

Abstract
The molecular mechanism of thrombin activation by Na+ remains elusive. Its kinetic formulation requires extension of the classical Botts-Morales theory for the action of a modifier on an enzyme to correctly account for the contribution of the E*, E, and E:Na+ forms. The extended scheme establishes that analysis of kcat unequivocally identifies allosteric transduction of Na+ binding into enhanced catalytic activity. The thrombin mutant N143P features no Na+-dependent enhancement of kcat yet binds Na+ with an affinity comparable to that of wild type. Crystal structures of the mutant in the presence and absence of Na+ confirm that Pro143 abrogates the important H-bond between the backbone N atom of residue 143 and the carbonyl O atom of Glu192, which in turn controls the orientation of the Glu192-Gly193 peptide bond and the correct architecture of the oxyanion hole. We conclude that Na+ activates thrombin by securing the correct orientation of the Glu192-Gly193 peptide bond, which is likely flipped in the absence of cation. Absolute conservation of the 143–192 H-bond in trypsin-like proteases and the importance of the oxyanion hole in protease function suggest that this mechanism of Na+ activation is present in all Na+-activated trypsin-like proteases.
Introduction
Regulation of activity through metal ion complexation plays a key role in many enzyme-catalyzed reactions, and over one-third of known proteins are metalloproteins (1–3). The earliest evidence for monovalent cation (M+) activation of enzymes was provided by Boyer (4) with the discovery of the absolute requirement of K+ by pyruvate kinase (5). Monod demonstrated Na+-dependent catalytic rate enhancement in β-galactosidase (6). Following these discoveries, many enzymes were observed to display increased activity in the presence of M+ (7). A recent classification of M+-activated enzymes groups them based on the selectivity of the effect, as established by kinetic studies, and the mechanism of activation, as shown from structural analysis (8). The mechanism of activation can be established from crystal structures as cofactor-like or allosteric. In the former case, the M+ anchors the substrate to the active site of the enzyme, often acting in tandem with a divalent cation like Mg2+. In this mechanism of activation, called Type I, the M+ is absolutely required for catalysis. In the latter, called Type II, the M+ enhances enzyme activity through conformational transitions triggered upon binding to a site where the M+ makes no direct contact with substrate. In this case, that is most relevant to the clotting enzyme thrombin and related trypsin-like proteases (9, 10), the M+ acts as an allosteric effector and is not absolutely required for catalysis.
Earlier kinetic studies on the hydrolysis of chromogenic substrates by Orthner and Kosow revealed that thrombin is optimally active in the presence of Na+ (11), an effect originally observed in factor Xa by the same group (12) and in activated protein C by Steiner and Castellino (13–16). Curiously, the original observation on thrombin was at odds with the accepted view at the time that salts, and Na+ in particular, actually acted as inhibitors of thrombin hydrolysis of synthetic substrates (17, 18). In fact, the inhibitory effect of Na+ was rationalized as a direct perturbation of the catalytic His57 that would reduce effectiveness of the charge relay system (18). Wells and Di Cera demonstrated that the Na+ activation of thrombin is specific and allosteric and involves the transition of the enzyme between two active forms (19) leading to an increase in kcat and a decrease in Km. These forms are E (Na+-free) with low activity and E:Na+ (Na+-bound) with high activity, originally defined as the slow and fast forms (19). The E and E:Na+ forms are significantly (2:3 ratio) populated under physiological conditions, because the Kd for Na+ binding is 110 mm at 37 °C (19–23) and the physiological [NaCl] = 140 mm is not sufficient for saturation. Na+ is the major driving force behind the procoagulant, prothrombotic, and signaling functions of thrombin in the blood (24), because it is required for optimal cleavage of fibrinogen (24–26), PAR1, PAR3, and PAR4 (27, 28) and activation of factors V (29), VIII (30), and XI (31). Importantly, Na+ binding is dispensable for cleavage of the anticoagulant protein C with or without thrombomodulin (25, 26). This explains why several naturally occurring mutations of the prothrombin gene, like prothrombin Frankfurt (32), Salakta (33), Greenville (34), Scranton (35), Copenhagen (36), and Saint Denis (37), that affect residues linked to Na+ binding (38) are associated with bleeding and why all anticoagulant thrombin mutants engineered to date are defective for Na+ binding (26, 39–43). After being overlooked in earlier structures (44–46), the Na+ binding site of thrombin was identified unequivocally using Rb+ replacement (47) and facilitated the subsequent identification of the analogous Na+ binding sites in factor Xa (48, 49), factor VIIa (50), activated protein C (51), factor IXa (52), and the thrombin precursor meizothrombin-desF1 (53). Na+ has a significant influence on the activity of factors VIIa (54, 55), IXa (52), Xa (12, 56–61), activated protein C (13–16, 62), and meizothrombin-desF1 (53, 63). Therefore, studies on the molecular mechanism of Na+ binding to thrombin are key to understand the function and regulation of this and many other clotting enzymes.
The role of two active species in equilibrium, E and E:Na+, can be cast mathematically in terms of the well known Botts-Morales scheme for the action of a “modifier” on enzyme function (64). In this scheme, binding of Na+ to thrombin is expected to produce a single kinetic phase reflecting the conversion of E into E:Na+. Recent findings, however, indicate that this is not the case. Na+ binding to thrombin and meizothrombin-desF1 gives rise to two kinetic phases, one fast (in the microsecond time scale) due to Na+ binding to E to produce E:Na+ (23, 65) and another considerably slower (in the millisecond time scale) that reflects a pre-equilibrium between E and another form, E*, that is unable to bind Na+ or substrate to the active site (23, 66). The three forms of thrombin lead to the kinetic scheme (Scheme 1).
SCHEME 1.
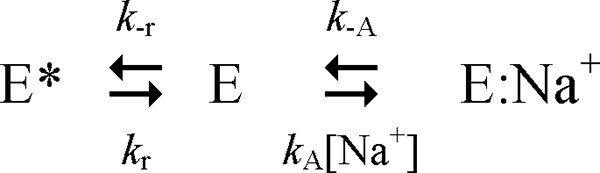
Two Na+-free forms E* and E interconvert with kinetic rate constants kr and k−r, and E interacts with Na+ with a rate constant kA to populate E:Na+, that may dissociate into the parent components with a rate constant k−A. The presence of E* redefines the slow form (19) as a mixture of two conformations in equilibrium, E* and E, which requires extension of the Botts-Morales scheme to ensure a rigorous treatment of the allosteric effect of Na+ on thrombin. In addition, more information is needed on the molecular events that translate Na+ binding into enhanced catalytic activity as reported by the values of kcat and kcat/Km. Site-directed mutagenesis may become advantageous if mutants defective for allosteric transduction of the Na+ effect can be identified and crystallized in the presence and absence of Na+. One such mutant is presented in this study and helps define a major structural transition linked to Na+ binding to thrombin.
MATERIALS AND METHODS
The human thrombin mutant N143P was constructed, expressed, and purified to homogeneity as reported elsewhere (20, 26, 38), using the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) in a HPC4-modified pNUT expression vector containing the human prethrombin-1 gene. Values of s = kcat/Km and kcat for the hydrolysis of the chromogenic substrates H-d-Phe-Pro-Phe-p-nitroanilide (FPF),2H-d-Phe-Pro-Lys-p-nitroanilide (FPK), and H-d-Phe-Pro-Arg-p-nitroanilide (FPR) were determined from analysis of progress curves and corrected for product inhibition as detailed elsewhere (67) under experimental conditions of 5 mm Tris, 0.1% PEG 8000, pH 8.0, at 25 °C, in the presence of 200 mm NaCl or choline chloride (ChCl). Values of s = kcat/Km for the hydrolysis of the chromogenic substrates H-d-Phe-Gly-Arg-p-nitroanilide (FGR), FPF, FPK, and FPR, the release of fibrinopeptide A from fibrinogen, cleavage of the protease-activated receptors PAR1, PAR3, and PAR4, and activation of protein C in the absence or presence of 100 nm thrombomodulin and 5 mm CaCl2 were determined as reported elsewhere (26, 28, 68) under physiological experimental conditions of 5 mm Tris, 0.1% PEG 8000, 145 mm NaCl, pH 7.4, at 37 °C.
Stopped-flow fluorescence measurements were carried out with an Applied Photophysics SX20 spectrometer, with excitation at 280 nm and a cutoff filter at 305 nm (23). Samples of wild-type or N143P thrombin at a final concentration of 50 nm in 5 mm Tris, 0.1% PEG 8000, pH 8.0, at 15 °C were mixed 1:1 with 60-μl solutions of the same buffer containing variable amounts of NaCl (up to 400 mm) kept at constant ionic strength of 400 mm with ChCl. The baseline was measured with 400 mm ChCl in the mixing syringe. Each trace was determined in quadruplicate. Na+ binding was also studied in the presence of saturating amounts (1 mm) of the active site inhibitor benzamidine to establish the contribution of the E*-E interconversion to the total fluorescence change detected in the absence of inhibitor. Under these conditions, Scheme 1 predicts that binding of benzamidine would shift the E*-E equilibrium in favor of E, so that binding of Na+ would only cause the transition of E to E:Na+. Accordingly, the total fluorescence change in the presence of benzamidine was ∼50% of that measured in the absence of inhibitor and was not due to an inner filter effect of benzamidine estimated at <5% after proper correction (69). Similar results were obtained when the E*-E equilibrium was perturbed with saturating amounts of the active site inhibitor argatroban, featuring a negligible inner filter effect.
Binding of the active site inhibitor argatroban was studied directly by isothermal titration calorimetry under experimental conditions of 5 mm Tris, 0.1% PEG 8000, 200 mm NaCl, or ChCl, pH 8.0, at 25 °C, using an iTC200 calorimeter (MicroCal Inc., Northampton, MA) with the sample cell containing thrombin and the syringe injecting argatroban (70). The sample volume for iTC200 was 204.6 μl, and the total volume of injected ligand was 39.7 μl. The thermal equilibration step at 25 °C was followed by an initial 60-s delay step and subsequently an initial 0.2-μl injection. Typically, 19 serial injections of 2 μl and 1 last injection of 1.5 μl of ligand were performed at an interval of 180 s. The stirring speed was maintained at 1000 rpm, and the reference power was kept constant at 5 microcalories/s. The heat associated with each injection of ligand was integrated and plotted against the molar ratio of ligand to macromolecule. Thermodynamic parameters were extracted from a curve fit to the data using the software (Origin 7.0) provided by MicroCal according to a one-site binding model.
Crystals of the thrombin mutant N143P were obtained at 22 °C using the hanging drop, vapor-diffusion method, with each reservoir containing 500 μl. Equal volumes (1 μl) of the protein sample and reservoir solution (see Table 1) were mixed to prepare the hanging drops. Diffraction quality crystals grew in 2 weeks and were cryoprotected in a solution similar to the reservoir solution but containing 15% (v/v) glycerol prior to flash freezing. X-ray diffraction data for the N143P mutant in the E:Na+ form were collected at the Advanced Photon Source (beamline Biocars 14-BMC, Argonne National Laboratory, Argonne, IL), and those for the N143P mutant in the E* form were collected with a home source Mar345 detector (MarResearch, Germany). The data were indexed, integrated, and scaled with the HKL2000 software package (71). The structures were solved by molecular replacement using MOLREP from the CCP4 suite (72) and PDB accession codes 1SHH (38) and 3BEI (73) as a search model for the N143P mutant in the E:Na+ and E* forms, respectively. Refinement and electron density generation were performed with Crystallography and NMR System (74) for the mutant N143P in the E:Na+ form or REFMAC (75) for the mutant N143P in the E* form, and 5% of the reflections were randomly selected as a test set for cross-validation. Ramachandran plots were calculated using PROCHECK (76). Statistics for data collection and refinement are summarized in Table 1. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession codes: 3JZ1 for N143P in the E:Na+ form; 3JZ2 for N143P in the E* form).
TABLE 1.
Crystallographic data for the thrombin mutant N143P
| E:Na+ form | E* form | |
|---|---|---|
| Buffer/salt | 0.2 m NaNO3 | 0.1 m imidazole |
| PEG | 3350 (20%) | 8000 (7%) |
| PDB ID | 3JZ1 | 3JZ2 |
| Data collection | APS 14-BM-C | Home Mar345 |
| Wavelength (Å) | 0.9 | 1.54 |
| Space group | C2 | P43 |
| Unit cell dimensions (Å) | a = 120.0 | a = 57.9 |
| b = 47.7 | b = 57.9 | |
| c = 50.7 | c = 119.8 | |
| β = 92.3° | ||
| Molecules/asymmetric unit | 1 | 1 |
| Resolution range (Å) | 40–1.6 | 40–2.4 |
| Observations | 150,267 | 83,891 |
| Unique observations | 36,932 | 14,942 |
| Completeness (%) | 98.6 (97.0) | 96.7 (77.8) |
| Rsym (%) | 6.1 (32.0) | 7.6 (40.8) |
| I/σ(I) | 16.9 (3.1) | 19.0 (2.2) |
| Refinement | CNS | REFMAC |
| Resolution (Å) | 40-1.6 | 40-2.4 |
| |F|/σ(|F|) | >0 | No cutoff |
| Rcryst, Rfree | 0.203, 0.216 | 0.189, 0.246 |
| Reflections (working/test) | 34,471/1,818 | 14,167/747 |
| Protein atoms | 2,239 | 2242 |
| Na+ ions | 2 | 0 |
| Solvent molecules | 214 | 116 |
| r.m.s.d. bond lengthsa (Å) | 0.008 | 0.012 |
| r.m.s.d. anglesa (°) | 1.7 | 1.4 |
| r.m.s.d. ΔB (Å2) (mm/ms/ss)b | 2.39/2.80/4.07 | 2.02/1.36/2.45 |
| 〈B〉 protein (Å2) | 28.1 | 34.1 |
| 〈B〉 Na+ (Å2) | 24.6 | - |
| 〈B〉 solvent (Å2) | 42.8 | 20.3 |
| Ramachandran plot | ||
| Most favored (%) | 98.4 | 100 |
| Generously allowed (%) | 1.1 | 0 |
| Disallowed (%) | 0.5 | 0 |
a Root mean squared deviation (r.m.s.d.) from ideal bond lengths and angles and r.m.s.d. in B-factors of bonded atoms.
b mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain.
RESULTS
Correct analysis of the allosteric effect of Na+ on thrombin requires extension of the classical Botts-Morales scheme (64) in terms of the E*, E, and E:Na+ forms in Scheme 1. The presence of E* produces an extended scheme as given in Scheme 2.
SCHEME 2.

The active forms E and E:Na+ feature different values of kinetic rate constants for binding (k1,0, k1,1) and dissociation (k−1,0, k−1,1) of substrate S, as well as rates of substrate conversion into product (k2,0, k2,1). The parameters KA = kA/k−A and KA′ = kA′/k−A′ are the equilibrium association constants for Na+ binding to E and ES, respectively. The ratio r = k−r/kr = [E*]/[E] is the equilibrium constant for the E*-E interconversion and removal of E* (r = 0) converts Scheme 2 into the well known Botts-Morales scheme. To derive the expressions for the independent parameters s = kcat/Km and kcat, Scheme 2 must be solved for the velocity of product formation at steady state, v, per unit active enzyme concentration, etot. The solution is known in the case of the Botts-Morales scheme (9, 64, 77). Hence, solution of Scheme 2 can be obtained in terms of the known solution of the Botts-Morales scheme perturbed by the presence of E*.
The expression for v/etot depends on the total number of trajectories at steady state for Scheme 2. Each trajectory leading to a species must contain the product of four rate constants; because of the five intermediates in Scheme 2 only four are independent due to conservation of mass. The sum of trajectories toward each species defines the contribution of that species at steady state (78). The expression for the velocity of product formation at steady state is therefore,
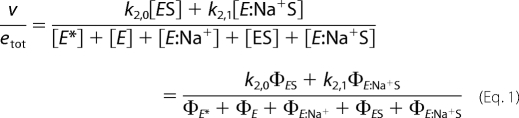 |
where ΦEX is the sum of trajectories toward species EX in Scheme 2 (EX = E*, E, ES, E:Na+, or E:Na+S). The contribution of E* is derived as follows. All trajectories leading to a species other than E* must contain the rate constant kr, and therefore ΦEX = krΣEX (EX = E, ES, E:Na+, or E:Na+S), where the Σ values (listed in Table 2) refer to the sum of trajectories of the Botts-Morales scheme obtained when E* is omitted in Scheme 2. This is because E* can only participate to the flux of trajectories by generating E. All trajectories leading to E*, on the other hand, must contain the rate constant k−r, and therefore ΦE* = k−rΣE. This is because E* can only be generated from E. With these important simplifications, Equation 1 becomes,
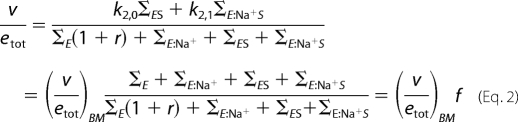 |
and the solution of the steady-state properties of Scheme 2 is the same as that of the Botts-Morales scheme, (v/etot)BM, which does not depend on r, corrected by a factor f that depends on r, [Na+], and [S]. Under saturating concentrations of substrate, one has f = 1 (see Table 2) in Equation 2 and,
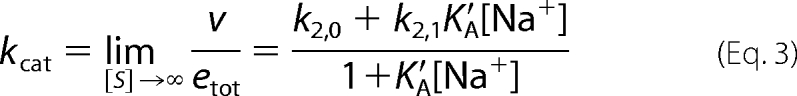 |
The expression for kcat in Scheme 2 is the same as that in the Botts-Morales scheme. The presence of E* has no influence on the value of kcat, as expected, because this parameter is a property of the enzyme-substrate complex and not of the free enzyme. Three independent parameters k2,0, k2,1, and KA′ are resolved from measurements of kcat as a function of [Na+]. The value of s has a more complicated expression (see Table 2), i.e.,
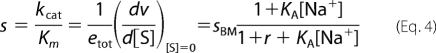 |
and contains the value sBM in the Botts-Morales scheme (9, 64, 77) corrected by the contribution of r. The algebraic form of Equation 4 is such that the parameters KA reflecting the Na+ binding affinity of the free enzyme and r cannot be resolved independently. These parameters can only be resolved by stopped-flow measurements of Na+ binding (23). The presence of E* has a profound influence on the value of s, contrary to what is observed in the case of kcat. As r increases because the population of E* increases, the value of s decreases without limits. A mutation of thrombin that stabilizes E* has the potential to abrogate activity even in the absence of perturbation of Na+ binding (KA) or specific effects on the values of s for the E form s0 = k1,0k2,0/(k−1,0 + k2,0) or the E:Na+ form s1 = k1,1k2,1/(k−1,1 + k2,1). That is a basic difference with respect to the Botts- Morales mechanism for which s is bound from s0 to s1.
TABLE 2.
Coefficients in the velocity expression (Equation 2) for the kinetics of Na+ activation
Because these coefficients refer to the Botts-Morales scheme, obtained from Scheme 2 when E* is not present, they do not depend on the equilibrium constant r.

The allosteric transduction of Na+ binding into enhanced catalytic activity of thrombin can be quantified from Equations 3 and 4 by taking the ratio between values at [Na+] = ∞ and [Na+] = 0, i.e.,
 |
 |
where the value of β measures the enhancement of kcat and does not depend on r. This parameter only measures the difference in kcat between the two active forms of thrombin, E:Na+S and ES, and does not depend on E*. Hence, a mutation that selectively changes the value of r has no effect on the value of β. The value of α, on the other hand, depends explicitly on r and can increase even when s1 and s0 remain constant if E* becomes more populated.
Of particular interest is the case where allosteric transduction of Na+ binding into enhanced catalytic activity is compromised. Residues involved in Na+ binding need not be the same as those transducing this event into enhanced catalytic activity. Binding and catalysis in Scheme 2 are thermodynamically independent, because the rate constants reflecting catalysis are not subject to detailed balancing (64, 78). Indeed, although many residues of thrombin influencing Na+ binding have been identified (38, 54), no residue controlling allosteric transduction has so far emerged from mutagenesis studies. In the presence of Na+ binding, a mutation that abrogates allosteric transduction into enhanced catalytic activity leads to k2,0 = k2,1 and s0 = s1, or β = 1 and α = 1 + r > 1. The condition α > 1 in Scheme 2 is very different from the condition α = 1 predicted by the Botts-Morales scheme when s1 = s0. Therefore, conclusions on the mechanism of Na+ binding to thrombin based only on analysis of s = kcat/Km data can be misleading. For example, a value of α = 22 measured for the hydrolysis of FPR by wild-type thrombin (Table 3) can also be obtained for a mutant that binds Na+ normally but does not transduce at all, provided r is large enough. Without the knowledge gleaned from Scheme 2 and Equation 5, such mutant would be deemed no different from wild type, when in fact it holds important information on the mechanism of Na+ activation. Because the value of α cannot be used to establish the extent or lack of allosteric transduction, such conclusions must rely on the value of β, especially when no independent information is available on the value of r. A value of β = 1 implies that E and E:Na+ turn over substrate at the same rate and that Na+ has no transducing effect on the enzyme. In conclusion, when Na+ binds to thrombin but produces no enhancement of either kcat or s = kcat/Km for the active E and E:Na+ forms, the resulting value of kcat will not change with [Na+], but the value of s will still be higher in the presence of Na+ due to the contribution of the inactive form E*.
TABLE 3.
Kinetic properties (s = kcat/Km, Km, and kcat) of wild-type and mutant N143P thrombin toward chromogenic substrates
| Enzyme | Substrate | sa | Kma | kcata | sb | Kmb | kcatb | α | β |
|---|---|---|---|---|---|---|---|---|---|
| μm−1 s−1 | μm | s−1 | μm−1 s−1 | μm | s−1 | ||||
| wt | FPF | 0.013 ± 0.001 | 110 ± 10 | 1.4 ± 0.1 | 0.48 ± 0.01 | 17 ± 1 | 8.3 ± 0.3 | 37 ± 1 | 5.9 ± 0.2 |
| wt | FPK | 0.30 ± 0.01 | 40 ± 1 | 12 ± 1 | 7.0 ± 0.2 | 4.0 ± 0.2 | 28 ± 1 | 23 ± 1 | 2.3 ± 0.1 |
| wt | FPR | 4.0 ± 0.2 | 0.88 ± 0.04 | 3.5 ± 0.1 | 88 ± 6 | 0.33 ± 0.03 | 29 ± 1 | 22 ± 1 | 8.3 ± 0.2 |
| N143P | FPF | 0.0058 ± 0.0001 | 40 ± 1 | 0.23 ± 0.01 | 0.0054 ± 0.0001 | 33 ± 1 | 0.18 ± 0.01 | 0.93 ± 0.02 | 0.78 ± 0.02 |
| N143P | FPK | 0.0036 ± 0.0001 | 72 ± 2 | 0.26 ± 0.01 | 0.028 ± 0.001 | 6.4 ± 0.1 | 0.18 ± 0.01 | 7.8 ± 0.2 | 0.69 ± 0.02 |
| N143P | FPR | 0.56 ± 0.01 | 13 ± 1 | 7.5 ± 0.1 | 2.1 ± 0.1 | 2.9 ± 0.1 | 6.0 ± 0.1 | 3.8 ± 0.2 | 0.80 ± 0.02 |
a Experimental conditions: 5 mm Tris, 0.1% PEG 8000, pH 8.0, 25°C, 200 mm ChCl.
b Experimental conditions: 5 mm Tris, 0.1% PEG 8000, pH 8.0, 25°C, 200 mm NaCl.
All three forms of thrombin have been identified crystallographically (9), but they provide few details on the molecular mechanism of Na+ activation. E and E:Na+ are very similar (38), underscoring their active conformation. E*, on the other hand, features a collapsed active site and a Na+ site environment that cannot retain the cation (43, 73, 79), consistent with its inactive nature. The presence of E* increases the apparent Km for substrate binding, and the binding of Na+ may overcome this effect by shifting the equilibrium toward E:Na+. However, the Na+ effect on kcat cannot be explained with the presence of E*, because this form is by definition never populated under saturating concentrations of substrate where kcat is measured. The enhancement in kcat must be explained in terms of the differences between E and E:Na+ for which existing structures offer little insight. Mutations that abrogate the Na+ effect on kcat in the presence of Na+ binding are therefore crucial to decouple the mechanism of thrombin activation by Na+.
Residue Asn143 plays a key role in the function of thrombin and the entire class of serine proteases to which thrombin belongs (10). Trypsin-like proteases stabilize substrate binding to the active site using a network of H-bonds, five of which are highly conserved and ensure the efficient hydrolysis of amide bonds (80, 81). Of these H-bonds, two stabilize an anti-parallel β-sheet between Gly216 and the P3 residue of substrate, and the other three involve the backbone N and O atoms of the P1 residue of substrate. The N atom engages the backbone O atom of Ser214. The O atom of the P1 residue, on the other hand, makes two H-bonding interactions with the backbone N atoms of the catalytic Ser195 and Gly193 in the so-called “oxyanion hole.” The role of this region of the enzyme is to stabilize the developing negative charge on the O atom of substrate during formation of the tetrahedral intermediate (82) by 1.5–3.0 kcal/mol (83). The lack of a side chain at position 193 allows correct positioning of the amido hydrogen to form the requisite H-bond with the oxyanion substrate. Indeed, Gly is highly conserved at position 193 in serine proteases, but few exceptions do exist and are associated with perturbed substrate hydrolysis and resistance to inhibition (84–87). The conformation of the 192–193 peptide bond keeps the backbone of Gly193 in the correct orientation for formation of the oxyanion hole. This is ensured by a highly conserved H-bond between the carbonyl O atom of residue 192 and the backbone N atom of residue 143 on the adjacent β-strand. When this H-bond is broken, as in the E* form of thrombin (43, 73, 79), factor VIIa (50), complement factor B (88), and the arterivirus protease nsp4 (89), the 192–193 peptide bond flips and the oxyanion hole is disrupted. Our interest in residue Asn143 of thrombin was to establish the role of the H-bond with Glu192 in the allosteric transitions of the enzyme. The N143P mutation was made to selectively abrogate the H-bonding capabilities of the backbone N atom, as successfully done in other systems (90–92), and produce a constitutively flipped Glu192-Gly193 peptide bond. We expected the mutation to stabilize the E* form and perhaps afford an anticoagulant effect as recently observed for the Δ146–149e mutant (43).
The properties of the N143P mutant were tested under physiological conditions against a panel of chromogenic and macromolecular substrates (Fig. 1). The mutation changes the value of kcat/Km to a different extent depending on the substrate used, arguing against an exclusive stabilization of the E* form that would have produced a similar drop in activity for all substrates tested, as recently observed for the Δ146–149e mutant (43). Importantly, the N143P mutation does not abrogate activity, which hardly explains why Pro at position 143 is not documented in any trypsin-like protease sequenced to date (10). If the N143P mutant disrupts the oxyanion hole, as expected, then the energetic consequences do not abrogate activity altogether and depend on the particular enzyme-substrate complex formed. This is consistent with the results obtained with direct perturbation of Gly193 (83) and studies on clotting factor VIIa where active site ligands were found to reconstitute the correct architecture of the oxyanion hole disrupted by the flip in the 192–193 peptide bond (50). Binding of thrombomodulin to exosite I of N143P does not ameliorate the energetic penalty on substrate recognition by the active site, even in the case of the anticoagulant substrate protein C. Although the data in Fig. 1 obviously do not rule out that the N143P mutation stabilizes the E* form, they do demonstrate a specific perturbation of the intrinsic kinetic properties of the enzyme in addition to possible changes in the allosteric equilibria involving E*, E, and E:Na+. However, no relevant anticoagulant propensity is gained by the mutant when Pro is substituted at position 143.
FIGURE 1.

Functional properties of the thrombin mutant N143P. Shown are the values of s = kcat/Km for the hydrolysis of chromogenic substrates FGR, FPF, FPK, and FPR, fibrinogen (FpA), PAR1, PAR3, PAR4, protein C (PC), and protein C (PC+TM) or FPR (S+TM) in the presence of 100 nm thrombomodulin and 5 mm CaCl2 for the thrombin mutant N143P (smut) relative to wild-type (swt). Each substrate experiences a different loss of activity that cannot be reconciled with an exclusive perturbation of the E*-E equilibrium in favor of the inactive form E*, as recently reported for the Δ146–149e mutant (43). Hence, different substrates probe the oxyanion hole of the enzyme in different ways leading to distinct perturbed energetics. Thrombomodulin is unable to restore activity toward chromogenic substrate or protein C. Experimental conditions are: 5 mm Tris, 0.1% PEG 8000, 145 mm NaCl, pH 7.4, at 37 °C. The values of swt are: 0.52 ± 0.05 μm−1s−1 (FGR), 0.28 ± 0.03 μm−1s−1 (FPF), 4.2 ± 0.2 μm−1s−1 (FPK), 37 ± 1 μm−1s−1 (FPR), 17 ± 1 μm−1s−1 (FpA), 39 ± 1 μm−1s−1 (PAR1), 0.35 ± 0.02 μm−1s−1 (PAR3), 0.34 ± 0.01 μm−1s−1 (PAR4), 59 ± 3 m−1s−1 (PC), 0.22 ± 0.01 μm−1s−1 (PC+TM), and 34 ± 2 μm−1s−1 (S+TM).
Having established that the N143P mutation affects specifically the catalytic properties of thrombin, we addressed whether this perturbation was linked to the binding of Na+. The catalytic activity of N143P is compromised up to two orders of magnitude toward the chromogenic substrates FPF, FPK, and FPR that differ in their P1 residue (Table 3). In the wild type, the kcat/Km values of FPF, FPK, and FPR differ up to three orders of magnitude and are significantly enhanced by Na+ with an increase in kcat that ranges form 2- (FPK) to 8-fold (FPR). In the N143P mutant, the values of kcat change little in the presence of Na+, and they are actually slightly higher in the absence of cation (Table 3). Values of α are significantly smaller than those measured for wild type. Importantly, these changes in the N143P mutant are observed for substrates with large values of α in the wild type due to an effect of Na+ predominantly on kcat (FPR), Km (FPK), or both kcat and Km (FPF). Because these may be signatures of perturbation of the allosteric transduction, Na+ binding to N143P was studied directly by stopped-flow, and the results are shown in Fig. 2. A large fluorescence increase upon binding of Na+ documents the interaction of the cation with the N143P mutant. However, contrary to what is observed in the wild-type, the mutant lacks the slow phase of fluorescence enhancement. This phase is due to the E*-E interconversion of the enzyme (23), and its loss in the N143P mutant can be attributed either to absence of the E* form, or to a change in the kinetic rate constants kr and k−r that define the rate of interconversion. The values of β ≈ 1 in Table 3 coupled to the presence of Na+ binding strongly suggest a lack of allosteric transduction for the N143P mutant. Hence, if the mutation causes a flip of the Glu192-Gly193 peptide bond with disruption of the oxyanion hole, it can be concluded that the oxyanion hole is disrupted in the E form due to a flip of the Glu192-Gly193 peptide bond. Restoration of the correct orientation of the Glu192-Gly193 peptide bond would represent a key signature of the allosteric transduction of Na+ binding into enhanced kcat values for thrombin.
FIGURE 2.

Na+ binding to thrombin wild-type and N143P. A, kinetic traces of Na+ binding to wild-type thrombin in the 0–250 ms time scale. Shown are the traces obtained at 0 (open circles) and 50 mm Na+ in the absence (black circles) or presence (gray circles) of 1 mm benzamidine. Notice how the binding of Na+ obeys a two-step mechanism, with a fast phase completed within the dead time (<0.5 ms) of the spectrometer, followed by a single-exponential slow phase. The presence of benzamidine abolishes the slow phase and yields a rapid phase whose amplitude is comparable to that observed in the absence of benzamidine. Experimental conditions are: 50 nm thrombin, 5 mm Tris, 0.1% PEG 8000, pH 8.0, at 15 °C. The [Na+] was changed by keeping the ionic strength constant at 400 mm with ChCl. Continuous lines were drawn using the expression b − a exp(−kobst) with best-fit parameter values: open circles, a = 0 ± 0 V, kobs = 0 ± 0 s−1, b = 8.11 ± 0.01 V; black circles, a = 0.27 ± 0.02 V, kobs = 130 ± 10 s−1, b = 8.98 ± 0.02 V; and gray circles, a = 0 ± 0 V, kobs = 0 ± 0 s−1, b = 8.63 ± 0.01 V. These values are in excellent agreement with those reported previously in the absence of benzamidine (23). B, kinetic traces of Na+ binding to the thrombin mutant N143P in the 0–250 ms time scale. Shown are the traces obtained at 0 (open circles) and 50 mm Na+ in the absence (black circles) or presence (gray circles) of 1 mm benzamidine. Notice how the binding of Na+ to N143P produces only a rapid phase of fluorescence enhancement comparable in total amplitude to that observed in the wild type. The presence of benzamidine reduces the amplitude of the fast phase and gives a total amplitude again comparable to that seen in the wild type. Experimental conditions are: 50 nm thrombin, 5 mm Tris, 0.1% PEG 8000, pH 8.0, at 15 °C. Continuous lines were drawn using the expression b − a exp(−kobst) with best-fit parameter values: open circles, a = 0 ± 0 V, kobs = 0 ± 0 s−1, b = 8.09 ± 0.01 V; black circles, a = 0 ± 0 V, kobs = 0 ± 0 s−1, b = 8.87 ± 0.01 V; and gray circles, a = 0 ± 0 V, kobs = 0 ± 0 s−1, b = 8.51 ± 0.01 V. C, Na+ binding curves of thrombin wild-type (open circles) and N143P (black circles) obtained from the total change in intrinsic fluorescence determined by stopped-flow kinetics. Experimental conditions are: 50 nm thrombin, 5 mm Tris, 0.1% PEG 8000, pH 8.0, at 15 °C. The [Na+] was changed by keeping the ionic strength constant at 400 mm with ChCl. Continuous lines were drawn according to the equation, F = (F0 + F1Kapp[Na+])/(1 + Kapp[Na+]), where F0 and F1 are, respectively, the limiting values of intrinsic fluorescence in the absence and under saturating [Na+] and Kapp = KA/(1 + r) is the apparent Na+ binding affinity (23). The best-fit parameter values are: wild-type, F0 = 8.07 ± 0.04 V, F1 = 9.12 ± 0.03 V, Kapp = 120 ± 20 m−1; N143P, F0 = 8.10 ± 0.02 V, F1 = 9.19 ± 0.02 V, Kapp = 52 ± 5 m−1.
The values of α in Table 3 are perturbed relative to wild type, which brings about the role of E* in the N143P mutant. If E* is absent in this mutant (r = 0), then the values of α > 1 should reflect the effect of Na+ on the Km, because no enhancement is seen for kcat. Furthermore, the total fluorescence change reported in Fig. 2 should reflect the transition of E to E:Na+ for this mutant and would have twice the amplitude of the same transition in the wild type. The contribution of the E-E* interconversion can be abrogated by measuring Na+ binding with a ligand bound to the active site. Scheme 1 predicts that binding of a ligand to the active site under saturating conditions would shift the equilibrium from E* to E completely, because E* has the active site occluded. Binding of Na+ under these conditions should give only a fast phase of fluorescence increase reflecting the conversion of E to E:Na+ with an amplitude similar to that of the fast phase observed in the absence of active site ligand. The data in Fig. 2 confirm this expectation for wild type. When Na+ binding is measured in the presence of the active site inhibitor benzamidine, whose fluorescence properties do not change when free or bound to the enzyme (93), only a rapid phase of fluorescence increase is observed whose amplitude is comparable to that of the fast phase measured in the absence of benzamidine. When the same experiment is carried out with the mutant N143P, a fast phase is observed that is smaller than that seen in the absence of benzamidine, thereby demonstrating that some of the rapid fluorescence change detected upon Na+ binding to the N143P mutant in the absence of benzamidine is due to the E*-E interconversion that has become too fast to resolve. Amplitude analysis (94) suggests that the E*:E ratio in the N143P mutant is close to 1:1 and comparable to that seen in the wild type (23). In conclusion, the N143P mutant partitions between E and E* and binds Na+ like wild type, but does not transduce this event into enhanced catalytic activity. Restoration of the oxyanion hole is therefore a major determinant of the mechanism allowing Na+ to activate thrombin.
The lack of allosteric transduction in the N143P mutant is further supported by the equilibrium components of ligand recognition. Binding of the active site inhibitor argatroban to wild type increases 6-fold in the presence of Na+ due to a massive (6.7 kcal/mol) entropic contribution (Fig. 3). Most of this entropic enhancement is lost in the N143P mutant, and the affinity for argatroban increases only 2-fold in the presence of Na+. It is straightforward to show that the ratio of affinities for a ligand binding to the active site in the presence versus absence of Na+ has the same form as Equation 5, i.e.,
 |
where K0 and K1 are the equilibrium binding constants of the ligand to the E and E:Na+ forms, respectively. A value of γ = 6.2 is obtained for argatroban binding to wild type. Because r = 0.15 for wild type under the same experimental conditions (23), the ratio K1/K0 = 5.4 indicates preferential binding of argatroban to the E:Na+ form relative to the E form. In the case of the mutant N143P, the value of r cannot be accessed experimentally because of the lack of the slow phase linked to Na+ binding (see Fig. 2). Assuming that r is comparable to the value measured for wild type, it can be concluded that binding of Na+ to N143P increases the affinity for argatroban at most by 1.7-fold. Na+ binding to thrombin would therefore control the correct architecture of the oxyanion hole in both the ground and transition states, thereby allowing optimization of both substrate binding and catalysis by the enzyme.
FIGURE 3.

Argatroban binding to thrombin wild type and N143P. Argatroban binding to thrombin wild type and N143P in the presence or absence of Na+, as indicated, measured by isothermal titration calorimetry. The top panel shows the heat exchanged in each individual titration. The bottom panel is the integration of the data to yield the overall heat exchanged as a function of the ligand/protein molar ratio. Experimental conditions were 5 mm Tris, 0.1% PEG 8000, pH 8.0, 25 °C, 200 mm NaCl or ChCl, as indicated. The thrombin and argatroban concentrations were 13.44 μm and 140 μm (wild-type, Na+), 13.44 μm and 140 μm (wild-type, Ch+), 52.5 μm and 777 μm (N143P, Na+), and 52.5 μm and 777 μm (N143P, Ch+). Titration curves were fit using the Origin software of the iTC200, with best-fit parameter values (N is the stoichiometric constant): wild-type, Na+: n = 0.97 ± 0.01, K = 1.8 ± 0.4 108 m−1, ΔG = −11.2 ± 0.1 kcal/mol, ΔH = −7.6 ± 0.1 kcal/mol, TΔS = 3.6 ± 0.1 kcal/mol; wild-type, Ch+: n = 1.06 ± 0.01, K = 2.9 ± 0.3 107 m−1, ΔG = −10.1 ± 0.1 kcal/mol, ΔH = −13.2 ± 0.1 kcal/mol, TΔS = −3.1 ± 0.1 kcal/mol; Ν143P, Na+: n = 1.03 ± 0.01, K = 2.0 ± 0.5 106 m−1, ΔG = −8.6 ± 0.1 kcal/mol, ΔH = −5.9 ± 0.1 kcal/mol, TΔS = 2.7 ± 0.1 kcal/mol; Ν143P, Ch+: n = 1.04 ± 0.01, K = 1.0 ± 0.1 106 m−1, ΔG = −8.2 ± 0.1 kcal/mol, ΔH = −7.2 ± 0.1 kcal/mol, TΔS = 1.0 ± 0.1 kcal/mol.
To further dissect the molecular basis of the properties documented by kinetic and equilibrium measurements, crystal structures of the N143P mutant were solved in the presence and absence of Na+. The structure in the absence of Na+ removes any doubts about the contribution of E* for this mutant, called into question by the lack of slow phase in the Na+ binding data reported in Fig. 2. The structure is practically identical (r.m.s.d. = 0.15 Å) to the E* form identified from two other mutations (43, 73, 79). The presence of the N143P mutation abrogates the H-bond with the carbonyl O atom of Glu192 and causes a flip of the Glu192-Gly193 peptide bond with disruption of the oxyanion hole (Fig. 4). The mutant folds in a self-inhibited conformation (Fig. 4) due to a collapse of the 215–217 β-strand into the active site that moves the indole ring of Trp215 >10 Å to engage the catalytic His57 on the opposite side of the active site cleft. Hence, the thrombin mutants D102N, Δ146–149e, and N143P crystallize in the same E* form, notwithstanding differences in sequence and crystallization conditions. In the presence of Na+ the collapse of the 215–217 β-strand into the active site is corrected, and Trp215 moves back into the position observed in the E:Na+ form (Fig. 4). The Na+ site is restored and binds the cation with the same coordination as wild type. However, the flip of the Glu192-Gly193 peptide bond is retained because of the presence of Pro143.
FIGURE 4.

X-ray crystal structure of the thrombin mutant N143P. Left: ribbon representation of the structure of the thrombin mutant N143P (Pro143 is shown as a stick model and indicated by arrow) in the E* (cyan) and E:Na+ (gold) forms. The 215–217 β-strand in the mutant collapses into the primary specificity pocket (red arrows), with the side chain of Trp215 (stick model) repositioned into the active site (residues of the catalytic triad His57, Asp102, and Ser195 are shown as stick models) in hydrophobic interaction with Trp60d, Tyr60a, Leu99, and His57. This represents a drastic change (r.m.s.d. 0.384 Å) from the E:Na+ conformation of the mutant N143P (Na+ shown as a purple ball) where the side chain of Trp215 is positioned 10.5 Å away and leaves the active site accessible to substrate. Right: details of the disruption of the oxyanion hole in the thrombin mutant N143P in the E:Na+ form (CPK, gold). The conformation of the same residues in the E* form is shown by comparison (CPK, cyan). The Glu192-Gly193 peptide bond is flipped (red arrow), because the presence of Pro143 abolishes the H-bond between the backbone N atom of residue 143 and the backbone O atom of Glu192. The 2Fo − Fc electron density map (light green mesh) refers to the E:Na+ form and is contoured at 1.0 σ.
DISCUSSION
The extension of the Botts-Morales scheme presented in this study provides a rigorous framework to understand the mechanism of thrombin activation by Na+. A major property of the extended scheme is that kcat offers unequivocal evidence of the presence of allosteric transduction of Na+ binding. Hence, any mechanism of Na+ activation must involve discussion of the properties of the active forms E and E:Na+. The inactive E* form cannot be invoked to explain the mechanism of Na+ activation of thrombin pertaining to kcat. The kinetic constant kcat is a property of the enzyme-substrate complex, which cannot form when thrombin assumes the inactive E* form. This refutes recent naive suggestions that Na+ activates thrombin by switching the enzyme from an inactive to an active conformation (95, 96) and that also lead to the untenable prediction of kcat/Km = 0 in the absence of Na+. Thrombin exists in the E* and E forms in the absence of Na+. The presence of E explains why thrombin is not inactive in the absence of Na+ (kcat/Km > 0), as documented by three decades of experimental data (9, 11, 19). The presence of E* explains why Na+ binding to thrombin obeys a two-step kinetic mechanism (Scheme 1) (23) and why the activity of some mutants defective for Na+ binding is orders of magnitude lower than that of the E form of wild-type (43, 70, 97).
Analysis of the structures of E and E:Na+ bound to PPACK reveals that they are practically identical to each other (r.m.s. = 0.19 Å) except for a small (1 Å) upward shift in the 60-loop in the absence of Na+ (38) that hardly explains the difference in kcat observed experimentally. The E and E:Na+ forms are also very similar when free (38) except for the Arg187–Asp222 ion pair that is broken in the E form, a rotation of Asp189 in the primary specificity pocket, the conformation of Ser195 that breaks the H-bond with His57 in the E form, and the architecture of the water network spanning >20 Å from the Na+ site to the active site (38). Mutagenesis of Arg187, Asp189, and Asp222 supports the role of these residues in the E-E:Na+ interconversion, because their replacement to Ala weakens Na+ binding (38, 98). The S195A mutant has little effect on Na+ binding (65), but its role on allosteric transduction cannot be established because of its lack of catalytic activity. The water network links the β-strands 215–219, 225–227, and 191–193 that define the Na+ site and the walls of the primary specificity pocket and likely provides the long range connectivity needed to allosterically communicate information from the Na+ site to the active site Ser195 and to residues involved in substrate recognition, such as Asp189. However, the role of the water network is hard to probe by mutagenesis, because no side chains from the protein are involved, other than Asp189, Glu192, and Ser195. Hence, residues of thrombin responsible for transducing Na+ binding into enhanced catalytic activity measured by kcat have remained elusive. The mutant N143P reported in this study provides direct evidence that the architecture of the oxyanion hole is under the control of Na+ binding and ensures optimal catalysis in the E:Na+ form. The mutation causes the Glu192-Gly193 peptide bond to flip and disrupts the correct orientation of the backbone N atom of Gly193. Kinetically, the kcat for hydrolysis of FPF, FPK, or FPR becomes independent of [Na+], and the value of α is reduced compared with wild-type. Hence, the Glu192-Gly193 peptide bond is most likely flipped in the E form and assumes the correct orientation in the E:Na+ form. These changes are not documented by existing structures of E and E:Na+ (38) most likely because the Glu192-Gly193 peptide bond has intrinsic flexibility in the absence of Na+ and can assume alternative conformations depending on solution and packing conditions. However, the Glu192-Gly193 peptide bond is flipped in the K+-bound structure of thrombin (99), which could explain why this cation is less effective than Na+ in activating the enzyme (19, 54).
The mutant N143P in the Na+-free form crystallizes in the inactive E* form, as observed for two other mutants of thrombin (43, 73, 79). The structural architecture of the E* form is now well established and is completely distinct from that of the zymogen form of the protease, contrary to some recent unsubstantiated claims (95, 96). Activation of many trypsin-like proteases requires proteolytic processing of an inactive zymogen precursor (100). Cleavage of the proprotein precursor occurs at the identical position in all known members of the family, i.e. between residues 15 and 16. The nascent N terminus induces conformational change in the enzyme through formation of an ion pair with the highly conserved Asp194 that organizes both the oxyanion hole and substrate binding site (101, 102). The ion pair between Ile16 and Asp194 remains intact in the E* form, with a distance of only 2.6 Å between the N terminus of Ile16 and the Oδ2 atom of Asp194. On the other hand, the ion pair is broken in existing structures of the zymogen forms of trypsin (102), chymotrypsin (103), and chymase (104). Finally, stopped-flow experiments demonstrate that the E*-E interconversion takes place on a time scale <10 ms (23), as opposed to the zymogen-protease conversion that evolves over a much longer (100–1000 ms) time scale (94, 105).
The value of r for the N143P mutant must be high enough to enable detection of the E* conformation by crystallographic studies. Yet, the E form also exists for this mutant because both s0 and k2,0 have finite values (Table 3) and future x-ray studies should be able to trap this conformation of the enzyme under suitable conditions. Because the values of α for this mutant depend on the substrate used and also differ from the value of γ for argatroban binding, it must be concluded that the N143P mutation abrogates the Na+-induced enhancement of kcat but influences to different extent the binding of substrates and ligands to the active site. Hence, the mutation must perturb the primary specificity pocket and possibly other recognition sites within the active site in a way that is probed differently by FPF, FPK, FPR, and argatroban.
The combination of kinetic and structural data enables us to conclude that a major difference between the E and E:Na+ forms of thrombin is the conformation of the Glu192-Gly193 peptide bond controlling the architecture of the oxyanion hole. The architecture is disrupted in the E form and correctly formed in the E:Na+ form, thereby accounting for the higher kcat upon Na+ binding. The difference between the inactive E* form and the active E form, both lacking Na+ bound at its site, likely resides in the conformation of Trp215 and the 215–217 β-strand. In the E* form, the β-strand collapses into the active site and precludes substrate binding, but in the E form the β-strand is correctly positioned to allow substrate diffusion into the active site. Furthermore, rearrangement of the 186- and 220-loops in the E form relative to the E* form enables Na+ binding and conversion into the more active E:Na+ form. Evidence that Na+ binding plays an important structural and functional role in other clotting enzymes is overwhelming (12–16, 48–63). Absolute conservation of the 143–192 H-bond in trypsin-like proteases (10) and the importance of the oxyanion hole in protease function (80, 81) suggest that the mechanism of Na+ activation revealed by the N143P mutant of thrombin is present in all Na+-activated trypsin-like proteases.
This work was supported, in whole or in part, by National Institutes of Health Grants HL49413, HL58141 HL73813, and HL95315 (to E. D. C.). This work was also supported by a postdoctoral fellowship from the American Heart Association (to W. N.)
The atomic coordinates and structure factors (codes 3JZ1 and 3JZ2) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- FPF
- H-d-Phe-Pro-Phe-p-nitroanilide
- FPK
- H-d-Phe-Pro-Lys-p-nitroanilide
- FPR
- H-d-Phe-Pro-Arg-p-nitroanilide
- FGR
- H-d-Phe-Gly-Arg-p-nitroanilide
- PEG
- polyethylene glycol
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1.Ibers J. A., Holm R. H. (1980) Science 209, 223–235 [DOI] [PubMed] [Google Scholar]
- 2.Castagnetto J. M., Hennessy S. W., Roberts V. A., Getzoff E. D., Tainer J. A., Pique M. E. (2002) Nucleic Acids Res. 30, 379–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tainer J. A., Roberts V. A., Getzoff E. D. (1992) Curr. Opin. Biotechnol. 3, 378–387 [DOI] [PubMed] [Google Scholar]
- 4.Boyer P. D., Lardy H. A., Phillips P. H. (1942) J. Biol. Chem. 146, 673–681 [Google Scholar]
- 5.Kachmar J. F., Boyer P. D. (1953) J. Biol. Chem. 200, 669–682 [PubMed] [Google Scholar]
- 6.Cohn M., Monod J. (1951) Biochim. Biophys. Acta 7, 153–174 [DOI] [PubMed] [Google Scholar]
- 7.Suelter C. H. (1970) Science 168, 789–795 [DOI] [PubMed] [Google Scholar]
- 8.Di Cera E. (2006) J. Biol. Chem. 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 9.Di Cera E. (2008) Mol. Aspects Med. 29, 203–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Page M. J., Di Cera E. (2008) Cell Mol. Life Sci. 65, 1220–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orthner C. L., Kosow D. P. (1980) Arch. Biochem. Biophys. 202, 63–75 [DOI] [PubMed] [Google Scholar]
- 12.Orthner C. L., Kosow D. P. (1978) Arch. Biochem. Biophys. 185, 400–406 [DOI] [PubMed] [Google Scholar]
- 13.Steiner S. A., Amphlett G. W., Castellino F. J. (1980) Biochem. Biophys. Res. Commun. 94, 340–347 [DOI] [PubMed] [Google Scholar]
- 14.Steiner S. A., Castellino F. J. (1982) Biochemistry 21, 4609–4614 [DOI] [PubMed] [Google Scholar]
- 15.Steiner S. A., Castellino F. J. (1985) Biochemistry 24, 1136–1141 [DOI] [PubMed] [Google Scholar]
- 16.Steiner S. A., Castellino F. J. (1985) Biochemistry 24, 609–617 [DOI] [PubMed] [Google Scholar]
- 17.Curragh E. F., Elmore D. T. (1964) Biochem. J. 93, 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Workman E. F., Lundblad R. L. (1978) Arch. Biochem. Biophys. 185, 544–548 [DOI] [PubMed] [Google Scholar]
- 19.Wells C. M., Di Cera E. (1992) Biochemistry 31, 11721–11730 [DOI] [PubMed] [Google Scholar]
- 20.Prasad S., Wright K. J., Banerjee, Roy D., Bush L. A., Cantwell A. M., Di Cera E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13785–13790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffon N., Di Stasio E. (2001) Biophys. Chem. 90, 89–96 [DOI] [PubMed] [Google Scholar]
- 22.Guinto E. R., Di Cera E. (1996) Biochemistry 35, 8800–8804 [DOI] [PubMed] [Google Scholar]
- 23.Bah A., Garvey L. C., Ge J., Di Cera E. (2006) J. Biol. Chem. 281, 40049–40056 [DOI] [PubMed] [Google Scholar]
- 24.Di Cera E., Page M. J., Bah A., Bush-Pelc L. A., Garvey L. C. (2007) Phys. Chem. Chem. Phys. 9, 1292–1306 [DOI] [PubMed] [Google Scholar]
- 25.Dang O. D., Vindigni A., Di Cera E. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 5977–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang Q. D., Guinto E. R., Di Cera E. (1997) Nat. Biotechnol. 15, 146–149 [DOI] [PubMed] [Google Scholar]
- 27.Di Cera E., Dang Q. D., Ayala Y. M. (1997) Cell Mol. Life Sci. 53, 701–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayala Y. M., Cantwell A. M., Rose T., Bush L. A., Arosio D., Di Cera E. (2001) Proteins 45, 107–116 [DOI] [PubMed] [Google Scholar]
- 29.Myles T., Yun T. H., Hall S. W., Leung L. L. (2001) J. Biol. Chem. 276, 25143–25149 [DOI] [PubMed] [Google Scholar]
- 30.Nogami K., Zhou Q., Myles T., Leung L. L., Wakabayashi H., Fay P. J. (2005) J. Biol. Chem. 280, 18476–18487 [DOI] [PubMed] [Google Scholar]
- 31.Yun T. H., Baglia F. A., Myles T., Navaneetham D., López J. A., Walsh P. N., Leung L. L. (2003) J. Biol. Chem. 278, 48112–48119 [DOI] [PubMed] [Google Scholar]
- 32.Degen S. J., McDowell S. A., Sparks L. M., Scharrer I. (1995) Thromb. Haemost. 73, 203–209 [PubMed] [Google Scholar]
- 33.Miyata T., Aruga R., Umeyama H., Bezeaud A., Guillin M. C., Iwanaga S. (1992) Biochemistry 31, 7457–7462 [DOI] [PubMed] [Google Scholar]
- 34.Henriksen R. A., Dunham C. K., Miller L. D., Casey J. T., Menke J. B., Knupp C. L., Usala S. J. (1998) Blood 91, 2026–2031 [PubMed] [Google Scholar]
- 35.Sun W. Y., Smirnow D., Jenkins M. L., Degen S. J. (2001) Thromb. Haemost. 85, 651–654 [PubMed] [Google Scholar]
- 36.Stanchev H., Philips M., Villoutreix B. O., Aksglaede L., Lethagen S., Thorsen S. (2006) Thromb. Haemost. 95, 195–198 [PubMed] [Google Scholar]
- 37.Rouy S., Vidaud D., Alessandri J. L., Dautzenberg M. D., Venisse L., Guillin M. C., Bezeaud A. (2006) Br. J. Haematol. 132, 770–773 [DOI] [PubMed] [Google Scholar]
- 38.Pineda A. O., Carrell C. J., Bush L. A., Prasad S., Caccia S., Chen Z. W., Mathews F. S., Di Cera E. (2004) J. Biol. Chem. 279, 31842–31853 [DOI] [PubMed] [Google Scholar]
- 39.Cantwell A. M., Di Cera E. (2000) J. Biol. Chem. 275, 39827–39830 [DOI] [PubMed] [Google Scholar]
- 40.Dang Q. D., Sabetta M., Di Cera E. (1997) J. Biol. Chem. 272, 19649–19651 [DOI] [PubMed] [Google Scholar]
- 41.Gibbs C. S., Coutré S. E., Tsiang M., Li W. X., Jain A. K., Dunn K. E., Law V. S., Mao C. T., Matsumura S. Y., Mejza S. J. (1995) Nature 378, 413–416 [DOI] [PubMed] [Google Scholar]
- 42.Tsiang M., Paborsky L. R., Li W. X., Jain A. K., Mao C. T., Dunn K. E., Lee D. W., Matsumura S. Y., Matteucci M. D., Coutré S. E., Leung L. L., Gibbs C. S. (1996) Biochemistry 35, 16449–16457 [DOI] [PubMed] [Google Scholar]
- 43.Bah A., Carrell C. J., Chen Z., Gandhi P. S., Di Cera E. (2009) J. Biol. Chem. 284, 20034–20040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bode W., Turk D., Karshikov A. (1992) Protein Sci. 1, 426–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rydel T. J., Tulinsky A., Bode W., Huber R. (1991) J. Mol. Biol. 221, 583–601 [DOI] [PubMed] [Google Scholar]
- 46.Banner D. W., Hadváry P. (1991) J. Biol. Chem. 266, 20085–20093 [PubMed] [Google Scholar]
- 47.Di Cera E., Guinto E. R., Vindigni A., Dang Q. D., Ayala Y. M., Wuyi M., Tulinsky A. (1995) J. Biol. Chem. 270, 22089–22092 [DOI] [PubMed] [Google Scholar]
- 48.Zhang E., Tulinsky A. (1997) Biophys. Chem. 63, 185–200 [DOI] [PubMed] [Google Scholar]
- 49.Schärer K., Morgenthaler M., Paulini R., Obst-Sander U., Banner D. W., Schlatter D., Benz J., Stihle M., Diederich F. (2005) Angew Chem. Int. Ed. Engl. 44, 4400–4404 [DOI] [PubMed] [Google Scholar]
- 50.Bajaj S. P., Schmidt A. E., Agah S., Bajaj M. S., Padmanabhan K. (2006) J. Biol. Chem. 281, 24873–24888 [DOI] [PubMed] [Google Scholar]
- 51.Schmidt A. E., Padmanabhan K., Underwood M. C., Bode W., Mather T., Bajaj S. P. (2002) J. Biol. Chem. 277, 28987–28995 [DOI] [PubMed] [Google Scholar]
- 52.Schmidt A. E., Stewart J. E., Mathur A., Krishnaswamy S., Bajaj S. P. (2005) J. Mol. Biol. 350, 78–91 [DOI] [PubMed] [Google Scholar]
- 53.Papaconstantinou M. E., Gandhi P. S., Chen Z., Bah A., Di Cera E. (2008) Cell Mol. Life Sci. 65, 3688–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dang Q. D., Di Cera E. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10653–10656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petrovan R. J., Ruf W. (2000) Biochemistry 39, 14457–14463 [DOI] [PubMed] [Google Scholar]
- 56.Rezaie A. R., He X. (2000) Biochemistry 39, 1817–1825 [DOI] [PubMed] [Google Scholar]
- 57.Rezaie A. R., Kittur F. S. (2004) J. Biol. Chem. 279, 48262–48269 [DOI] [PubMed] [Google Scholar]
- 58.Monnaie D., Arosio D., Griffon N., Rose T., Rezaie A. R., Di Cera E. (2000) Biochemistry 39, 5349–5354 [DOI] [PubMed] [Google Scholar]
- 59.Underwood M. C., Zhong D., Mathur A., Heyduk T., Bajaj S. P. (2000) J. Biol. Chem. 275, 36876–36884 [DOI] [PubMed] [Google Scholar]
- 60.Camire R. M. (2002) J. Biol. Chem. 277, 37863–37870 [DOI] [PubMed] [Google Scholar]
- 61.Levigne S., Thiec F., Cherel S., Irving J. A., Fribourg C., Christophe O. D. (2007) J. Biol. Chem. 282, 31569–31579 [DOI] [PubMed] [Google Scholar]
- 62.He X., Rezaie A. R. (1999) J. Biol. Chem. 274, 4970–4976 [DOI] [PubMed] [Google Scholar]
- 63.Kroh H. K., Tans G., Nicolaes G. A., Rosing J., Bock P. E. (2007) J. Biol. Chem. 282, 16095–16104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Botts J., Morales M. (1953) Trans. Faraday Soc. 49, 696–707 [Google Scholar]
- 65.Gianni S., Ivarsson Y., Bah A., Bush-Pelc L. A., Di Cera E. (2007) Biophys. Chem. 131, 111–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lai M. T., Di Cera E., Shafer J. A. (1997) J. Biol. Chem. 272, 30275–30282 [DOI] [PubMed] [Google Scholar]
- 67.Krem M. M., Di Cera E. (2003) Biophys. Chem. 100, 315–323 [DOI] [PubMed] [Google Scholar]
- 68.Vindigni A., Di Cera E. (1996) Biochemistry 35, 4417–4426 [DOI] [PubMed] [Google Scholar]
- 69.Liu Y., Kati W., Chen C. M., Tripathi R., Molla A., Kohlbrenner W. (1999) Anal. Biochem. 267, 331–335 [DOI] [PubMed] [Google Scholar]
- 70.Gandhi P. S., Page M. J., Chen Z., Bush-Pelc L., Di Cera E. (2009) J. Biol. Chem. 284, 24098–24105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 72.Bailey S. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 73.Gandhi P. S., Chen Z., Mathews F. S., Di Cera E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1832–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 75.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 76.Morris A. L., MacArthur M. W., Hutchinson E. G., Thornton J. M. (1992) Proteins 12, 345–364 [DOI] [PubMed] [Google Scholar]
- 77.Page M. J., Di Cera E. (2006) Physiol. Rev. 86, 1049–1092 [DOI] [PubMed] [Google Scholar]
- 78.Hill T. L. (1977) Free Energy Transduction in Biology, Academic Press, New York [Google Scholar]
- 79.Pineda A. O., Chen Z. W., Bah A., Garvey L. C., Mathews F. S., Di Cera E. (2006) J. Biol. Chem. 281, 32922–32928 [DOI] [PubMed] [Google Scholar]
- 80.Hedstrom L. (2002) Chem. Rev. 102, 4501–4524 [DOI] [PubMed] [Google Scholar]
- 81.Perona J. J., Craik C. S. (1995) Protein Sci. 4, 337–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Birktoft J. J., Blow D. M. (1972) J. Mol. Biol. 68, 187–240 [DOI] [PubMed] [Google Scholar]
- 83.Bobofchak K. M., Pineda A. O., Mathews F. S., Di Cera E. (2005) J. Biol. Chem. 280, 25644–25650 [DOI] [PubMed] [Google Scholar]
- 84.Timm D. E. (1997) Protein Sci. 6, 1418–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Katona G., Berglund G. I., Hajdu J., Gráf L., Szilágyi L. (2002) J. Mol. Biol. 315, 1209–1218 [DOI] [PubMed] [Google Scholar]
- 86.Parry M. A., Jacob U., Huber R., Wisner A., Bon C., Bode W. (1998) Structure 6, 1195–1206 [DOI] [PubMed] [Google Scholar]
- 87.Schmidt A. E., Ogawa T., Gailani D., Bajaj S. P. (2004) J. Biol. Chem. 279, 29485–29492 [DOI] [PubMed] [Google Scholar]
- 88.Ponnuraj K., Xu Y., Macon K., Moore D., Volanakis J. E., Narayana S. V. (2004) Mol. Cell 14, 17–28 [DOI] [PubMed] [Google Scholar]
- 89.Barrette-Ng I. H., Ng K. K., Mark B. L., Van Aken D., Cherney M. M., Garen C., Kolodenko Y., Gorbalenya A. E., Snijder E. J., James M. N. (2002) J. Biol. Chem. 277, 39960–39966 [DOI] [PubMed] [Google Scholar]
- 90.Korostelev A., Asahara H., Lancaster L., Laurberg M., Hirschi A., Zhu J., Trakhanov S., Scott W. G., Noller H. F. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19684–19689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu G., Liu J., Terzyan S., Zhai P., Li G., Zhang X. C. (2003) J. Biol. Chem. 278, 2452–2460 [DOI] [PubMed] [Google Scholar]
- 92.Marciano D. C., Pennington J. M., Wang X., Wang J., Chen Y., Thomas V. L., Shoichet B. K., Palzkill T. (2008) J. Mol. Biol. 384, 151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Evans S. A., Olson S. T., Shore J. D. (1982) J. Biol. Chem. 257, 3014–3017 [PubMed] [Google Scholar]
- 94.Fersht A. R., Requena Y. (1971) J. Mol. Biol. 60, 279–290 [DOI] [PubMed] [Google Scholar]
- 95.Johnson D. J., Adams T. E., Li W., Huntington J. A. (2005) Biochem. J. 392, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Filippis V., De Dea E., Lucatello F., Frasson R. (2005) Biochem. J. 390, 485–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guinto E. R., Caccia S., Rose T., Fütterer K., Waksman G., Di Cera E. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prasad S., Cantwell A. M., Bush L. A., Shih P., Xu H., Di Cera E. (2004) J. Biol. Chem. 279, 10103–10108 [DOI] [PubMed] [Google Scholar]
- 99.Carrell C. J., Bush L. A., Mathews F. S., Di Cera E. (2006) Biophys. Chem. 121, 177–184 [DOI] [PubMed] [Google Scholar]
- 100.Neurath H., Dixon G. H. (1957) Fed. Proc. 16, 791–801 [PubMed] [Google Scholar]
- 101.Fehlhammer H., Bode W. (1975) J. Mol. Biol. 98, 683–692 [DOI] [PubMed] [Google Scholar]
- 102.Fehlhammer H., Bode W., Huber R. (1977) J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 103.Wang D., Bode W., Huber R. (1985) J. Mol. Biol. 185, 595–624 [DOI] [PubMed] [Google Scholar]
- 104.Reiling K. K., Krucinski J., Miercke L. J., Raymond W. W., Caughey G. H., Stroud R. M. (2003) Biochemistry 42, 2616–2624 [DOI] [PubMed] [Google Scholar]
- 105.Fersht A. R. (1972) J. Mol. Biol. 64, 497–509 [DOI] [PubMed] [Google Scholar]