher, 1988). High levels of NO, on the other hand, are toxic. This antimicrobial effect is utilized by the mammalian host defence when the inducible NO synthase of macrophages generates toxic amounts of NO in an attempt to kill off invading microorganisms (Nathan, 1992; Fang, 1997). To counteract this hazard, bacteria have developed a number of defence systems that allow them to quickly respond to nitrosative stress and to protect them against nitrosative damage (Fang, 1997; Poole, 2005)
The direct cellular effects of NO are not yet fully understood. This is in part because other, partly even more reactive NO-derived reactive nitrogen species (RNS), including peroxynitrite (ONOO−), nitroxyl anion (NO−) and nitrogen dioxide (NO2), are breakdown products of NO and contribute to the spectrum of NO-mediated damage to cells (Stamler et al., 1992). Nevertheless, it has been clearly demonstrated that NO and NO-derived RNS modulate the activity of various enzymes (Fang, 1997) and damage critical cell processes, including protein synthesis and DNA replication (Kim et al., 1998; Bundy et al., 2000).
RNS-mediated protein modifications involve the nitration of tyrosines (Beckman, 1996) as well as the reversible binding of NO to metal centres, including the haem–iron of guanylate cyclase, the [2Fe–2S]2+ of SoxR as well as the [4Fe–4S] cluster of FNR (Arnold et al., 1977; Nunoshiba et al., 1993; Cruz-Ramos et al., 2002). Moreover, in the presence of oxygen, endogenously produced NO or NO released from compounds such as DEANO appear to rapidly cause S-nitrosylation of protein thiols (SNO) and glutathione (GSNO), presumably via the formation of RNS such as N2O3, NO2· and thiyl radicals (Schrammel et al., 2003). GSNO, which has been described as the most abundant source of NO in the cell (Sun et al., 2006), causes the direct formation of S-nitrosothiols in proteins. S-nitrosothiols in proteins are then often hydrolysed to highly reactive sulphenic acids, which, in turn, either are stabilized or react with reduced glutathione to become glutathionylated or form disulphide bonds with nearby cysteines (Poole et al., 2004). One demonstration for the multifaceted effects of NO on protein thiols came from studies with glyceraldehyde-3-phosphate dehydrogenase (GapA). The active-site cysteine of GapA can become S-nitrosylated, oxidized to sulphenic acid or glutathionylated upon treatment with GSNO, peroxynitrite or DEANO respectively (Ishii et al., 1999; Mohr et al., 1999). Other targets of RNS-mediated thiol modifications are cysteine residues of antioxidant enzymes (e.g. catalase and 902 N. Brandes, A. Rinck, L. I. Leichert and U. Jakob glutathione peroxidase), kinases (e.g. Akt/PKB), proteases (e.g. caspase) and transcription factors (e.g. HIF-1) (reviewed in Sun et al., 2006). Depending on the individual proteins, these thiol modifications lead to either the activation or inactivation of the respective protein function.
The few studies that have so far been conducted to identify proteins with RNS-sensitive cysteines used tools to specifically enrich for and identify proteins that harbour S-nitrosylated cysteines (Rhee et al., 2005). These studies potentially miss, however, all those proteins, in which S-nitrosothiols are hydrolysed and further modified. We, therefore, decided to use our recently established differential thiol trapping technique, which allows us to identify proteins, whose cysteines undergo a wide range of different reversible oxidative thiol modifications in response to NO treatment. By using this technique, we identified 10 different Escherichia coli proteins that harbour RNS-sensitive thiol groups. Interestingly, the identified proteins are largely distinct from bacterial proteins that harbour H2O2-sensitive cysteines. They included a large proportion of genes that are essential for E. coli growth under our cultivation conditions. Media supplementation studies suggested that RNS-mediated modification of AceF, as well as of enzymes in the branched-chain amino acid biosynthesis pathway, is responsible for the observed growth defect of NO-treated E. coli cells. To characterize the specificity of oxidative modifications in greater detail, we focused our subsequent studies on one of our identified proteins, the small subunit of glutamate synthase. In vivo activity assays revealed that glutamate synthase is reversibly inhibited by NO treatment but not by other ROS, such as H2O2 or superoxide. Similar results were obtained with the purified enzyme, indicating that glutamate synthase and probably many of our other identified proteins contain cysteines, which are specifically sensitive to treatment with RNS.
Results and discussion
Global analysis of the thiol-disulphide state of E. coli proteins upon DEANO treatment
Reactive nitrogen species (RNS) are potent antimicrobial agents released by host immune cells upon stimulation (Nathan, 1992; Fang, 1997). To identify potential RNS-sensitive target proteins in E. coli, we applied a differential thiol trapping technique that we recently developed (Leichert and Jakob, 2004). To determine the concentrations of NO that affect the growth of E. coli, we cultivated wild-type E. coli MG1655 in minimal media. We then exposed exponentially growing bacteria to increasing concentrations of the NO-donor diethylamine NONOate (DEANO) and further monitored cell growth at 37°C (Fig. 1). Under these temperature and pH conditions, the half-life of DEANO is 2 min and DEANO liberates 1.5 mol of NO per mole of parent compound (Maragos et al., 1991). We found that DEANO treatment transiently affected the growth rate of E. coli without killing the cells (Fig. 1). This result served as good indication that the original redox status of proteins and cellular compartments can be restored over time, and excluded the possibility that we are following the effects of oxidative stress-induced cell death. Our results were in good agreement with previous studies that analysed the transcriptional effects of DEANO treatment in bacteria (Hyduke et al., 2007). In contrast to the studies by Hyduke and coworkers, however, in which 8 μM DEANO was sufficient to induce pronounced bacteriostasis, at least 5-fold higher DEANO concentrations were needed in our studies to substantially affect cell growth. These differences are likely due to differences in media composition, which might influence the effective NO concentration that diffuses into bacteria, and/or due to the different strain backgrounds that were used in these studies.
Fig. 1.
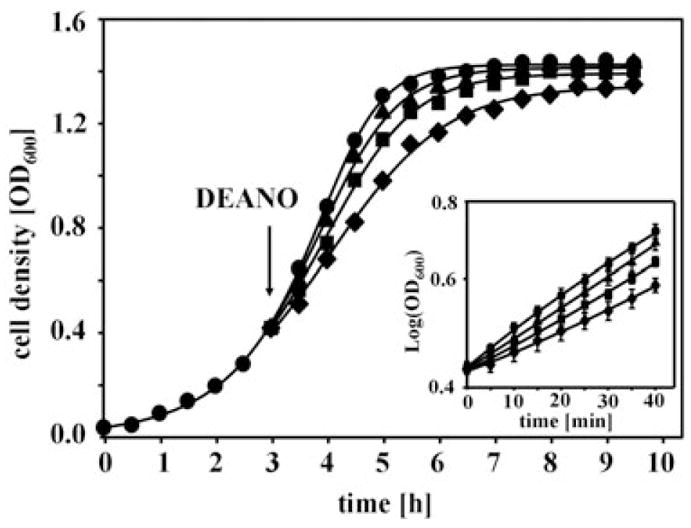
Influence of DEANO treatment on E. coli growth. MG1655 cells were grown at 37°C in MOPS minimal medium to OD600 of 0.4 and treated with either (●) 0 mM DEANO, (▲) 10 μM DEANO, (■) 50 μM DEANO or (◆) 0.5 mM DEANO at 37°C. Inset: E. coli growth during the first 40 min after addition of increasing concentrations of DEANO is shown.
To analyse the effects of NO treatment on the thiol status of the E. coli proteome, we treated E. coli with 0.05 and 0.5 mM DEANO and removed aliquots before as well as 10 min after the DEANO treatment. We then utilized a slightly modified differential thiol trapping technique (see Experimental procedures), which is based on the sequential reaction of two variants of iodoacetamide (IAM) with accessible cysteine residues in proteins (Leichert and Jakob, 2004). In the first step of this labelling method, all reduced cysteines in proteins are irreversibly carbamidomethylated with cold IAM. In a second step, all reversible thiol modifications, including S-nitrosothiols, disulphide bonds and sulphenic acids, are reduced using DTT. Then all newly accessible cysteines are modified using 14C-labelled IAM. At the end of this trapping protocol, the thiol groups in all proteins are irreversibly carbamidomethylated, but only those proteins that originally contained thiol modifications will be radioactive, allowing their identification and quantification. The proteins are then separated on 2D gels, and the extent of 14C-incorporation is determined by autoradiography. High ratios of 14C-activity/protein occur in proteins with in vivo thiol modifications, while low ratios of 14C-activity/protein are found in proteins whose thiol groups are not significantly modified (Leichert and Jakob, 2004).
Analysis of the 100 most thiol-modified proteins in E. coli cells treated with 0.5 mM DEANO for 10 min (Fig. 2) showed that only 10 proteins responded to these severe nitrosative stress conditions, with a more than 1.4-fold increase in their thiol oxidation state. Most other thiol-modified proteins are periplasmic proteins, which are fully oxidized in aerobically growing E. coli cells (e.g. OmpA, DsbA) and whose thiol groups are not affected by NO treatment (Fig. 2) (Leichert and Jakob, 2004). The extent of thiol modifications in the 10 proteins that apparently harbour RNS-sensitive thiol groups was, in each case, dependent on the DEANO concentration used (Table 1). Importantly, we noted that a large proportion (60%) of our identified proteins have been described to be conditionally essential for E. coli under the conditions used in this study, possibly explaining the antimicrobial effects of NO treatment (Baba et al., 2006; Joyce et al., 2006). This result was in excellent agreement with a recently conducted nitrosoproteome study in the Gram-positive Mycobacterium tuberculosis (Rhee et al., 2005). Upon treatment of M. tuberculosis with millimolar concentrations of the NO donor-acidified nitrite, 29 S-nitroso proteins were identified. A total of 62% of these proteins were found to be encoded by genes that are predicted to be essential or required for optimal growth of M. tuberculosis (Rhee et al., 2005).
Fig. 2.
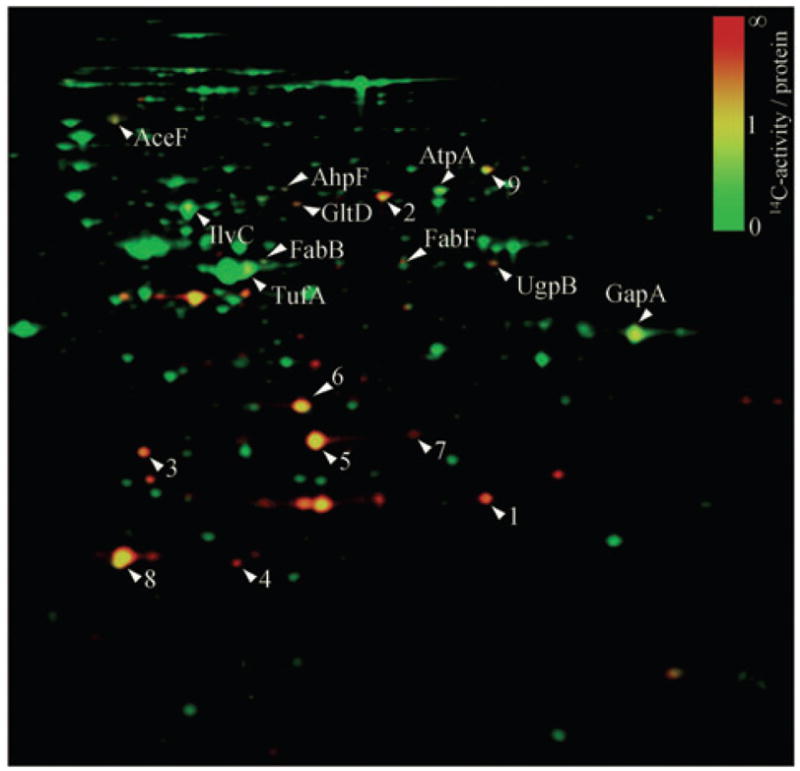
DEANO treatment affects the thiol state of only a selected group of E. coli proteins. False coloured overlay of the Coomassie blue-stained 2D gel (shown in green) and the autoradiograph (shown in red) of a differentially thiol-trapped protein extract from exponentially growing E. coli cells 10 min after treatment with 0.5 mM DEANO. The colour code represents the relative oxidation status of proteins, with green spots representing proteins whose thiol groups are largely reduced in vivo, while red spots represent proteins whose thiol groups are oxidized in vivo. The 10 labelled E. coli proteins have a more than 1.4-fold higher ratio of 14C-radioactivity/protein in DEANO-treated cells as compared with untreated control cells and are considered to harbour NO-sensitive thiol groups. A selected group of proteins, which are already oxidized under control conditions, are used as reference and indicated with a number: (1) ArtJ, (2) DppA, (3) HisJ, (4) DsbA, (5) OmpA IF 1, (6) OmpA IF 2, (7) OmpA IF, (8) AhpC and (9) OppA.
Table 1.
Nitrosative stress-sensitive proteins in E. coli.
| Fold change in 14C-activity/protein after 10 min of DEANO treatmenta |
|||||
|---|---|---|---|---|---|
| Gene | Protein | # of cysteines | 0.05 mM | 0.5 mM | Location of cysteines |
| aceF | Dihydrolipoamide S-acetyltransferase | 1 | 1.6 | 1.7 | Prosthetic group: thiol-containing lipoamideb |
| ahpF | Alkyl hydroperoxide reductase, large subunit F | 6 | 1.7 | 2.3 | Two redox-active disulphides per subunit (C129/C132 and C345/C348)c |
| atpA | ATP synthase subunit alpha | 4 | 1.1 | 1.6 | C90 in conserved patchd |
| fabB | 3-oxoacyl-(acyl carrier protein) synthase I | 6 | 1.3 | 2.0 | C163 in active sitee |
| fabF | 3-oxoacyl-(acyl-carrier-protein) synthase II | 4 | 1.2 | 1.7 | C164 in active sitef |
| gapA | Glyceraldehyde 3-P dehydrogenase | 3 | 1.2 | 1.9 | C149 in active siteg |
| gltD | Glutamate synthase, small subunit | 11 | 1.1 | 1.9 | Two [4Fe–4S]1+2+clustersh |
| ilvC | Ketol-acid reductoisomerase | 6 | 0.9 | 1.4 | C45/156 in conserved patch, C226 highly conservedd |
| tufA/B | Translation elongation factor EF-Tu | 3 | 1.0 | 1.5 | C81 associated with aminoacyl-tRNA binding; C137 associated with GTP bindingi |
| ugpB | sn-glycerol 3-phosphate transport protein | 2 | 1.1 | 1.6 | C264 in conserved patchd |
The activity/protein ratio of the given protein spot on gels from differentially trapped extracts from E. coli MG1655 (wild type) treated with different concentrations of DEANO for 10 min was divided by the corresponding ratio on gels from differentially trapped extracts from untreated E. coli MG1655. The data presented are the average of two different experiments, each of which included two separate samples. The experiments were performed in the presence of chloramphenicol to block de novo protein synthesis.
NO treatment causes oxidative thiol modification in a distinct set of E. coli proteins
Three of our identified proteins, glyceraldehyde-3-phosphate dehydrogenase (GapA), the alpha subunit of ATP synthase (AtpA) and the elongation factor Tu (Ef-Tu) have been previously shown to harbour RNS-sensitive cysteines (Rhee et al., 2005; Hara et al., 2006) (Table 1). While the reported thiol modifications of GapA’s active-site cysteine included S-nitrosylation, sulphenic acid formation or glutathionylation, depending on the type of RNS used (Ishii et al., 1999; Mohr et al., 1999), AtpA and Ef-Tu were both identified to be S-nitrosylated upon treatment of M. tuberculosis with acidified nitrite (Rhee et al., 2005). To investigate the nature of our observed thiol modifications, we applied the biotin switch method, which was designed to identify nitrosylated proteins in cell lysates (Jaffrey et al., 2001). We did not obtain any conclusive results with the biotin switch method possibly for the reasons described elsewhere (Huang and Chen, 2006; Karala and Ruddock, 2007). Thus, we developed a variation of our differential thiol trapping technique, in which we used ascorbic acid instead of DTT to specifically reduce and then 14C-IAM label S-nitrosylated cysteines in our DEANO-treated samples. Ascorbic acid has been previously used to reduce nitrosothiol groups (Jaffrey et al., 2001). We were unable to detect any significantly S-nitrosylated proteins in our DEANO-treated E. coli cells (data not shown). This can either be due to the lack of suitable methods that are able to detect S-nitrosylated proteins, or might indicate that most S-nitrosylations are unstable and rapidly convert to other oxidative thiol modifications either in vivo or during our sample preparation in vitro. Because the biotin switch method is based on the specific biotinylation and purification of only those proteins, whose cysteines are S-nitrosylated and therefore sensitive to ascorbic acid treatment, it enriches for S-nitrosylated proteins (Jaffrey et al., 2001). This, however, makes it impossible to quantify the proportion of proteins that harbour S-nitrosothiols. A recently developed computer program, which simulates metal-independent RNS chemistry, suggests that nitrosothiols might not be the end-product but rather intermediates that only transiently accumulate in vivo (Lancaster, 2006). Our data agree with this simulation and suggest that the majority of our identified RNS-sensitive proteins do not contain S-nitrosothiols but other reversible oxidative thiol modifications.
All remaining proteins that came out of our screen appear to be newly identified bacterial RNS-target proteins. One of these proteins is dihydrolipoyl transacetylase AceF, the E2 component of pyruvate dehydrogenase (PDH) (Langley and Guest, 1977). RNS most likely modify the two vicinal thiol groups in the lipoyl group of AceF, which have been previously suggested to be the target of environmental toxins such as arsenic (Knowles, 1985). RNS treatment of M. tuberculosis has recently been shown to lead to the nitrosylation of the E3 component of PDH (Lpd), whose two active-site cysteines are essential for the reoxidation of AceF’s lipoyl group (Rhee et al., 2005). We were unable to detect Lpd as significantly oxidized protein in DEANO-treated E. coli cells.
Another RNS-sensitive protein, which is encoded by a conditionally essential E. coli gene, is the 3-oxoacyl (-acyl carrier protein) synthase I (FabB). FabB plays a critical role in the elongation cycle of type II fatty acid biosynthesis. Like the closely related 3-oxoacyl (-acyl carrier protein) synthase II (FabF), which we also identified as RNS-sensitive target protein (Table 1), FabB uses one highly conserved cysteine for its catalytic activity. Modification of this active-site cysteine in both FabB and FabF would substantially interrupt fatty acid synthesis in bacteria, as well as impact the thermoregulation of membrane fluidity (de Mendoza et al., 1983).
The ketol-acid reductoisomerase IlvC was the last protein in our subset of RNS-sensitive proteins, which are encoded by conditionally essential E. coli genes. IlvC plays a central role in the biosynthesis of isoleucine and valine (Joyce et al., 2006). Targets of oxidative thiol modification in IlvC could be either a cysteine pair (Cys145/Cys156) located within a highly conserved region of the sequence and/or a very highly conserved single Cys226 (Liu et al., 2006). Noteworthy, dihydroxyacid dehydratase IlvD, which uses the reaction product of IlvC as its substrate in the Ile/Val biosynthetic pathway, has only recently been suggested to contain a NO-sensitive [Fe–S] cluster (Hyduke et al., 2007).
Alkylhydroperoxide reductase AhpF was one of the remaining proteins that we found to be increasingly thiol modified in response to NO treatment. AhpF acts as dedicated reductase for alkylhydroperoxidase AhpC, which uses disulphide bond formation to detoxify alkyl hydroperoxides in E. coli (Poole, 2005). While the C-terminus of AhpF contains the FAD/NAD binding domain, the N-terminal domain is homologous to protein disulphide isomerase (PDI), a protein that is involved in the oxidative protein folding in the endoplasmic reticulum (Murzin et al., 1995). In eukaryotes, PDI has recently been shown to have S-denitrosation activity, which in turn leads to disulphide bond formation in both of the active-site CXXC motifs of PDI (Sliskovic et al., 2005). Our finding that AhpF accumulates in the oxidized state while no apparent changes were detected in the oxidation status of AhpC suggested that E. coli AhpF might have AhpC-independent denitrosation activity similar to that found in PDI.
Finally, we identified the glycerol 3-phosphate transport protein UgpB as well as the small subunit (GltD) of glutamate synthase to be highly RNS-sensitive E. coli proteins. GltD turned out to be the only [Fe–S] cluster-containing protein in our set of proteins that harbour RNS-sensitive thiol groups. GltD contains at total of 11 cysteines, of which 8 are proposed to be involved in the formation of two oxidation stable [4Fe–4S]1+2+ clusters (Vanoni et al., 1992). That only one of our identified RNS-sensitive target proteins contain [Fe–S] clusters was remarkable, given that [Fe–S] clusters have been found as targets of RNS in several other proteins (Ding and Demple, 2000; Cruz-Ramos et al., 2002). This result might reflect, however, the preference of NO to oxidize the iron in the cluster rather than the cysteines that coordinate the clusters, an event that we would not detect using our differential thiol trapping method.
Identification of essential RNS-sensitive proteins involved in DEANO-induced growth inhibition
A significant proportion of our RNS-modified proteins are encoded by genes that have previously been shown to be essential for E. coli under our growth conditions used (Baba et al., 2006; Joyce et al., 2006). To investigate whether RNS-mediated thiol modification of one or more of these essential proteins is responsible for the observed growth arrest, we performed media supplementation experiments. To investigate whether the growth inhibitory effects of DEANO treatment are caused by the transient inactivation of AceF, one of the three enzymes constituting the PDH complex, for instance, we analysed the DEANO sensitivity of MG1655 in MOPS minimal media supplemented with 0.6% acetate as carbon source. The PDH complex catalyses the oxidative decarboxylation of pyruvate to acetyl-CoA, a key reaction that connects glycolysis with the TCA cycle. Inactivation of any of the three PDH components leads to the cessation of flux to acetyl-CoA and causes growth inhibition on glucose. Growth on acetate, which can be directly converted to acetyl-CoA, allows E. coli to bypass this key reaction provided that gluconeogenic enzymes such as GapA are available to form C6 precursor molecules. Therefore, DEANO treatment of MG1655 in media supplemented with acetate should no longer affect E. coli growth if RNS-induced inactivation of AceF was indeed the major cause of the observed DEANO-induced growth inhibition. Likewise, we supplemented media with branched-chain amino acids to investigate the role of IlvC’s thiol modification in DEANO-induced growth inhibition. Supplementation with branched-chain amino acids has recently been shown to alleviate the DEANO-induced growth arrest in BW25113 cells (Hyduke et al., 2007). Like before, we exposed the cells to the various concentrations of DEANO and monitored their growth. As shown in Fig. 3B. E. coli cells grown in media supplemented with 0.6% acetate as carbon source were no longer affected by 10 μM DEANO and were reproducibly less affected by higher concentrations of DEANO than E. coli cells grown in glucose-containing media (Fig. 3A). This result suggests that the DEANO-induced changes in E. coli growth observed at low concentrations of DEANO are caused by the inactivation of PDH and largely excluded a major involvement of GapA’s oxidative modification. This result was in excellent agreement with our in vivo thiol trapping data, which revealed that AceF shows one of the highest degrees of oxidative thiol modification of any protein at these low DEANO concentrations (Table 1). Supplementation with branched-chain amino acids Ile/Leu/Val was found to reduce the growth inhibitory effect of DEANO treatment, although it was unable to fully prevent the growth inhibition especially at high DEANO concentrations (Fig. 3C). E. coli cells cultivated in media supplemented with both acetate and branched-chain amino acids, on the other hand, were completely resistant to DEANO concentrations of up to 50 μM (Fig. 3D). We concluded from these results that oxidative modification of PDH as well as of enzymes of the branched-chain amino acid biosynthesis contributes to the growth inhibitory effect that low micromolar concentrations of DEANO exert on E. coli cells grown in MOPS minimal media. Interestingly, media supplementation with acetate and branched-chain amino acids was not sufficient to prevent growth inhibition at 0.5 mM DEANO. This result suggested that under these extreme nitrosative stress conditions, modification of other protein(s), such as Ef-Tu or ATP synthase, might become responsible for the inhibition of E. coli growth.
Fig. 3.
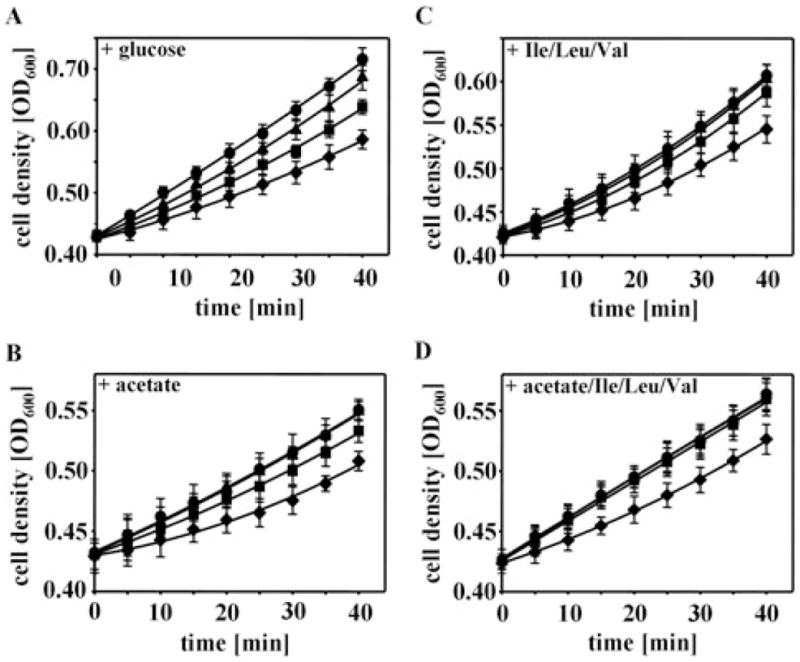
Identification of essential proteins involved in DEANO-induced growth inhibition. MG1655 wild-type cells were grown in MOPS minimal medium supplemented with (A) 0.2% glucose, (B) 0.6% acetate, (C) 0.2% glucose and Ile/Leu/Val (0.35 mM/0.35 mM/0.3 mM), or (D) 0.6% acetate and Ile/Leu/Val until OD600 of 0.4 was reached. The cultures were split and treated with either (●) 0 mM DEANO, (▲) 10 μM DEANO, (■) 50 μM DEANO or (◆) 0.5 mM DEANO at 37°C. The mean and standard deviation of three independent experiments is shown.
Activity of IlvC is affected by DEANO treatment in vivo
Escherichia coli cells cultivated in the absence of branched-chain amino acids were significantly more sensitive to micromolar concentrations of DEANO than E. coli cells grown in their presence (compare Fig. 3A and C). This result has been observed before and was attributed to the inactivation of the [Fe–S]-containing dihydroxyacid dehydratase IlvD, an enzyme essential in the Ile/Val biosynthesis pathway (Hyduke et al., 2007). This conclusion was based on IlvD activity assays, which revealed that treatment of E. coli cell lysates with 8 μM DEANO causes a 40% decrease in IlvD activity. Furthermore, in vivo studies showed that IlvD overexpression prevents the growth inhibitory effect that 8 μM DEANO exert on BW25113 cells (Hyduke et al., 2007). Our identification that IlvC, the second essential enzyme of this pathway, is substantially modified by DEANO treatment in vivo, suggested that inactivation of IlvC might also contribute to the observed growth inhibitory effect of DEANO. To test the effects of DEANO on IlvC activity in vivo, we treated MG1655 with the indicated concentrations of DEANO. We then washed and lysed the cells and determined IlvC’s enzyme activity. As shown in Fig. 4, IlvC activity gradually decreases with increasing concentrations of DEANO. This result suggests that RNS targets two enzymes of the same amino acid pathway. RNS-mediated inactivation of these enzymes eventually depletes the pool of Ile/Val, which apparently contributes to the growth inhibition observed during DEANO treatment.
Fig. 4.
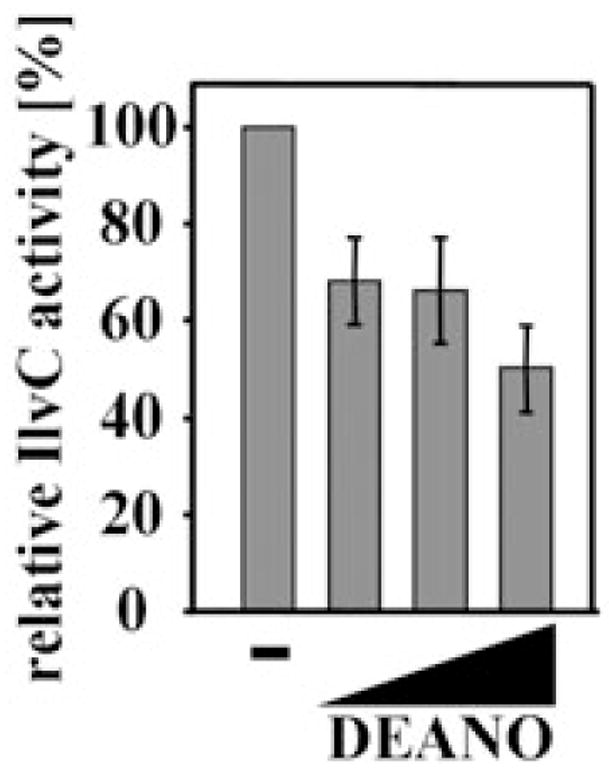
Ketol-acid reductoisomerase IlvC is sensitive to nitric oxide treatment in vivo. MG1655 wild-type cells were grown in MOPS minimal medium to OD600 of 0.4 and treated with increasing concentrations of DEANO (0.01, 0.1, 0.5 mM). After 10 min of incubation, aliquots were removed and tested for IlvC activity. IlvC activity in the absence of DEANO treatment was set to 100%. The mean and standard deviation of three independent experiments is shown.
Interestingly, changes in IlvC’s enzymatic activity were already observed at DEANO concentrations that did not apparently alter the thiol-disulphide status of IlvC as judged by our differential thiol trapping technique (Table 1). This result might indicate that functionally important residues other than cysteines are targeted by RNS as well. Alternatively, this result might illustrate the limits of the differential thiol trapping technique in proteins with numerous cysteines (Leichert and Jakob, 2004). The differential thiol trapping technique is based on a ratio approach where the largest change in oxidation ratio is observed for proteins in which all cysteine residues are completely reduced under non-stress conditions and fully oxidized under stress conditions. Proteins that harbour cysteines that are in equilibrium with their redox environment, on the other hand, might not reveal dramatic changes in the overall oxidation ratio upon oxidative stress treatment, although the oxidation status of one individual, potentially functionally important, cysteine might be dramatically altered.
Glutamate synthase – a protein specifically sensitive to nitrosative stress
Comparison of the proteins that harbour RNS-sensitive cysteines with the proteins that we have previously found to harbour H2O2-sensitive cysteines (Leichert and Jakob, 2004) revealed that the two sets of proteins are largely non-overlapping. H2O2-sensitive proteins that we previously identified included the thiol peroxidase Tpx, which is involved in detoxifying H2O2 in vivo, methionine synthase MetE, which has been shown to become rapidly glutathionylated upon H2O2 stress both in vivo and in vitro (Hondorp and Matthews, 2004), and the oxidation-sensitive GTP cyclohydrolase FolE, as well as the metabolic enzymes GlyA and SerA (Leichert and Jakob, 2004). To investigate whether the identified proteins in this study have indeed thiol groups that are specifically sensitive towards thiol modifications induced by RNS, we analysed the functional consequences of nitrosative and oxidative stress treatment on one of our identified target proteins. We decided to focus on GltD, the small (β) subunit of glutamate synthase GOGAT (i.e. NADPH-dependent glutamine: 2-oxoglutarate amidotransferase), which we found to be increasingly thiol modified in response to increasing concentrations of DEANO (Table 1). GltD was the only protein from our list of RNS-sensitive target proteins that contains both, cysteines involved in [4Fe–4S] clusters and non-metal coordinating cysteines. Besides the eight cysteines that potentially coordinate the two [4Fe–4S] clusters in GltD, the protein has three additional cysteines, which we found to be conserved within a subgroup of about 60 GltD homologues (Vanoni et al., 1992; Liu et al., 2006).
GOGAT, which belongs to the family of eubacterial NADPH-dependent glutamate synthases, is part of the cyclic two-step glutamine synthetase (GS)–glutamate synthase pathway that synthesizes glutamate under conditions of ammonia-limited growth (Tempest et al., 1970). It catalyses the transfer of the amido group from glutamine to α-ketoglutarate in a process that yields two molecules of glutamate (Reitzer, 2003). The GOGAT holoenzyme is composed of four dimers, with each dimer consisting of a large (α, GltB) and small (β, GltD) subunit (Miller and Stadtman, 1972). While the α-subunit contains the FMN cofactor and one [3Fe–4S]0,+1 cluster, the β-subunit (GltD) contains the single FAD cofactor and two [4Fe–4S]+1,+2 clusters. Recently it has been shown that the two [4Fe–4S] clusters are not present in isolated GltD subunits, suggesting that they are located at or near the interface between the α- and β-subunits (Agnelli et al., 2005).
To investigate whether NO-induced thiol modifications have any effect on the in vivo activity of GOGAT, and to test whether this is specific for RNS treatment or represents a general redox-sensitivity, we determined the activity of GOGAT in E. coli cells treated with various RNS and reactive oxygen species (ROS). Like before, we cultivated wild-type E. coli cells to midlogarithmic growth phase and exposed them to various concentrations of the NO donors DEANO and GSNO, as well as to H2O2 and the superoxide generating reagent paraquat. Ten minutes after the treatment, we harvested the cells and measured the in vivo GOGAT activity. As shown in Fig. 5A, we found that the in vivo activity of GOGAT dramatically decreased in response to increasing concentrations of DEANO or GSNO but was essentially unchanged by H2O2 or paraquat treatment (Fig. 5A). Within 10 min of treatment with 0.5 mM DEANO, less than 20% of the original GOGAT activity was observed. Very similar results were observed with GSNO, although significantly higher concentrations were necessary to achieve similar effects. This is probably due to a slower uptake of GSNO into E. coli cells. While the lipophilic NO released from DEANO rapidly passes through E. coli membranes (Lancaster, 1997), GSNO relies on dipeptide permeases (De Groote et al., 1995), whose expression might become limiting in minimal media (Spiro, 2006).
Fig. 5. Glutamate synthase (GOGAT) is specifically sensitive to nitric oxide treatment.

A. MG1655 wild-type cells were grown in MOPS minimal medium to OD600 of 0.4 and treated with increasing concentrations of DEANO (0.01, 0.1, 0.5 mM), GSNO (1, 10 mM), H2O2 (0.2, 2 mM) or paraquat (0.5, 5 mM). After 10 min of incubation, aliquots were removed and tested for GOGAT activity. GOGAT activity of untreated cells was set to 100%. No GOGAT activity was detected in the ΔgltD-deletion strain. Values are mean values from three independent measurements.
B. Purified GOGAT (200 nM) in 20 mM PPB was treated with increasing concentrations (0.01, 0.1, 0.5, 1, 5 mM) of DEANO, GSNO or H2O2 for 10 min at 30°C. Then, GOGAT was diluted 1:20 into the GOGAT activity assay. GOGAT activity in the absence of any treatment was set to 100%. The mean and standard deviation of three independent experiments is shown.
C. Purified GOGAT (4 μM) in 20 mM PPB was either left untreated (trace a), or was treated with 1.5 mM DEANO for 15 min at 30°C (trace b). After removal of DEANO using desalting columns, the absorbance spectra were recorded and normalized for the absorption at 280 nm.
We found that the sensitivity of glutamate synthase is specific for NO and RNS because neither high concentration of H2O2 nor of the superoxide producing paraquat substantially affected GOGAT activity in vivo. These results agreed with our previous in vivo thiol trapping studies that did not reveal GltD as a particularly H2O2-sensitive E. coli protein (Leichert and Jakob, 2004). Our results suggested therefore that GOGAT is a protein, whose thiol groups and functional activity are specifically sensitive to nitrosative stress agents. To ascertain that the components of our activity assay are not affected by the NO treatment, we conducted a GOGAT activity assay using DEANO-treated cell lysates, which showed the expected reduction in GOGAT activity. We then added wild-type lysate directly to this assay mixture and found the expected high GOGAT activity of untreated cell lysates (data not shown), indicating that DEANO does not affect any components of the GOGAT activity assay. These experiments showed that it is indeed the specific sensitivity of GOGAT towards NO treatment that causes the observed inactivation of glutamate synthase in vivo.
Nitrosative stress treatment causes [4Fe–4S] cluster disassembly of GOGAT in vitro
To investigate the influence of nitrosative stress treatment on GOGAT activity in more detail, we purified the wild-type protein after overexpression from E. coli cells. We then incubated the purified protein in various concentrations of DEANO, GSNO or H2O2 and analysed the in vitro GOGAT activity. Like in vivo, we observed that increasing concentrations of DEANO or GSNO cause increasing inactivation of GOGAT while H2O2 treatment had small effects on GOGAT activity only at very high concentrations (Fig. 5B). These results nicely replicated our in vivo data and showed that GOGAT is particularly sensitive to nitrosative stress treatment. Interestingly, however, we did notice that the in vitro treatment of GOGAT with DEANO was substantially less effective than the in vivo treatment (Fig. 5A). While 0.01 mM DEANO was sufficient to reduce the in vivo activity of GOGAT to about 50%, 1 mM DEANO was necessary to achieve the same effects in vitro (Fig. 5B). This decreased efficiency in the reactivity of DEANO in vitro is likely to be due to the in vivo conversion of NO into more thiol-reactive species (Stamler et al., 1992).
To investigate the effects of DEANO treatment on the Fe–S clusters in GOGAT, we then analysed the UV/VIS spectra of GOGAT in vitro upon 15 min of treatment in 1.5 mM DEANO. This experiment should allow us to monitor the stability and possible nitrosative modifications of GOGAT’s [Fe–S] clusters in response to NO treatment. Purified GOGAT showed the previously described three peaks at 278, 380 and 431 nm as well as a broad shoulder at 480 nm (Fig. 5B), which is typical for [Fe–S] cluster-containing proteins (Miller and Stadtman, 1972). These signals are contributed by the two [4Fe–4S] clusters of the small subunit and the one [3F-4S] cluster of the large subunit (Orme-Johnson and Orme-Johnson, 1978). Upon incubation of GOGAT in 1.5 mM DEANO and removal of any residual DEANO, a significant decrease over the complete visible absorption range of the [Fe–S] clusters was observed (Fig. 5C). In contrast to the redox-sensitive [4Fe–4S] cluster of FNR, where NO treatment causes the formation of dinitrosyl–iron–cysteine complexes that absorb light around 360 nm (Cruz-Ramos et al., 2002), no apparent increase in absorption between 300 and 360 nm was observed in our sample. This result suggested that at least in our in vitro experiments, NO treatment causes the disassembly of GOGAT’s [Fe–S] clusters, possibly by modifying one or more of the cysteines involved in cluster assembly. Similar effects of RNS treatment on [4Fe–4S]2+ clusters have, to our knowledge, only been observed in aconitase, where peroxynitrite treatment was found to cause cluster disassembly by oxidizing one of the cluster-coordinating cysteines (Han et al., 2005).
RNS targets cysteines of GOGAT’s [Fe–S] cluster
To investigate whether RNS-mediated cysteine modifications are indeed responsible for the observed [Fe–S] cluster disassembly, we determined the redox status of the individual cysteines in DEANO-treated GOGAT using mass spectrometry. To visualize and compare the redox status of the individual cysteines in untreated and DEANO-oxidized GOGAT, we incubated both proteins with the alkylating agent N-ethylmaleimide (NEM) under denaturing conditions. NEM irreversibly binds to all reduced thiols and leads to the mass addition of 125 Da per cysteine. In the next step, all reversible thiol modifications such as disulphide bonds or sulphenic acids present in the GOGAT preparations were reduced with DTT. These newly exposed thiol groups were then alkylated with the thiol-reactive reagent NEM IAM, which adds 52 Da per cysteine. After this differential labelling process, proteins were digested with trypsin and peptides were analysed by mass spectrometry. Based on this trapping protocol, peptides that harbour originally reduced cysteines will reveal their peptide mass, including a 125 Da mass addition per cysteine, while the same peptide with originally oxidized cysteines will only have a 52 Da mass addition per cysteine (Fig. 6). Although this method is not quantitative, it allows us to directly monitor the relative changes in the distribution of reduced and oxidized peptides. We found five of GOGAT’s 11 cysteines to be heavily oxidatively modified upon DEANO treatment in vitro (Fig. 6). Cysteines C94, C104 and C108 are known to be involved in the formation of one of the two [Fe–S] clusters in GOGAT (Vanoni and Curti, 1999). Oxidative modification of these cysteines supports our spectroscopic studies that show the disassembly of GOGAT’s [Fe–S] cluster(s) (Fig. 5C). Our finding that substantial reactivation of GOGAT can be observed upon recovery of E. coli cells from DEANO treatment in the presence of chloramphenicol (data not shown) suggests, however, that GOGAT’s [Fe–S] clusters can be repaired in aerobically growing E. coli cells. In addition to the three cysteines that are involved in [Fe–S] cluster formation, we also found two of the three non-cluster-associated cysteines of GOGAT (C161, C302) to be significantly oxidatively modified (Fig. 6). All other cysteines were either found to be DEANO-resistant (C59), or were not identified in our mass spectrometric data. These results indicate that RNS-mediated thiol modifications lead to [Fe–S] cluster disassembly in GOGAT. It remains to be determined whether the cysteines that coordinate the [Fe–S] cluster(s) are the primary RNS targets, or whether they only become targets once the [Fe–S] cluster has disassembled.
Fig. 6.

Analysis of GOGAT’s thiol status using mass spectrometry. Purified GOGAT (4 μM) in 20 mM PPB buffer that was either left untreated or oxidized with 0.5 mM DEANO for 15 min at 30°C were first incubated with NEM (125 Da per cysteine), followed by DTT reduction and IAM (57 Da per cysteine) treatment. Mass spectra of the differentially labelled tryptic peptides were obtained by MALDI-MS, and specific regions of these spectra are presented for the tryptic peptides, which were found to harbour significantly oxidized cysteines upon DEANO treatment. Cys161 and C302 are both non-[Fe–S] cluster-associated cysteines, while C94, C104 and Cys108 coordinate one of the two [Fe–S] clusters in GOGAT. For Cys161 (aa 149–167), m/z value of 1894.3 represents fully reduced peptide while m/z value of 1826.2 corresponds to fully oxidized peptide. For Cys302 (aa 290–304), m/z value of 1617.9 represents fully reduced peptide while m/z value of 1549.8 corresponds to fully oxidized peptide. For C94 (aa 77–96), m/z value of 2341.6 represents fully reduced peptide while m/z value of 2273.5 corresponds to fully oxidized peptide. For C104/C108 (aa 103–124), m/z of 2592.9 represents fully reduced peptide while m/z of 2456.7 corresponds to the fully oxidized peptide and m/z of 2524.8 represents the peptide harbouring one NEM and one IAM label.
Conclusions
We show here that sublethal concentrations of NO cause the oxidative modification of a specific subset of thiol-containing E. coli proteins, many of which have been previously shown to be encoded by genes that are essential under our growth conditions (Gerdes et al., 2003). Media supplementation studies provided evidence that the growth inhibitory effect of NO is due to oxidative modification of enzymes of the PDH complex as well as enzymes of the branched-chain amino acid biosynthesis pathway. While the nitrosative stress conditions that E. coli cells experience in our laboratory setting are short term and the effects are largely reversible, bacteria that become targets of the mammalian host defence presumably experience high concentrations of RNS over longer periods of time (Kroncke et al., 2002). We assume that, at a minimum, the same set of bacterial proteins will be affected in these cells. Long-term inactivation of these essential proteins, potential overoxidation of the affected cysteines, or additional non-thiol modifications might then lead to irreversible protein damage and to the lethal effects of RNS.
Interestingly, our RNS-sensitive proteins are largely distinct from the set of proteins that we previously identified to harbour H2O2-sensitive cysteines. This specificity appears to make physiological sense, because reactive oxygen and nitrogen species differ in their chemistry and require distinct cellular responses and defence systems (Klatt and Lamas, 2000; Kroncke et al., 2002; Leichert and Jakob, 2004). It will be our future challenge to determine what makes these proteins particularly sensitive to oxidative and nitrosative stressors and distinguishes them from the hundreds of cysteine-containing E. coli proteins, whose thiol groups are not affected by ROS and RNS. Specific nitrosylation motifs have been suggested (Stamler et al., 1997; Ascenzi et al., 2000); however, only one of our identified proteins (EF-Tu) was found to harbour a cysteine in such motif, and many abundant proteins that harbour these motifs have not been identified in our study (data not shown). It is therefore very likely that amino acids or prosthetic groups such as [Fe–S] clusters, which are nearby in the tertiary structure, are even more important in determining the reactivity of individual cysteine residues. This conclusion also agrees with earlier studies that showed that the structural micro-environment of the cysteine Sγ atom is absolutely critical (Ascenzi et al., 2000). Therefore, unless we have the three-dimensional structure of many of these NO- and H2O2-sensitive proteins, it will be very difficult to predict redox-regulated proteins simply based on their amino acid sequence. We can, however, exploit their specific oxidative and nitrosative stress sensitivity for diagnostic purposes by determining marker proteins that are specifically sensitive to distinct reactive oxygen and nitrogen species. Identification of such marker proteins in cells and tissues can thus be used as direct read-out for the presence of specific reactive oxygen or nitrogen species.
Experimental procedures
Bacterial strains and culture conditions
The E. coli strain K-12 MG1655 (F− λ−ilvG−rfb-50 rph-1) was used for differential thiol trapping and enzyme assays. The deletion strain MG1655 ΔgltD (JW3180) was obtained from the University of Wisconsin E. coli Genome Project (Kang et al., 2004). Bacterial cultures were grown at 37°C either in Luria–Bertani (LB) medium or in MOPS minimal medium (Neidhardt et al., 1974) containing 10 μM thiamine. For media supplementation studies, MG1655 was grown in MOPS minimal medium using 0.2% glucose, 0.6% acetate as carbon source. To supplement the media with branched-chain amino acids, 0.35 mM Ile, 0.35 mM Leu and 0.3 mM Val was added. Media for plasmid selection and maintenance were supplemented with ampicillin (200 μg ml−1) or kanamycin (25 μg ml−1). NO solutions were freshly prepared by either dissolving diethylamine NONOate (DEANO) in 10 mM KOH or S-nitroso-L-glutathione (GSNO) in H2O (both from Cayman Chemicals, Ann Arbor). The stability of DEANO in growth media was monitored by its characteristic UV absorbance at 250 nm (Maragos et al., 1991). No influence of the culture conditions on the DEANO stability was observed. H2O2 solutions and paraquat were obtained from Fisher Scientific and MP Biochemicals respectively.
Plasmid construction
Plasmids were constructed using standard recombinant DNA techniques. Polymerase chain reactions (PCR) were performed using Pfu Turbo DNA polymerase according to the manufacturer’s manual, and DNA was purified using the PCR Purification Kit (Qiagen). Plasmids were isolated using the SV Miniprep plasmid kit (Promega). All DNA-modifying enzymes were from Promega. The 6 kb coding region of E. coli gltB, encoding the α-subunit of GOGAT and gltD, which encodes the β-subunit of GOGAT, was amplified by PCR from E. coli K-12 MG1655 genomic DNA using the primers 5′-CGTTCTAGAATGACACGCAAACCC-3′ and 5′-ATAGGATCCACTCATTAAACTTCCAGCC-3′ respectively, which incorporated XbaI and BamHI restriction sites. The amplified genes were cloned into the expression vector pET11a (Novagen), thereby creating plasmid pET11gltBD. The correct sequence of the plasmid was confirmed by sequencing.
Differential thiol trapping
Sample preparation and differential thiol trapping
MG1655 E. coli cells were grown at 37°C in MOPS minimal media until an OD600 of 0.4 was reached. The translation inhibitor chloramphenicol (200 μg ml−1) was added to the bacterial cell culture 10 min prior to the stress treatment. The cell culture was split and treated with different concentrations of NO, ranging from 0 to 0.5 mM DEANO. After 10 min of incubation at 37°C, 1.8 ml of the cell culture was harvested directly onto 200 μl TE-buffer supplemented with 1 M IAM. Then, the thiol trapping protocol established by Leichert and Jakob (2004) was followed with slight modifications, which concerned mainly the avoidance of TCA during the trapping procedure. This was necessary to prevent uncontrolled NO formation during sample preparation, which can be generated when nitrite, one of the possible end-products of NO treatment, is acidified (Dykhuizen et al., 1996). Cells were pelleted by centrifugation (5000 g, 4°C, 1 min) and resuspended in 100 μl denaturing buffer [6 M Urea, 200 mM Tris-HCl, pH 8.5, 10 mM EDTA, 0.5% (w/v) SDS] supplemented with 100 mM IAM. This first alkylation procedure irreversibly modified all free thiol groups that were made accessible by urea and SDS denaturation of the proteins. After 10 min of incubation at 25°C, the reaction was stopped by adding 500 μl ice-cold 100% v/v acetone. After incubation at −20°C for 2 h, the precipitated and alkylated proteins were pelleted (13 000 g, 4°C, 30 min) and the protein pellet was washed with 80% acetone. The remaining steps of the thiol trapping, as well as the subsequent 2D gel electrophoresis, were conducted as previously described (Leichert and Jakob, 2004).
Data analysis and identification of proteins from 2D gels
For each of the described experiments, at least two individually trapped samples of cultures were prepared from at least two independent cell cultures. The data analysis was conducted as previously described (Leichert and Jakob, 2004). For a protein to be considered significantly thiol modified, the average of the 14C-activity/protein ratio for a given protein spot had to be at least 1.4-fold above the average of the 14C-activity/protein ratio of this protein under control conditions. Thiol-trapped samples using non-radioactive IAM in both alkylation steps were separated on 2D gels and used to excise proteins of interest. These proteins were identified by Peptide Mass Fingerprinting at the Michigan Proteome Consortium (http://www.proteomeconsortium.org).
Purification of glutamate synthase (GOGAT)
Escherichia coli BL21(DE3) cells were transformed with the plasmid pET11gltBD and grown aerobically at 37°C in LB medium containing 0.2 mg ml−1 ampicillin and 0.05 mg ml−1 kanamycin until an OD600 of 0.5 was reached. Then, expression of the two glutamate synthase subunits GltB and GltD was induced by adding 1 mM IPTG. Two hours after IPTG induction, cells were harvested by centrifugation (4000 g, 4°C, 20 min) and resuspended in 20 mM potassium phosphate buffer (PPB), 1 mM EDTA, pH 7.6 at a ratio of 1 g of cells per 2 ml of buffer. All following steps were carried out at 4°C. The purification of GOGAT was based on a protocol described by Hashim et al. (2004) with slight modifications that will be reported elsewhere.
Enzyme assays
MG1655 was grown at 37°C in MOPS minimal medium to OD600 of 0.5 and treated with the indicated concentrations of oxidants (DEANO, GSNO, H2O2, paraquat) for 10 min. Cells were harvested by centrifugation (6000 g, 5 min, RT), washed twice with 1% (w/v) KCl, and resuspended at 10-fold concentration in 1% KCl (Goss et al., 2001). The concentrated cell extract was used for GOGAT and IlvC in vivo activity assays, which were performed as previously described (Meers et al., 1970; Aulabaugh and Schloss, 1990). Both activities were assayed at 30°C by measuring the initial rates of NADPH oxidation at 340 nm. For GOGAT activity assays, 50 μl of concentrated cells was added to the reaction cocktail [0.15 mM NADPH, 50 mM Tris, pH 7.6, 5 mM α-ketoglutarate, 0.1% (w/v) cetyl trimethyl ammonium bromide (CTAB), 0.01% sodium deoxycholate (NaDOC) in 0.8 ml H2O]. Reactions were started by addition of 5 mM L-glutamine for GOGAT activity measurements. The specific activities are expressed as nanomoles of NADPH oxidized per minute per milligram of protein. To determine IlvC activity, 200 μl of concentrated cells was added to the reaction cocktail [0.2 mM NADPH, 100 mM Tris, pH 7.6, 10 mM MgCl2, 1 mM acetolactate, 0.1% (w/v) CTAB, 0.01% NaDOC in 0.7 ml H2O]. Racemic 2-acetolactate was prepared by alkaline hydrolysis of 2-hydroxy-2-methyl-3-ketobutyrate (Sigma-Aldrich) (Durner et al., 1993), and concentration of synthesized acetolactate was determined according to the method of Westerfeld (1945). Reactions were started by addition of 0.2 mM NADPH. To determine the influence of oxidants on the in vitro activity of purified GOGAT, 0.2 μM GOGAT in 20 mM PPB, pH 7.6 was incubated with the indicated concentrations of DEANO, GSNO or H2O2 at 30°C. At defined time points, 50 μl of this solution was mixed with the reaction cocktail (excluding NaDOC and CTAB) to a final GOGAT concentration of 10 nM. Five millimolar L-glutamine was added to start the reaction, and the initial rates of NADPH oxidation were measured as described above.
UV absorbance and analysis of GOGAT’s thiol status using mass spectrometry
To prepare oxidized GOGAT, 4 μM purified GOGAT in 20 mM PPB was incubated with 1.5 mM DEANO for 15 min at 30°C. Then, DEANO was removed using a NAP5 column (Amersham Biosciences). The UV/Vis spectra were recorded and normalized for the absorption at 280 nm using a CARY 100 Bio UV/VIS spectrophotometer (Varian Instruments, USA) interfaced to a computer. To analyse the thiol status of GOGAT, 30 μl of a 4 μM solution of purified GOGAT or GOGAT oxidized with 0.5 mM DEANO for 15 min at 30°C was precipitated with 10% (w/v) TCA (Ilbert et al., 2007). The pellets were resuspended in 20 μl of DAB buffer [6 M urea, 200 mM Tris-HCl (pH 8.5), 10 mM EDTA, 0.5% (w/v) SDS] containing 100 mM NEM for 1 h under vigorous shaking. The proteins were then precipitated again with TCA (10% w/v) and resuspended in 20 μl DAB buffer supplemented with 10 mM DTT to reduce all existing oxidative thiol modifications. After 1 h at 25°C, 20 μl of 50 mM IAM was added and all newly accessible thiol groups were alkylated for 1 h at 25°C. All small molecules were removed with C4-reversed phase material deposited in gel-loader pipette tips (Millipore). Tryptic digests of GOGAT and MALDI-MS analysis of the peptides were conducted at the Michigan Proteome consortium (http://www.proteomeconsortium.org).
Acknowledgments
We are grateful to Prasanthi Geda for her help in initiating this study. We are very thankful for the many insightful discussions with Dr Robert Bender, and thank Dr J. Bardwell for critically reading this manuscript. We thank the Michigan Proteomics Consortium for conducting the mass spectrometry. This work was supported by the National Institute of Health RO1 Grant GM065318.
References
- Agnelli P, Dossena L, Colombi P, Mulazzi S, Morandi P, Tedeschi G, et al. The unexpected structural role of glutamate synthase [4Fe-4S] (+1,+2) clusters as demonstrated by site-directed mutagenesis of conserved C residues at the N-terminus of the enzyme beta subunit. Arch Biochem Biophys. 2005;436:355–366. doi: 10.1016/j.abb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Anborgh PH, Parmeggiani A, Jonak J. Site-directed mutagenesis of elongation factor Tu. The functional and structural role of residue Cys81. Eur J Biochem. 1992;208:251–257. doi: 10.1111/j.1432-1033.1992.tb17180.x. [DOI] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′: 5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascenzi P, Colasanti M, Persichini T, Muolo M, Polticelli F, Venturini G, et al. Re-evaluation of amino acid sequence and structural consensus rules for cysteine-nitric oxide reactivity. Biol Chem. 2000;381:623–627. doi: 10.1515/BC.2000.081. [DOI] [PubMed] [Google Scholar]
- Aulabaugh A, Schloss JV. Oxalyl hydroxamates as reaction-intermediate analogues for ketol-acid reductoisomerase. Biochemistry. 1990;29:2824–2830. doi: 10.1021/bi00463a027. [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- Bundy RE, Marczin N, Chester AH, Yacoub M. A redox-based mechanism for nitric oxide-induced inhibition of DNA synthesis in human vascular smooth muscle cells. Br J Pharmacol. 2000;129:1513–1521. doi: 10.1038/sj.bjp.0703240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Ramos H, Crack J, Wu G, Hughes MN, Scott C, Thomson AJ, et al. NO sensing by FNR: regulation of the Escherichia coli NO-detoxifying flavohaemoglobin, Hmp. EMBO J. 2002;21:3235–3244. doi: 10.1093/emboj/cdf339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groote MA, Granger D, Xu Y, Campbell G, Prince R, Fang FC. Genetic and redox determinants of nitric oxide cytotoxicity in a Salmonella typhimurium model. Proc Natl Acad Sci USA. 1995;92:6399–6403. doi: 10.1073/pnas.92.14.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Demple B. Direct nitric oxide signal transduction via nitrosylation of iron-sulfur centers in the SoxR transcription activator. Proc Natl Acad Sci USA. 2000;97:5146–5150. doi: 10.1073/pnas.97.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durner J, Knorzer OC, Boger P. Ketol-acid reductoisomerase from barley (Hordeum vulgare) (purification, properties, and specific inhibition) Plant Physiol. 1993;103:903–910. doi: 10.1104/pp.103.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen RS, Frazer R, Duncan C, Smith CC, Golden M, Benjamin N, Leifert C. Antimicrobial effect of acidified nitrite on gut pathogens: importance of dietary nitrate in host defense. Antimicrob Agents Chemother. 1996;40:1422–1425. doi: 10.1128/aac.40.6.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang FC. Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, et al. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185:5673–5684. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss TJ, Perez-Matos A, Bender RA. Roles of glutamate synthase, gltBD, and gltF in nitrogen metabolism of Escherichia coli and Klebsiella aerogenes. J Bacteriol. 2001;183:6607–6619. doi: 10.1128/JB.183.22.6607-6619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44:11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- Hara MR, Cascio MB, Sawa A. GAPDH as a sensor of NO stress. Biochim Biophys Acta. 2006;1762:502–509. doi: 10.1016/j.bbadis.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Hashim S, Kwon DH, Abdelal A, Lu CD. The arginine regulatory protein mediates repression by arginine of the operons encoding glutamate synthase and anabolic glutamate dehydrogenase in Pseudomonas aeruginosa. J Bacteriol. 2004;186:3848–3854. doi: 10.1128/JB.186.12.3848-3854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondorp ER, Matthews RG. Oxidative stress inactivates cobalamin-independent methionine synthase (MetE) in Escherichia coli. PLoS Biol. 2004;2:e336. doi: 10.1371/journal.pbio.0020336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Chen C. An ascorbate-dependent artifact that interferes with the interpretation of the biotin switch assay. Free Radic Biol Med. 2006;41:562–567. doi: 10.1016/j.freeradbiomed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Hyduke DR, Jarboe LR, Tran LM, Chou KJ, Liao JC. Integrated network analysis identifies nitric oxide response networks and dihydroxyacid dehydratase as a crucial target in Escherichia coli. Proc Natl Acad Sci USA. 2007;104:8484–8489. doi: 10.1073/pnas.0610888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilbert M, Horst J, Ahrens S, Winter J, Graf PC, Lilie H, Jakob U. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat Struct Mol Biol. 2007;14:556–563. doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Sunami O, Nakajima H, Nishio H, Takeuchi T, Hata F. Critical role of sulfenic acid formation of thiols in the inactivation of glyceraldehyde-3-phosphate dehydrogenase by nitric oxide. Biochem Pharmacol. 1999;58:133–143. doi: 10.1016/s0006-2952(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Ji XB, Hollocher TC. Reduction of nitrite to nitric oxide by enteric bacteria. Biochem Biophys Res Commun. 1988;157:106–108. doi: 10.1016/s0006-291x(88)80018-4. [DOI] [PubMed] [Google Scholar]
- Joyce AR, Reed JL, White A, Edwards R, Osterman A, Baba T, et al. Experimental and computational assessment of conditionally essential genes in Escherichia coli. J Bacteriol. 2006;188:8259–8271. doi: 10.1128/JB.00740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Durfee T, Glasner JD, Qiu Y, Frisch D, Winterberg KM, Blattner FR. Systematic mutagenesis of the Escherichia coli genome. J Bacteriol. 2004;186:4921–4930. doi: 10.1128/JB.186.15.4921-4930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karala AR, Ruddock LW. Does s-methyl methanethiosulfonate trap the thiol-disulfide state of proteins? Antioxid Redox Signal. 2007;9:527–531. doi: 10.1089/ars.2006.1473. [DOI] [PubMed] [Google Scholar]
- Karsten WE, Viola RE. Identification of an essential cysteine in the reaction catalyzed by aspartate-beta-semialdehyde dehydrogenase from Escherichia coli. Biochim Biophys Acta. 1992;1121:234–238. doi: 10.1016/0167-4838(92)90360-p. [DOI] [PubMed] [Google Scholar]
- Kim YM, Son K, Hong SJ, Green A, Chen JJ, Tzeng E, et al. Inhibition of protein synthesis by nitric oxide correlates with cytostatic activity: nitric oxide induces phosphorylation of initiation factor eIF-2 alpha. Mol Med. 1998;4:179–190. [PMC free article] [PubMed] [Google Scholar]
- Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- Knowles FC. Reactions of lipoamide dehydrogenase and glutathione reductase with arsonic acids and arsonous acids. Arch Biochem Biophys. 1985;242:1–10. doi: 10.1016/0003-9861(85)90472-2. [DOI] [PubMed] [Google Scholar]
- Kroncke KD, Klotz LO, Suschek CV, Sies H. Comparing nitrosative versus oxidative stress toward zinc finger-dependent transcription. Unique role for NO. J Biol Chem. 2002;277:13294–13301. doi: 10.1074/jbc.M111216200. [DOI] [PubMed] [Google Scholar]
- Lancaster JR., Jr A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- Lancaster JR., Jr Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chem Res Toxicol. 2006;19:1160–1174. doi: 10.1021/tx060061w. [DOI] [PubMed] [Google Scholar]
- Langley D, Guest JR. Biochemical genetics of the alpha-keto acid dehydrogenase complexes of Escherichia coli K12: isolation and biochemical properties of deletion mutants. J Gen Microbiol. 1977;99:263–276. doi: 10.1099/00221287-99-2-263. [DOI] [PubMed] [Google Scholar]
- Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Calzi M, Poole LB. Requirement for the two AhpF cystine disulfide centers in catalysis of peroxide reduction by alkyl hydroperoxide reductase. Biochemistry. 1997;36:13357–13364. doi: 10.1021/bi9713660. [DOI] [PubMed] [Google Scholar]
- Liu XI, Korde N, Jakob U, Leichert LI. CoSMoS: conserved sequence motif search in the proteome. BMC Bioinformatics. 2006;7:37. doi: 10.1186/1471-2105-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire KA, Siggaard-Andersen M, Bangera MG, Olsen JG, von Wettstein-Knowles P. beta-Ketoacyl-[acyl carrier protein] synthase I of Escherichia coli: aspects of the condensation mechanism revealed by analyses of mutations in the active site pocket. Biochemistry. 2001;40:9836–9845. doi: 10.1021/bi0105577. [DOI] [PubMed] [Google Scholar]
- Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, et al. Complexes of NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- Massey V, Veeger C. On the reaction mechanism of lipoyl dehydrogenase. Biochim Biophys Acta. 1960;40:184–185. doi: 10.1016/0006-3002(60)91337-8. [DOI] [PubMed] [Google Scholar]
- Meers JL, Tempest DW, Brown CM. Glutamine (amide): 2-oxoglutarate amino transferase oxido-reductase (NADP); an enzyme involved in the synthesis of glutamate by some bacteria. J Gen Microbiol. 1970;64:187–194. doi: 10.1099/00221287-64-2-187. [DOI] [PubMed] [Google Scholar]
- de Mendoza D, Klages Ulrich A, Cronan JE., Jr Thermal regulation of membrane fluidity in Escherichia coli. Effects of overproduction of beta-ketoacyl-acyl carrier protein synthase I. J Biol Chem. 1983;258:2098–2101. [PubMed] [Google Scholar]
- Miller RE, Stadtman ER. Glutamate synthase from Escherichia coli. An iron-sulfide flavoprotein. J Biol Chem. 1972;247:7407–7419. [PubMed] [Google Scholar]
- Mohr S, Hallak H, de Boitte A, Lapetina EG, Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoshiba T, deRojas-Walker T, Wishnok JS, Tannenbaum SR, Demple B. Activation by nitric oxide of an oxidative-stress response that defends Escherichia coli against activated macrophages. Proc Natl Acad Sci USA. 1993;90:9993–9997. doi: 10.1073/pnas.90.21.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme-Johnson WH, Orme-Johnson NR. Overview of iron–sulfur proteins. Methods Enzymol. 1978;53:259–268. doi: 10.1016/s0076-6879(78)53031-0. [DOI] [PubMed] [Google Scholar]
- Poole LB. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch Biochem Biophys. 2005;433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- Reitzer L. Nitrogen assimilation and global regulation in Escherichia coli. Annu Rev Microbiol. 2003;57:155–176. doi: 10.1146/annurev.micro.57.030502.090820. [DOI] [PubMed] [Google Scholar]
- Rhee KY, Erdjument-Bromage H, Tempst P, Nathan CF. S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci USA. 2005;102:467–472. doi: 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrammel A, Gorren AC, Schmidt K, Pfeiffer S, Mayer B. S-nitrosation of glutathione by nitric oxide, peroxynitrite, and (*)NO/O(2)(*-) Free Radic Biol Med. 2003;34:1078–1088. doi: 10.1016/s0891-5849(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280:8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- Spiro S. Nitric oxide-sensing mechanisms in Escherichia coli. Biochem Soc Trans. 2006;34:200–202. doi: 10.1042/BST0340200. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S) NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempest DW, Meers JL, Brown CM. Synthesis of glutamate in Aerobacter aerogenes by a hitherto unknown route. Biochem J. 1970;117:405–407. doi: 10.1042/bj1170405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoni MA, Curti B. Glutamate synthase: a complex iron-sulfur flavoprotein. Cell Mol Life Sci. 1999;55:617–638. doi: 10.1007/s000180050319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoni MA, Edmondson DE, Zanetti G, Curti B. Characterization of the flavins and the iron-sulfur centers of glutamate synthase from Azospirillum brasilense by absorption, circular dichroism, and electron paramagnetic resonance spectroscopies. Biochemistry. 1992;31:4613–4623. doi: 10.1021/bi00134a011. [DOI] [PubMed] [Google Scholar]
- Vanoni MA, Verzotti E, Zanetti G, Curti B. Properties of the recombinant beta subunit of glutamate synthase. Eur J Biochem. 1996;236:937–946. doi: 10.1111/j.1432-1033.1996.00937.x. [DOI] [PubMed] [Google Scholar]
- Westerfeld WW. A colorimetric determination of blood acetoin. J Biol Chem. 1945;161:495–502. [PubMed] [Google Scholar]
- Witkowski A, Joshi AK, Smith S. Mechanism of the beta-ketoacyl synthase reaction catalyzed by the animal fatty acid synthase. Biochemistry. 2002;41:10877–10887. doi: 10.1021/bi0259047. [DOI] [PubMed] [Google Scholar]