

Abstract
Dominant missense mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most common known genetic cause of Parkinson disease. LRRK2 encodes a serine/threonine protein kinase, and pathogenic mutations may increase kinase activity. Intrinsic GTP binding in the GTPase domain may govern kinase activity through an internal signal transduction cascade. As with many protein kinases, LRRK2 self-interacts through mechanisms that may regulate enzymatic activity. We find that the disruption of either GTPase or kinase activity enhances the formation of high molecular weight oligomers and prevents the formation of LRRK2 dimer structures. In addition, brief application of the broad spectrum kinase inhibitor staurosporine ablates LRRK2 dimers and promotes LRRK2 high molecular weight oligomers. LRRK2 interactions with other proteins in cell lines are kinase-independent and include chaperones and cell cytoskeleton components, suggesting that LRRK2 self-assembly principally dictates complex size. To further explore the mechanics of kinase activation, we separate soluble LRRK2 protein that encodes the pathogenic G2019S mutation into high molecular weight oligomers, dimers, and monomers and find that kinase activity resides with dimeric LRRK2. Some PD-associated mutations that increase kinase activity in vitro significantly increase the proportion of dimer structures relative to total LRRK2 protein, providing additional insight into how pathogenic mutations may alter normal enzymatic regulation. Targeting and tracking LRRK2 dimerization may provide a clear way to observe LRRK2 kinase activity in living cells, and disruption of dimeric LRRK2 through kinase inhibition or other means may attenuate pathogenic increases in LRRK2 enzymatic output.
Introduction
Parkinson disease (PD)2 encompasses a complex spectrum of symptoms and pathologies together with a largely undefined etiology (1, 2). The identification of genes important for disease susceptibility presents an opportunity to explore the molecular basis of the neuronal dysfunction and degeneration associated with the disease and discovery of potential therapeutic targets and strategies. Dominant missense mutations in the leucine-rich repeat kinase 2 gene (LRRK2) are a common cause of late onset PD in some populations (e.g. in North African Arabs, where the G2019S mutation can cause up to 30% of sporadic PD) (3, 4). In most Western populations, the commonest known mutation G2019S underlies between 1 and 5% of cases (5). A G2385R polymorphism strongly associates with PD in Eastern Asian populations (6, 7). Mutations in LRRK2 associate with disease in clinical populations difficult to distinguish from typical idiopathic late onset PD (i.e. LRRK2-negative disease). Some well described families with LRRK2 mutations, particularly those other than the G2019S mutation, demonstrate pleomorphic pathology that includes variable α-synuclein and Tau structures, whereas the majority of LRRK2 cases analyzed on a pathological level are consistent with typical pathological staging and idiopathic PD (8).
LRRK2 encodes a unique arrangement of conserved protein domains, exemplified by the presence of a functional GTPase and kinase domain within the same molecule. The G2019S mutation occurs in the kinase activation loop in subdomain VII. In vitro analyses suggest that mutations in LRRK2 cause subtle but highly significant alterations in kinase activity, and the G2019S mutation consistently induces ∼2–3-fold increases in output in various studies and kinase assay protocols (reviewed in Ref. 9). In full-length protein derived from mammalian cells, artificial mutations that ablate GTPase activity completely inhibit kinase activity, whereas mutations that ablate kinase activity appear to have little effect on GTP binding activity, at least in vitro (10, 11). PD-associated mutations in or near the GTPase domain may alter GTP binding and or hydrolysis activity (10, 12). LRRK2 autophosphorylates the GTP-binding pocket of the ROC (GTPase) domain, suggesting a potential feed-back or feed-forward regulatory loop (13). The accessory proteins required for LRRK2 GTPase activity or binding (GTPase-activating protein or guanine exchange factor) or native LRRK2 kinase substrates are not yet known.
Recognition of the native mechanisms of LRRK2 enzyme function will provide a foundation to understand the effects of pathologic LRRK2 mutations and the determination of whether LRRK2 activities are abnormal in PD cases. Protein kinases that bear a semblance to the encoded LRRK2 kinase domain both on a sequence and phylogenetic level (e.g. mixed lineage kinase 3) require protein dimerization for kinase activation (14, 15). Dimerization and oligomerization of protein kinases can play regulatory roles for a number of characterized serine/threonine kinases (16). Components of the mitogen-activated protein kinase signaling cascade, a potential target for LRRK2 kinase activity (17), are also regulated in part by kinase dimerization (18–20).
LRRK2 self-association has been documented (21), with evidence of kinase-dependent protein dimerization (22). LRRK2 self-associates through multiple interfaces across the protein, with an indication that pathogenic mutations might alter self-interaction (23). Herein, we further characterize the effects of pathogenic and activity-ablating mutations on LRRK2 dimerization and oligomerization. Although LRRK2 distribution, solubility and protein-interactions in cells seem independent from kinase activity, the formation of dimer-sized LRRK2 structures, distinguished from high molecular weight LRRK2 oligomers, closely tracks with kinase activity, and LRRK2 dimers represent the kinase-active LRRK2 structure.
EXPERIMENTAL PROCEDURES
Plasmids, Cell Culture, and Transfection
Mammalian expression constructs encoding human LRRK2 on the pcDNA3.1- Myc/His backbone (Invitrogen) were a generous gift from Ted and Valina Dawson (The Johns Hopkins University, Baltimore, MD) and were described previously (10). Individual maxiscale plasmid preparations (Qiagen) were assessed for equivalent LRRK2 expression by Western blot (SDS-PAGE, described below) and resequenced in their entirety prior to usage in experiments. A construct encoding wild-type LRRK2 on the pCMV-Myc vector backbone was a generous gift from Mark Cookson (NIA, National Institutes of Health, Bethesda, MD). To construct LRRK2 plasmids for tandem affinity purifications, the streptavidin-binding peptide-calmodulin-binding peptide tandem tag from the pCTAP construct (Stratagene) was amplified by PCR with primers encoding flanking XhoI sites and inserted into existing LRRK2 constructs on the pcDNA3.1- Myc/His backbone. Most experiments were carried out in HEK-293FT cells, cultured in Opti-MEM medium supplemented with 10% fetal bovine serum (Invitrogen). One day prior to transfection, cells were passaged and transfection was carried out at ∼80% confluence in Opti-MEM medium supplemented with 2% fetal bovine serum using FugeneHD reagent at a 1:3 ratio of plasmid to FugeneHD (Roche Applied Science), and cells were harvested 48 h post-transfection. HEK-293FT cells were discarded after 10 passages (after receipt from manufacturer; Invitrogen). Lysates from four lymphoblast cell lines previously confirmed positive for LRRK2 expression (24) and antibodies specific to human LRRK2 were a generous gift from Matt Farrer (Mayo Clinic Udall Center, Jacksonville, FL).
Protein Expression Analysis (Native PAGE/SDS-PAGE)
Transfected cells were gently harvested by a cell scraper, divided in half, and pelleted by centrifugation at 200 × g for 5 min at 4 °C. Cell pellets were resuspended with either native lysis buffer (phosphate-buffered saline (PBS), pH 7.4, 1× protease and phosphatase inhibitors (Roche Applied Science)), lysed by four cycles of freezing and thawing, and analyzed on a 3–12% bis-tris Blue native polyacrylamide gel (Invitrogen) or lysates combined with SDS-lysis buffer (PBS, pH 7.4, 1% SDS, 1× protease and phosphatase inhibitors (Roche Applied Science)) and sonicated with 10% power for 10 s on a Branson dismembranator and analyzed on a 7% Tris acetate SDS gel (Invitrogen). Prior to PAGE, lysates were centrifuged for 10 min at 20,000 × g, and protein content in supernatant was determined by a BCA assay (Pierce). Proteins resolved in acrylamide were transferred to PVDF membranes (Immobilon P, Millipore) at 4 °C overnight to ensure complete transfer of high molecular weight species. In most experiments, PVDF membranes were blocked with 5% nonfat milk in TBS-T (Tris-buffered saline Tween-20) for 1 h and incubated overnight in blocking buffer supplemented with anti-c-Myc HRP conjugate antibody (Roche Applied Science) and developed using electrochemical luminescence substrate (Pierce). For quantification of LRRK2 dimer intensity relative to total LRRK2 (see Fig. 3), PVDF membranes were probed with anti-c-Myc antibody (clone 9E10, Roche Applied Science) and the IRDye 800CW donkey anti-mouse antibody (LI-COR), and images were acquired and analyzed on a LI-COR Odyssey system.
FIGURE 3.

PD-associated LRRK2 mutations enhance the proportion of soluble LRRK2 dimer-sized and high molecular weight species. A, HEK-293T cells were transfected with constructs encoding human LRRK2 that harbors the indicated mutation, where GTPase-dead is K1347A-LRRK2 and kinase-dead is D1994A-LRRK2. A vector encoding WT-LRRK2 (Wild Type-NIH) derived independently from LRRK2-vectors used in this study was provided by Mark Cookson. Cell pellets were split equally for lysis by freeze/thaw cycles directly in PBS or lysis with 1% SDS and PBS with sonication, and 10 μg of protein lysate (as determined by BCA protein assay) was loaded onto a native gel (3–12% bis-tris) or an SDS gel (7% Tris acetate-SDS), respectively. Ponceau S stain was applied to PVDF membranes after transfer of protein complexes to PVDF to ensure even transfer, and the region near 500 kDa is shown and demonstrates the presence of protein across the membrane. LRRK2 complexes were visualized with the anti-c-Myc antibody by Western blot and imaged on a LI-COR Odyssey. Western blots representative of five independent experiments are shown. B, normalization of the LRRK2 dimer-sized structure (signal from ∼480 to ∼550 kDa) to total LRRK2 protein (SDS-solubilized) using densitometry analysis. Error bars, ±S.E. *, p < 0.009. n.s., nonsignificant in comparison with wild-type LRRK2, assessed by unpaired Student's t test.
Size Exclusion Chromatography
Cell pellets were combined with native buffer as above, lysed by four rounds of freeze/thaw, and centrifuged at 20,000 × g for 20 min to pellet large debris, and supernatant was concentrated to the column injection volume of 250 μl using Amicon Ultra 100 kDa cut-off membranes (Millipore). A Superdex 200 10/300 GL column (GE Healthcare) coupled in line with a BioLogix Duo-Flow system and digital fraction collector (Bio-Rad) was equilibrated with two column volumes of running buffer (PBS, pH 7.4, with 0.1× protease inhibitors (Roche Applied Science)) at a flow rate of 0.2 ml/min with a maintained pressure of ∼110 p.s.i. Prior to the injection of total cell lysate, column fractions were matched with molecular weight standards (Sigma FPLC marker set), and void volume was confirmed by in-line absorbance measurements at 280 nm and Bradford assay in initial fractions, whereby the fraction containing void volume was identified as the first fraction positive for protein. Fractions were analyzed by native PAGE analysis immediately following separation or processed for immunoprecipitation and kinase assays as described below.
LRRK2 Kinase Assays
Total cell lysates or FPLC fractions were combined with Protein G Dynabeads (Invitrogen) precoupled with anti-Myc antibody (clone 9E10, Roche Applied Science) and incubated at 4 °C on a rotor wheel. Dynabeads were washed five times with lysis buffer, beads were resuspended into kinase assay buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 5 mm EGTA, 20 mm β-glycerol phosphate, and 10 mm MBP), and kinase reactions were initiated with the addition of 10 μm ATP (0.5 μCi of [γ-32P]ATP (PerkinElmer Life Sciences)) and 20 mm MgCl2. The kinase reactions were incubated at 30 °C for 30 min with gentle shaking. The reaction was terminated by placing on ice and supplementing with 1× Laemmli buffer. Laemmli buffer was removed from the Dynabeads to separate the MBP fraction, and beads were resuspended into 2× Laemmli buffer and heated at 70 °C for 10 min with gentle shaking to elute and denature LRRK2 protein complexes. Kinase reactions were resolved with SDS-polyacrylamide gels and stained with Bio-Safe G-250 dye and dried onto Whatman paper. MBP phosphorylation and LRRK2 autophosphorylation signal were resolved with Kodak BioMax film, and intensity of spots was determined with NIH Image.
Recombinant WT-LRRK2 protein, truncated through the first 970 amino acids and fused with GST on the N terminus, was purchased from Invitrogen and combined into a kinase assay buffer that included PBS, pH 7.4, 5 mm EGTA, 20 mm β-glycerol phosphate, 10 μm ATP (0.5 μCi of [γ-32P]ATP (PerkinElmer Life Sciences)), 20 mm MgCl2, and varying concentrations of deoxycholate (Sigma). Reactions were incubated as above and analyzed by native PAGE and SDS-PAGE.
Confocal Microscopy
HEK-293FT cells were transfected with WT and mutant LRRK2 constructs, allowed to grow for 24 h post-transfection, and then replated on a 24-well plate with poly-l-lysine-coated glass coverslips. 48 h post-transfection, the cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min. Cells were washed twice in PBS and incubated in Blocking Buffer I (10% serum, 0.3% Triton X-100 in PBS) for 1 h at room temperature. Cells were then incubated overnight at 4 °C with the primary antibody (anti-Myc antibody in 10% serum, 0.3% Triton X-100 in PBS). The next day, the cells were washed three times with PBS and incubated in Blocking Buffer II (0.1% bovine serum albumin, 1% serum in PBS) for 1 h at room temperature. This was followed by incubation with anti-mouse Cy2 (Jackson Immunolabs; diluted in Blocking Buffer II) for 4 h at room temperature. Coverslips were washed three times with PBS and mounted on glass slides, and signal was resolved on a Leica confocal system.
Tandem Affinity Purifications and Mass Spectrometry Analysis
LRRK2 constructs encoding a c-terminal calmodulin binding peptide and a streptavidin binding peptide (from the pCTAP plasmid (Stratagene)) were prepared by Maxi-prep columns (Qiagen), and 200 μg of plasmid were used to transfect 800 cm2 of ∼80 confluent HEK-293FT cells (with FugeneHD as above) in Opti-MEM medium supplemented with 2% fetal bovine serum for each LRRK2 construct. Cells were collected and lysed by five cycles of freeze/thaw in native buffer, lysates were centrifuged at 20,000 × g for 10 min, and supernatant was combined with streptavidin-agarose beads (Stratagene) and incubated at 4 °C for 2 h. Beads were washed five times with lysis buffer and combined with buffer containing 10 mm biotin (Sigma). Complexes were eluted over the course of 30 min with gentle agitation at 16 °C, and supernatant was combined with calmodulin resin (Stratagene) and incubated at 4 °C for 2 h. Beads were washed seven times in lysis buffer, and complexes were eluted by EGTA and resuspended into 2× Laemmli buffer. SDS-PAGE was carried out on a 1.5-mm 10-well 7% Tris acetate gel (Invitrogen), and the gel was subsequently stained with G-250 Biosafe Coomassie dye (Bio-Rad) for 1 h, destained with water, wrapped in plastic, and imaged on a scanner. Visible bands were selected for removal with a razor blade, and individual samples were digested in gel with trypsin and analyzed by liquid chromatography-tandem mass spectrometry in the Taplin Mass Spectrometry Facility at Harvard Medical School.
RESULTS
G2019S-LRRK2 Protein Forms an Activity-dependent Dimer
LRRK2 self-interaction has been suggested through the use of differential fusion tags and co-immunoprecipitation (21), through yeast two-hybrid analysis and through the resolution of an LRRK2 species consistent with a dimer-sized complex (22, 23). Previously, endogenous LRRK2 protein was identified in lymphoblast cell lines, with some indication that LRRK2 may form dimer structures in these cells based on size exclusion separation (22, 24). We analyzed four lymphoblast cell lines that demonstrate various levels of LRRK2 protein expression by SDS-PAGE, normalized to glyceraldehyde-3-phosphate dehydrogenase expression, with LRRK2 detected by an antibody specific for human LRRK2 that detects a single band by SDS-PAGE Western blot but does not immunoprecipitate LRRK2 (antibody provided by Matthew Farrer) (Fig. 1A). Lysis of these cells by freeze/thaw cycles in PBS and lysate analyzed by native PAGE in an environment free of detergents revealed the presence of higher order LRRK2 structures, in addition to a prominent structure consistent with the expected size of a LRRK2 homodimer (Fig. 1A). The addition of detergents to lysis buffers, such as Triton X-100, SDS, digitonin, and deoxycholate, all affected LRRK2 oligomerization and dimerization visualized by native PAGE (data not shown); thus, detergents were excluded from buffers unless otherwise noted.
FIGURE 1.

LRRK2 forms kinase-sensitive high molecular weight conformations. A, cell pellets derived from four lymphoblast cell lines (labeled Lymph1 to -4) were split equally for lysis by freeze/thaw cycles directly in PBS or lysis with 1% SDS and PBS with sonication, and 10 μg of protein lysate (as determined by BCA protein assay) was loaded onto a native gel (3–12% bis-tris) or an SDS gel (7% Tris acetate-SDS), respectively. LRRK2 protein or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was visualized after transfer onto PVDF membrane and incubation with an antibody specific for LRRK2 or glyceraldehyde-3-phosphate dehydrogenase. Complexes consistent with the expected size of a LRRK2 homodimer and LRRK2 monomer are indicated. B, HEK-293T cells were transfected with constructs encoding human wild-type LRRK2, G2019S-LRRK2, or LRRK2 containing both the G2019S and D1994A (kinase-dead) mutations. Cell pellets were split equally for lysis by freeze/thaw cycles directly in PBS or lysis with 1% SDS and PBS with sonication, and 10 μg of protein lysate (as determined by BCA protein assay) was loaded onto a native gel (3–12% bis-tris) or an SDS gel (7% Tris acetate-SDS), respectively. LRRK2-positive complexes were visualized after transfer onto PVDF membrane and incubation with anti-Myc-HRP antibody. C, native lysates of LRRK2-transfected HEK-293FT cells derived from freeze/thaw lysis and subsequently treated with 5% β-ME and heat (65 °C for 10 min) as indicated were analyzed by native PAGE, and complexes consistent with the expected sizes of a LRRK2 homodimer and LRRK2 monomer are indicated.
To determine the characteristics of LRRK2 complexes present in HEK-293FT cells that we routinely use as a source for recombinant protein for in vitro kinase assays (10, 25), transiently transfected cells expressing human G2019S-LRRK2 protein lysed by freeze/thaw cycles in PBS and lysate analyzed by native PAGE show a nearly identical distribution of LRRK2 protein as compared with endogenous LRRK2 in lymphoblast cell lines. Transfer of complexes resolved by native PAGE to PVDF membranes cuts off near the highest molecular mass marker 1.2 MDa (e.g. the expected size of a complex of about four LRRK2 molecules), suggesting that protein complexes beyond this size do not efficiently enter and/or transfer from gel matrices. The presence of higher molecular weight but highly soluble LRRK2 oligomers can be assumed.
LRRK2 protein with a G2019S mutation showed a qualitatively similar native PAGE distribution compared with wild-type protein (Fig. 1B). The insertion of a kinase-inactivating mutation into kinase-overactive G2019S-LRRK2 (D1994A) dramatically reduced the signal resolved by native PAGE analysis compared with kinase-active LRRK2, despite equivalent amounts of total lysate loaded into each well and equivalent LRRK2 expression in SDS-solubilized cells, as determined by SDS-PAGE (Fig. 1B). In addition, native lysates analyzed by SDS-PAGE revealed comparably expressed LRRK2 protein levels in soluble fractions, independent of kinase activity, suggesting that kinase-inactive LRRK2 with a D1994A mutation in the kinase domain forms a higher proportion of high molecular weight oligomers that fail to resolve with native PAGE. In contrast, through analysis of multiple experiments, G2019S-LRRK2 tended to produce a higher proportion of lower molecular weight oligomers that included dimer structures, relative to wild-type LRRK2 when LRRK2 expression in total lysate was matched by SDS-PAGE (formally analyzed in Fig. 3). These results suggest that kinase activity may be required for the stabilization of lower molecular weight dimer conformations measurable by native PAGE analysis, consistent with the findings of Greggio et al. (22), who demonstrate that kinase-inactivating mutations increase the proportion of LRRK2 protein in the void volume fractions (e.g. >2 MDa) from FPLC separations.
Kinase dimerization and oligomerization is a common property to both receptor and non-receptor protein kinases. Although a LRRK2 dimer structure is suggested through the detection of a prominent complex by native PAGE near a marker of 480 kDa, the existence of an LRRK2 dimer has not been formally proved through structure analysis. Native cell lysate treated with 5% β-mercaptoethanol at 65 °C for 10 min immediately prior to electrophoresis dramatically increased the intensity of a band near the expected size of an LRRK2 monomer (Fig. 1C), suggesting that the prominent LRRK2 dimer-sized structure does not represent aberrantly migrating monomeric LRRK2. Thus, strongly reducing conditions and partial denaturation are capable of increasing the proportion of monomeric LRRK2 versus dimer-sized LRRK2 protein.
LRRK2 Distribution and Stable Interaction with Other Proteins Are Not Dependent on Kinase Activity
Strong precedent from structural analyses of other protein kinases suggests that kinase dimerization and oligomerization is a relatively common feature among protein kinases (16). Several LRRK2 protein interactors have been resolved, including HSP-90, that potentially confound the interpretation of LRRK2-positive complexes as dimeric or oligomeric (26, 27). Because LRRK2 protein bearing a kinase-dead mutation demonstrated a very different profile either through native PAGE (Figs. 1 and 3) or size exclusion chromatography (Fig. 4) (22) compared with kinase-active LRRK2, shifts in complex size may be due to interactions with other proteins and not solely self-assembly. Purification of LRRK2 complexes from lysates derived from cells transiently transfected with wild-type LRRK2, kinase-dead LRRK2 (D1994A), or PD-associated LRRK2 (G2019S) all fused to the streptavidin-binding peptide in concert with the calmodulin-binding peptide, as part of a tandem affinity purification strategy (Fig. 2A), demonstrate identical interacting profiles between kinase-active and kinase-dead LRRK2 protein. Gel slices were also dissected from tandem affinity purifications derived from cells transfected with only empty vector (no visible bands apparent, data not shown). Peptides were identified by mass spectrometry, and interacting proteins that correspond to two or more unique peptides of high quality are listed (Fig. 2A). No peptide matches of high quality were obtained from cells not expressing LRRK2 protein.
FIGURE 4.

Separation of stable and soluble LRRK2 conformations by size exclusion chromatography. Total soluble native protein lysates derived from cells expressing the indicated protein (kinase-active G2019S-LRRK2 or kinase-inactive G2019S/D1994A-LRRK2) were separated with a Superdex 200 10/300 GL column into seven 1-ml fractions and immediately loaded onto a native gel for analysis by Western blot with the anti-c-Myc-HRP antibody. The A280 chromatogram of protein standards (Sigma) previously analyzed through the Superdex column is aligned with fraction number and corresponds to samples loaded onto native gels, whereas native PAGE markers derive from the native mark protein ladder set (Invitrogen). Blue dextran defines the void volume of this column. HMW, high molecular weight.
FIGURE 2.

LRRK2 protein interactions and subcellular distribution are independent of LRRK2-kinase activity. A, LRRK2-positive protein complexes were purified from native lysates derived from HEK-293FT cells transfected with WT-LRRK2, kinase-dead (D1994A)-LRRK2, or kinase-overactive G2019S-LRRK2 constructs, through a tandem affinity purification strategy as illustrated. LRRK2 and interacting proteins were resolved from affinity beads, analyzed on a Tris acetate SDS gel, and stained with Coomassie dye, and the constituency of individual protein bands was identified by mass spectrometry. All unique proteins with more than two unique peptide hits of high quality are indicated, and in every case identified proteins matched the expected size in reference to the protein ladder. B, confocal microscopic analysis of HEK-293T cells transfected with the indicated plasmid. Cells were fixed, and LRRK2 expression was detected with anti-Myc-Cy2 antibody (shown as green signal), and cell images are representative. SBP, streptavidin-binding peptide; CBP, calmodulin-binding peptide.
LRRK2 probably resides in protein complexes composed primarily of other LRRK2 molecules, given the native PAGE profile and protein interaction profile, with minor or potentially labile interactions noted with HSP-90, HMMR, HSP-70 and Hsc-70, vimentin, and DnaJA1 in addition to the large proteins DNA-protein kinase and dynein heavy chain. Several of these interactions have been observed previously in other cell lines and tissue in other studies (26, 27). No differential interactions with other proteins or obvious changes in the proportion of protein interactors are found between kinase-active and kinase-dead LRRK2, suggesting that LRRK2 complexes (e.g. dimer-sized and high molecular weight) may not be due to protein interactions other than LRRK2 self-interaction.
Immunohistochemical analysis of transfected HEK-293FT cells demonstrated identical patterns of cytosolic distribution for wild-type, kinase-overactive, and kinase-dead LRRK2 protein, consistent with equivalent solubility measurements that demonstrated equal levels of protein in both soluble (native fractions) and insoluble (SDS-soluble) fractions when resolved by SDS-PAGE (Fig. 2B). In these transfected cells, measurements of toxicity, cell division and growth, or gross morphological analysis all suggested no significant effects due to LRRK2 or mutant (G2019S) LRRK2 expression compared with control (empty vector) transfection, and these cells did not develop LRRK2-positive puncta or ubiquitin-positive aggregates (data not shown). In contrast, fusion of enhanced green fluorescent protein to either the N or C terminus of LRRK2 caused a small proportion of mislocalization to the nucleus and the formation of prominent perinuclear aggregates in some cells (data not shown).
Pathogenic LRRK2 Mutations Enhance LRRK2 Dimer Formation
Because the introduction of a kinase activity-ablating mutation in LRRK2 (D1994A) greatly reduces the ability of LRRK2 to stabilize dimer-sized structures, other previously described mutations that may alter LRRK2 kinase activity would plausibly affect the proportion of LRRK2 residing in dimer structures. Combined analysis of several independent experiments suggests that some pathogenic mutations in LRRK2 increase the proportion of LRRK2 present in a dimer-sized structure normalized to total LRRK2 protein (SDS-solubilized) in comparison with wild-type LRRK2.
In comparing five separate experiments (individually run, with different plasmid preparations), the PD-associated mutations G2019S, I2020T, and I1122V demonstrated an increased proportion of LRRK2 dimer-sized structures normalized to total LRRK2 protein (SDS-solubilized) with respect to wild-type LRRK2 protein (p < 0.009, Student's two-tailed t test), whereas the mutation R1441C did not cause a significant increase (p = 0.0653), and no significant difference was detected between the Y1699C-LRRK2 and wild-type protein (Fig. 3). As an additional control, wild-type LRRK2 derived independently in the laboratory of Mark Cookson (NIA, National Institutes of Health) and present on a different vector backbone demonstrated no significant difference in dimer formation from wild-type LRRK2 on a pcDNA3.1 backbone.
As expected, a mutation in the GTPase domain that ablates GTP binding (K1347A) and therefore ablates downstream kinase activity showed the same reduction in dimer formation as the D1994A/G2019S double mutant protein. To ensure even loading, even LRRK2 expression in cells, and transfer in both native PAGE and SDS-PAGE blots, protein concentrations in individual lysates were determined by BCA assay, and 10 μg of total lysate were loaded per well. In addition, Ponceau S stain was applied to PVDF membranes, and uniformity of protein distribution was verified prior to membrane blocking and antibody application. A representative portion of the Ponceau S stain near the LRRK2 protein dimer molecular weight is given in Fig. 3A.
LRRK2 Dimers Possess Kinase Activity in Vitro
Because LRRK2 oligomerization resolved by native PAGE analyses showed that dimer-sized LRRK2 structures require kinase activity for stabilization, lower molecular weight LRRK2 complexes may represent the active form of LRRK2 protein, as compared with higher molecular weight oligomers. Cell lysate derived from freeze/thaw lysis of transfected cells and separated by size exclusion chromatography provides an efficient method for the separation of LRRK2 protein complexes directly in native lysates, with resolution from the void volume down to the 66 kDa marker on a Superdex 200 10/300 GL column (Fig. 4). Lysate from cells transfected with LRRK2 and separated by size exclusion chromatography with fractions immediately analyzed by native PAGE demonstrated effective separation of high molecular weight LRRK2 present in the void volume, LRRK2 dimer-sized structures, and a low amount of LRRK2 monomer-sized protein (in reference to native PAGE markers).
Relative to proteins of known size analyzed by size exclusion chromatography, LRRK2 complexes in cell lysates migrated in an unexpected manner, segregating as lower molecular weight molecules/complexes compared with standards used in native PAGE, suggesting an interaction under these conditions between LRRK2 and the Superdex matrix that acts to accelerate progression. LRRK2 complexes derived from cells transfected with kinase-dead G2019S/D1994A LRRK2 showed that much of the kinase-dead LRRK2 resides in extremely large but soluble complexes migrating with the void volume of the column, with less intensity in lower molecular weight fractions.
Lysate derived from G2019S-LRRK2-transfected cells (G2019S-LRRK2 used versus wild-type LRRK2 to enhance kinase signal versus background) separated by size exclusion chromatography and LRRK2 complexes immunoprecipitated with magnetic beads demonstrated that the majority of LRRK2 kinase activity in soluble LRRK2 pools derived from fractions enriched in LRRK2 dimers (Fig. 5). A portion of the eluted kinase reaction was analyzed by SDS-PAGE to determine input levels of LRRK2, and although monomeric LRRK2 was not present in the same quantity as dimeric or higher molecular weight LRRK2 complexes, normalization to input levels of either autophosphorylation activity or activity with MBP suggests a lack of activity in monomeric LRRK2 protein. The same experiment using equivalent amounts of cell lysate derived from empty vector-transfected cells or cells transfected with D1994A/G2019S LRRK2 produced no significant signal using identical exposure times and assay conditions in either autophosphorylation, as resolved by SDS-PAGE from eluted protein complexes, or signal against the generic kinase substrate MBP. These results suggest that contaminating kinases were not responsible for the activity observed in Fig. 5A. Thus, in soluble (non-membrane-bound) fractions, LRRK2 kinase activity principally derives from LRRK2 dimers.
FIGURE 5.
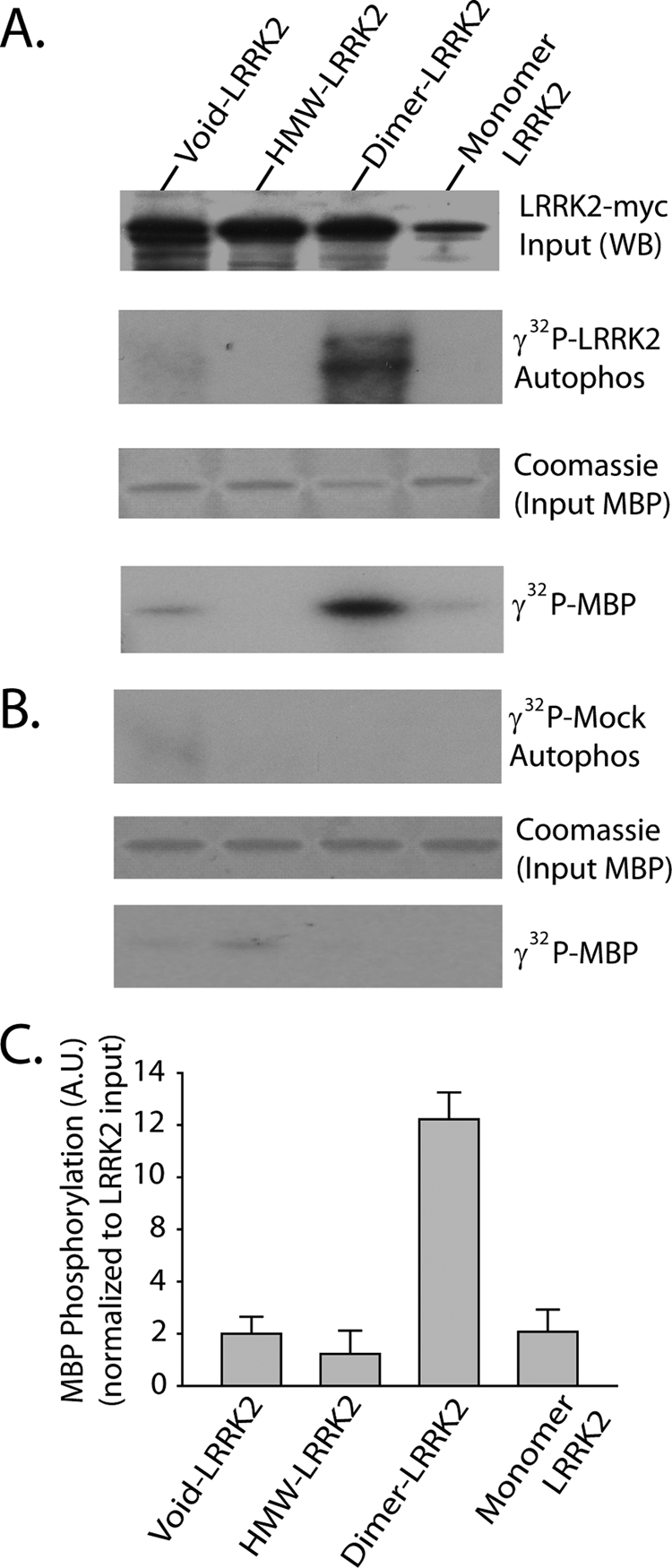
Dimer-sized G2019S-LRRK2 complexes are kinase-active conformations. A, total soluble native protein lysates derived from cells expressing G2019S-LRRK2 were separated with a Superdex 200 10/300 GL column. LRRK2 protein complexes from fractions 5 (void), 8 (dimer), or 10 (monomer) were immunoprecipitated with magnetic beads conjugated to anti-Myc antibody and combined into a kinase reaction containing dephosphorylated MBP protein that serves as an efficient LRRK2 substrate in vitro. Supernatant containing MBP was analyzed by SDS-PAGE and Coomassie stain. Eluted G2019S-LRRK2 protein was resolved onto SDS-gels, transferred to PVDF, and exposed to autoradiography film, and input levels from kinase reactions were determined with anti-Myc-HRP antibody. B, mock kinase reactions using fractionated lysate and immunoprecipitates from cells transected with empty vector and autoradiography films exposed for identical times as in A. C, quantification of LRRK2-mediated MBP phosphorylation via densitometry, adjusted for background and normalized to relative input levels of LRRK2 protein. Data are derived from three independent experiments, and error bars represent ±S.E. A.U., arbitrary units.
Previously, we described GTP-binding properties of mutant and wild-type LRRK2 protein using Sepharose beads cross-linked to GTPγS combined with LRRK2 containing whole-cell protein lysates (10). LRRK2 protein complexes separated by size exclusion chromatography were unable to interact with GTPγS (data not shown), presumably due to the dilution and removal of necessary guanine exchange factors that are otherwise present in whole-cell lysates.
Pharmacological Modulation of LRRK2 Dimerization
As critical components of signal transduction pathways, the activity of most protein kinases can be highly modulated by the cellular environment, either by stimulating events or suppressing events. LRRK2 dimer structures may represent a stabilized kinase-active conformation that is dependent on both GTPase and kinase activity. To identify small molecule inhibitors of LRRK2 kinase activity, a panel of broad spectrum kinase inhibitors was combined in a standard LRRK2 kinase assay (each at 100 nm; Fig. 6). Only staurosporine displays potent inhibitory activity against LRRK2 autophosphorylation (Fig. 6A), consistent with other published reports that suggest IC50 values lower than 10 nm in vitro (28, 29).
FIGURE 6.

Pharmacological inhibition of LRRK2 dimer-sized complexes. A, in vitro measurements of LRRK2 autophosphorylation in the presence of the indicated compound at a concentration of 100 nm or DMSO control (0.1% reaction volume). B, HEK-293T cells expressing human WT-LRRK2 protein were treated with the broad spectrum kinase inhibitor staurosporine at a concentration of 100 nm, as indicated, for 1 h prior to cell lysis via freeze/thaw cycles in native buffer (PBS); lysate was immediately analyzed by both native PAGE and SDS-PAGE; and LRRK2 expression was visualized by anti-Myc-HRP antibody. Results are representative of four independent experiments. C, native protein lysates prepared as in B were separated by size exclusion chromatography, and fractions enriched in high molecular weight (HMW) oligomer, dimer, or monomer are indicated, and LRRK2 levels in each fraction were determined by SDS-PAGE. Total LRRK2 protein levels in lysates before separation were also verified by SDS-PAGE, with total levels unaffected by staurosporine treatment.
A brief treatment of cells transfected with wild-type LRRK2 protein with 100 nm staurosporine immediately prior to lysis had a pronounced effect on the formation of LRRK2 dimer-sized complexes as visualized by native PAGE (Fig. 6B). In addition, separation of LRRK2 lysates into fractions enriched in high molecular weight oligomers, dimers, or monomers showed that staurosporine treatment reduced the amount of LRRK2 in dimer-enriched fractions and increased the amount of LRRK2 in void volume fractions (Fig. 6C). Total LRRK2 protein (SDS-soluble) was not affected by treatment of 100 nm staurosporine for 1 h as determined by SDS-PAGE and Western blot, and no cellular toxicity was caused by this dose of drug and time course (data not shown). Treatment of cells with 10 nm staurosporine had no effect on LRRK2 dimer formation, suggesting that this concentration of inhibitor is ineffective at targeting LRRK2 in living cells (data not shown).
Due to the broad cellular effects of staurosporine, it is possible that staurosporine-mediated inhibition of LRRK2 kinase activity and dimer formation may be a trans event through the inhibition of protein kinases upstream of LRRK2. To help clarify whether staurosporine is acting directly or indirectly on LRRK2 dimer formation, we obtained highly purified and active recombinant GST-fused WT-LRRK2 protein (truncated on the N terminus) and pharmacologically manipulated LRRK2 dimer formation in vitro (Fig. 7).
FIGURE 7.

LRRK2 autophosphorylation occurs in dimeric but not monomeric recombinant GST-WT-LRRK2 protein purified from baculovirus-infected insect cells. 200 ng of highly purified and active GST-tagged (N-terminal) truncated WT-LRRK2 protein (obtained from Invitrogen), with purity assessed by SDS-PAGE and Coomassie stain, was combined into a kinase reaction supplemented with varying concentrations of deoxycholate (DOC) that stabilize lower molecular weight LRRK2 oligomers visualized by native PAGE and staurosporine (100 nm) that prevents the stabilization of lower molecular weight LRRK2 oligomers but not monomers. Autophosphorylation (i.e. incorporation of 32P) activity is visualized through exposure of dried silver-stained gels to autoradiography film. Kinase activity correlates with dimeric and oligomeric LRRK2 protein but not inactive monomeric LRRK2. HMW, high molecular weight.
The inclusion of the zwitterionic detergent deoxycholate in recombinant LRRK2 kinase reactions at low concentrations enhanced the formation of lower molecular weight LRRK2 oligomers and dimers that can be resolved by native PAGE, whereas the inclusion of staurosporine completely ablated these structures (Fig. 7). Incorporation of phosphate (i.e. autophosphorylation) was observed in lower molecular weight oligomers and dimers but not monomeric LRRK2. Because LRRK2 autophosphorylation occurs in cis only (22), LRRK2 self-interaction must be required for autophosphorylation activity.
DISCUSSION
Because the phenotype associated with LRRK2 mutations closely mimics the phenotype of typical late onset PD, studies that provide insight into LRRK2 function may likewise highlight mechanisms important in the pathogenesis of PD. LRRK2 itself is a potential therapeutic target for neuroprotection because several potent and specific protein kinase inhibitors (to targets other than LRRK2) have been successfully implemented for the treatment of human disease. Most of what is understood regarding LRRK2 and PD has been formed from limited in vitro analyses, which surmise kinase-dependent neurotoxicity and pathological increases in kinase activity due to PD-linked mutations. Delineating the mechanics of LRRK2 kinase activity in cells will provide the foundation for the discovery and validation of specific and potent LRRK2 kinase inhibitors and understanding the link between cell death and LRRK2.
Recombinant LRRK2 protein derived from mammalian cell lines spurred a consensus story regarding GTPase control over kinase activity that appears to also hold true for LRRK1 and the LRRK2 homolog in slime mold GbpC (8). Several lines of evidence suggest that LRRK2 protein interacts with itself, although the functional implications of these interactions are poorly understood. Endogenous LRRK2 in lymphoblast cell lines and overexpressed LRRK2 in HEK cells demonstrate that LRRK2 is not restricted to the formation of dimer-sized complexes; rather, LRRK2 additionally forms higher order structures consistent with homomultimerization.
In native soluble lysate from transfected cells, a paucity of LRRK2 exists as a monomer, although monomeric LRRK2 can be generated under reduced conditions and heat-induced partial denaturation. Mutations that inactivate LRRK2 (GTPase or kinase activity) dramatically increase the size of LRRK2 complexes that become difficult to detect by conventional native PAGE and chromatography, because the D1994A mutation shifts LRRK2 to void volume-sized complexes but still highly soluble complexes. However, kinase-dead LRRK2 protein can be easily detected with relatively low concentrations of complex-busting ionic detergents in SDS-PAGE (10). These differences in complex formation are apparently not observable by light microscopy because fluorescent immunocytochemistry analysis demonstrates no difference in localization between kinase-dead and kinase-active LRRK2.
Because kinase-inactivated LRRK2 (either by staurosporine treatment or mutation in the kinase or GTPase domain) fails to form dimer-sized structures, the dimer itself becomes implicated as the stabilized active form of LRRK2 protein. Through size exclusion-based separation and subsequent immunoprecipitation, we derived significant kinase activity only from the fractions enriched in LRRK2 dimer-sized complexes. LRRK2 belongs to the tyrosine-kinase like protein kinase family, and examples of protein kinases in this family that dimerize during activation include Raf1 (30), MLK3 (31), and all receptor-serine kinases, and some kinases additionally oligomerize.
Although we have not formally defined the structure of LRRK2 complexes present in soluble fractions (e.g. through x-ray crystallography or NMR), strong precedent from other kinases and the migration patterns observed in this study suggest that LRRK2 forms a kinase-active dimer and may dynamically assemble (or disassemble) into trimeric, tetrameric, and higher molecular weight oligomers that have reduced kinase activity and may be unable to phosphorylate substrate proteins. Standard immunoprecipitations of soluble LRRK2 fractions capture both oligomeric and dimeric LRRK2 and are not capable of resolving inactive oligomeric LRRK2 from dimeric LRRK2 without further implementation of separation technology, such as native-PAGE or gel filtration.
The underlying mechanisms responsible for LRRK2 dimerization and kinase activation might include a cascade involving GTP binding, cis-autophosphorylation, or phosphorylation of LRRK2 by another protein kinase (Fig. 8). LRRK2 may dynamically shift back to inactive conformations due to the action of phosphatases and GTPase-activating proteins and potential interactions with chaperones and stochastic interactions with other LRRK2 oligomers. GTP binding often invokes conformation changes in GTPase proteins, and because the LRRK2 GTPase domain forms dimer structures resolved by crystal structure (32), GTP-bound LRRK2 may stabilize a dimer structure that may otherwise oligomerize into higher molecular weight structures (Fig. 8). A relatively small dimer structure may free the kinase domain to allow interaction with kinase substrates and enhanced autophosphorylation that further stabilizes the kinase-active conformation of LRRK2.
FIGURE 8.

Hypothetical model of LRRK2 kinase activation. A major fraction of LRRK2 protein in cells may reside in large oligomers with low or no kinase activity. LRRK2 oligomers are dissociated through conformational changes induced by GTP binding within the ROC GTPase domain, which may lead to the formation of a dimer structure initially stabilized by a ROC-ROC interaction that unmasks kinase activity and the potential for autophosphorylation. LRRK2 autophosphorylation activity may lead to the stabilization of the kinase-active dimer, which can be destabilized by competing phosphatase activity, GTPase hydrolytic activity, or stochastic interactions with LRRK2 oligomers. In this model, kinase activity is dependent on GTPase activity, whereas GTPase activity is not dependent on kinase activity but must be influenced by changes induced by autophosphorylation. Thus, this model predicts a reciprocal interaction whereby kinase activity and autophosphorylation stabilize lower molecular weight conformations and decrease the reversion to high molecular weight oligomers.
Previously, we demonstrated that LRRK2 resides in both freely soluble and SDS-soluble fractions in both human and mouse brain (33). It is tempting to speculate that as LRRK2 oligomerizes into higher molecular weight species, the complex demands the activities of HSP-70 and HSP-90 to maintain integrity. It is possible that patients that harbor the LRRK2 G2019S mutation have more LRRK2 present in dimer-sized complexes in susceptible cells as compared with the amount of wild-type LRRK2 protein dimers, relative to total LRRK2 protein. Trans acting factors that might include genetic variation and environmental exposures could increase LRRK2 activity, leading to neurodegeneration. Detection of LRRK2 dimer structures in lymphoblast cell lines derived from patients may provide an indirect LRRK2 activity assay and an additional opportunity to test LRRK2 kinase inhibitors directly on living cells.
Through size exclusion chromatography approaches in native lysates that exclude LRRK2 protein otherwise solubilized with SDS or other strong denaturing conditions, it is apparent that a fraction of total LRRK2 protein resides as a protein dimer. The reason that only a minor fraction of LRRK2 is in a kinase-active conformation is unknown, but it may be due to a lack of rate-limiting co-factors and/or inherent structural instability.
The results presented here measure LRRK2 protein only in soluble fractions and may not reflect the activities of insoluble LRRK2 protein. Unfortunately, the same conditions required to resolve total LRRK2 protein in cells (e.g. with SDS lysis buffer) may not be compatible with most enzymatic assays that demand properly folded protein. Since we have not described LRRK2 activity in insoluble fractions, it is impossible to say with certainty that the only LRRK2 complex that has kinase activity is the soluble protein dimer. In addition, we cannot rule out the notion that endogenous protein displays different characteristics than the exogenous LRRK2 protein analyzed in this study. Identification of a phosphorylated peptide in cells that is exquisitely dependent on LRRK2 activity will allow further insight into the mechanisms of LRRK2 enzymatic activity.
It is becoming clear that different pathogenic LRRK2 mutations differentially affect LRRK2 enzymatic activities. Some PD-associated LRRK2 mutations significantly enhance the proportion of LRRK2 protein present as a dimer-sized complex, although several pathogenic mutations failed to reach statistical significance versus wild-type LRRK2 dimer amounts. It is possible that some mutations (e.g. Y1699C) induce LRRK2 kinase activity outside of dimerization. Detailed profiles of individual mutant LRRK2 proteins will help determine the differences and similarities between pathogenic and normal LRRK2 activity and identify correct therapeutic targets. In addition, more refined biochemical assays may be capable of resolving benign versus pathogenic LRRK2 genetic variation, allowing more precise genetic counseling and diagnostics.
Acknowledgments
We thank the Dawson laboratories (The Johns Hopkins University Udall Center) and the laboratory of Mark Cookson (NIA, National Institutes of Health) for LRRK2 expression plasmids. We thank Matt Farrer (Mayo Jacksonville Udall Center) for the contribution of lysate derived from lymphoblast cell lines and antibodies specific for human LRRK2.
This work was supported, in whole or in part, by National Institutes of Health Grant R00 NS058111. This work was also supported by the Michael J. Fox Foundation for Parkinson's Research, the American Parkinson's Disease Association, and the benevolence of John A. and Ruth R. Jurenko.
- PD
- Parkinson disease
- PBS
- phosphate-buffered saline
- PVDF
- polyvinylidene difluoride
- HRP
- horseradish peroxidase
- FPLC
- flow pressure liquid chromatography
- MBP
- myelin basic protein
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- GST
- glutathione S-transferase
- WT-LRRK2
- wild-type LRRK2
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1.Cookson M. R. (2005) Annu. Rev. Biochem. 74, 29–52 [DOI] [PubMed] [Google Scholar]
- 2.Moore D. J., West A. B., Dawson V. L., Dawson T. M. (2005) Annu. Rev. Neurosci. 28, 57–87 [DOI] [PubMed] [Google Scholar]
- 3.Lesage S., Dürr A., Tazir M., Lohmann E., Leutenegger A. L., Janin S., Pollak P., Brice A. (2006) N. Engl. J. Med. 354, 422–423 [DOI] [PubMed] [Google Scholar]
- 4.Hulihan M. M., Ishihara-Paul L., Kachergus J., Warren L., Amouri R., Elango R., Prinjha R. K., Upmanyu R., Kefi M., Zouari M., Sassi S. B., Yahmed S. B., El Euch-Fayeche G., Matthews P. M., Middleton L. T., Gibson R. A., Hentati F., Farrer M. J. (2008) Lancet Neurol. 7, 591–594 [DOI] [PubMed] [Google Scholar]
- 5.Healy D. G., Falchi M., O'Sullivan S. S., Bonifati V., Durr A., Bressman S., Brice A., Aasly J., Zabetian C. P., Goldwurm S., Ferreira J. J., Tolosa E., Kay D. M., Klein C., Williams D. R., Marras C., Lang A. E., Wszolek Z. K., Berciano J., Schapira A. H., Lynch T., Bhatia K. P., Gasser T., Lees A. J., Wood N. W. (2008) Lancet Neurol. 7, 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funayama M., Li Y., Tomiyama H., Yoshino H., Imamichi Y., Yamamoto M., Murata M., Toda T., Mizuno Y., Hattori N. (2007) Neuroreport 18, 273–275 [DOI] [PubMed] [Google Scholar]
- 7.Zabetian C. P., Yamamoto M., Lopez A. N., Ujike H., Mata I. F., Izumi Y., Kaji R., Maruyama H., Morino H., Oda M., Hutter C. M., Edwards K. L., Schellenberg G. D., Tsuang D. W., Yearout D., Larson E. B., Kawakami H. (2009) Mov. Disord. 24, 1034–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biskup S., West A. B. (2008) Biochim. Biophys. Acta 1792, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greggio E., Cookson M. R. (2009) ASN Neuro. 1, e00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West A. B., Moore D. J., Choi C., Andrabi S. A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K. L., Dawson V. L., Dawson T. M. (2007) Hum. Mol. Genet. 16, 223–232 [DOI] [PubMed] [Google Scholar]
- 11.Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., Iwatsubo T. (2007) Biochemistry 46, 1380–1388 [DOI] [PubMed] [Google Scholar]
- 12.Lewis P. A., Greggio E., Beilina A., Jain S., Baker A., Cookson M. R. (2007) Biochem. Biophys. Res. Commun. 357, 668–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greggio E., Taymans J. M., Zhen E. Y., Ryder J., Vancraenenbroeck R., Beilina A., Sun P., Deng J., Jaffe H., Baekelandt V., Merchant K., Cookson M. R. (2009) Biochem. Biophys. Res. Commun. 389, 449–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung I. W., Lassam N. (1998) J. Biol. Chem. 273, 32408–32415 [DOI] [PubMed] [Google Scholar]
- 15.Leung I. W., Lassam N. (2001) J. Biol. Chem. 276, 1961–1967 [DOI] [PubMed] [Google Scholar]
- 16.Pelech S. (2006) J. Biol. 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gloeckner C. J., Schumacher A., Boldt K., Ueffing M. (2009) J. Neurochem. 109, 959–968 [DOI] [PubMed] [Google Scholar]
- 18.Cobb M. H., Goldsmith E. J. (2000) Trends Biochem. Sci. 25, 7–9 [DOI] [PubMed] [Google Scholar]
- 19.Cheng J., Yu L., Zhang D., Huang Q., Spencer D., Su B. (2005) J. Biol. Chem. 280, 13477–13482 [DOI] [PubMed] [Google Scholar]
- 20.Nitta R. T., Chu A. H., Wong A. J. (2008) J. Biol. Chem. 283, 34935–34945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gloeckner C. J., Kinkl N., Schumacher A., Braun R. J., O'Neill E., Meitinger T., Kolch W., Prokisch H., Ueffing M. (2006) Hum. Mol. Genet. 15, 223–232 [DOI] [PubMed] [Google Scholar]
- 22.Greggio E., Zambrano I., Kaganovich A., Beilina A., Taymans J. M., Daniëls V., Lewis P., Jain S., Ding J., Syed A., Thomas K. J., Baekelandt V., Cookson M. R. (2008) J. Biol. Chem. 283, 16906–16914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein C. L., Rovelli G., Springer W., Schall C., Gasser T., Kahle P. J. (2009) J. Neurochem. 111, 703–715 [DOI] [PubMed] [Google Scholar]
- 24.Melrose H. L., Kent C. B., Taylor J. P., Dachsel J. C., Hinkle K. M., Lincoln S. J., Mok S. S., Culvenor J. G., Masters C. L., Tyndall G. M., Bass D. I., Ahmed Z., Andorfer C. A., Ross O. A., Wszolek Z. K., Delldonne A., Dickson D. W., Farrer M. J. (2007) Neuroscience 147, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 25.West A. B., Moore D. J., Biskup S., Bugayenko A., Smith W. W., Ross C. A., Dawson V. L., Dawson T. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L., Xie C., Greggio E., Parisiadou L., Shim H., Sun L., Chandran J., Lin X., Lai C., Yang W. J., Moore D. J., Dawson T. M., Dawson V. L., Chiosis G., Cookson M. R., Cai H. (2008) J. Neurosci. 28, 3384–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dächsel J. C., Taylor J. P., Mok S. S., Ross O. A., Hinkle K. M., Bailey R. M., Hines J. H., Szutu J., Madden B., Petrucelli L., Farrer M. J. (2007) Parkinsonism Relat. Disord. 13, 382–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anand V. S., Reichling L. J., Lipinski K., Stochaj W., Duan W., Kelleher K., Pungaliya P., Brown E. L., Reinhart P. H., Somberg R., Hirst W. D., Riddle S. M., Braithwaite S. P. (2009) FEBS J. 276, 466–478 [DOI] [PubMed] [Google Scholar]
- 29.Covy J. P., Giasson B. I. (2009) Biochem. Biophys. Res. Commun. 378, 473–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorenz K., Schmitt J. P., Schmitteckert E. M., Lohse M. J. (2009) Nat. Med. 15, 75–83 [DOI] [PubMed] [Google Scholar]
- 31.Du Y., Böck B. C., Schachter K. A., Chao M., Gallo K. A. (2005) J. Biol. Chem. 280, 42984–42993 [DOI] [PubMed] [Google Scholar]
- 32.Deng J., Lewis P. A., Greggio E., Sluch E., Beilina A., Cookson M. R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1499–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biskup S., Moore D. J., Rea A., Lorenz-Deperieux B., Coombes C. E., Dawson V. L., Dawson T. M., West A. B. (2007) BMC Neurosci. 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]