

Abstract
Following exposure to various stresses (arsenite, UV, hyperthermia, and hypoxia), mRNAs are assembled into large cytoplasmic bodies known as “stress granules,” in which mRNAs and associated proteins may be processed by specific enzymes for different purposes like transient storing, sorting, silencing, or other still unknown processes. To limit mRNA damage during stress, the assembly of micrometric granules has to be rapid, and, indeed, it takes only ∼10–20 min in living cells. However, such a rapid assembly breaks the rules of hindered diffusion in the cytoplasm, which states that large cytoplasmic bodies are almost immobile. In the present work, using HeLa cells and YB-1 protein as a stress granule marker, we studied three hypotheses to understand how cells overcome the limitation of hindered diffusion: shuttling of small messenger ribonucleoprotein particles from small to large stress granules, sliding of messenger ribonucleoprotein particles along microtubules, microtubule-mediated stirring of large stress granules. Our data favor the two last hypotheses and underline that microtubule dynamic instability favors the formation of micrometric stress granules.
Introduction
In response to stress due to arsenite exposure (1), UV irradiation (2), hyperthermia (3), and hypoxia (4), eukaryotic cells rapidly reprogram their translational machinery to produce proteins necessary for cell survival, like heat shock proteins (5, 6). Because the maintenance of routine translation machinery would be hazardous under such conditions, stressed cells also stop the synthesis of “housekeeping” proteins (7, 8). Translational arrest mainly occurs after phosphorylation of the initiation factors eIF2 (9), even if other routes exist like the cleavage of eIF4G during viral infection (10). Following translational arrest, 60 S ribosomal subunit cannot be recruited to allow mRNA translation, which leads to the appearance of stalled 48 S pre-initiation complexes (referred to herein as mRNP3 particles). Stalled pre-initiation complexes containing mRNA are then redirected into large mRNP granules called stress granules (SGs) for still debated processes: repair, degradation, or delayed use after stress recovery (11, 12). To form SGs, self-aggregation between stalled pre-initiation complexes is mediated by specific proteins, in particular TIA-1 (9) and G3BP (13). This attraction is most probably of electrostatic origin due to the cationic domains of TIA-1 or G3BP and could also involve protein-protein interactions as TIA-I or G3BP have a tendency to self-aggregate (14). Other proteins than TIA-1 or G3BP are also essential for the formation and processing of SGs and are the subject of ongoing extensive research (see Ref. 15 and references therein).
In contrast to studies focused on protein functions, the kinetics and biophysical aspects of SG assembly have been the subject of rare studies up to now. In a pioneer study, Kedersha and co-workers observed that the shuttling of TIA-1 from the nucleus to the cytoplasm accompanies the mechanism of granule formation (14). The aggregation mechanism of the stalled pre-initiation complexes could be completed in ∼10 min and leads to the appearance of large SGs (>1 μm), which can further increase in size due to coalescence (14). Such a high rate of SG formation is particularly striking, because, in the viscous cytoplasm, obstacles like actin filaments considerably hinder thermal diffusion of particles larger than 50–100 nm (16–19). This means that mechanisms other than passive bulk diffusion come into play to overcome this limitation during stress. In the present study, we investigate how such rapid aggregation is possible. As recently reported (20–22), microtubules (MTs) are most probably implicated in the mechanism of SG formation and could serve to promote rapid SG assembly. However, their role in the mechanism of SG formation is not yet established. In a first report (20), total disruption of MTs in CV-1 cells by two different MT-disrupting agents, vinblastine or nocodazole, induced either the formation of more numerous SGs per cell or their disappearance, and more attention has been paid to the latter. Quantification was performed using the eukaryotic initiation factor 3 as a marker of SGs. In another report, MT-disrupting drugs in HeLa cells also induced the disappearance of SGs stained with anti-HDAC6 antibody (HDAC6 is a cytoplasmic deacetylase) (22). Interestingly, both eukaryotic initiation factor 3 and HDAC6 were found in tubulin immunoprecipitates. One conclusion of these studies was that MT-disrupting agents inhibit the formation of SGs. However, as noted by Kobolova et al. (21), another explanation for the disappearance of SGs could be that, after MT disruption, these proteins no longer localize in SGs due to their interaction with free tubulin. In agreement with this hypothesis, Kobolova et al. showed that MT disruption in HeLa cells leads to the formation of small and numerous SGs, but SGs do not disappear (21). In this case, SGs were labeled with anti-G3BP antibody, and G3BP was not present in tubulin immunoprecipitates (22).
In the present study, using an analytical model and numerical simulations, we first considered the mechanism of SG assembly in the cytoplasm and advanced three hypotheses to explain the rapid formation of micrometric SGs. 1) mRNP particles can shuttle from small to large SGs. This mechanism does not require MTs. 2) mRNP particles slide along MTs to form large SGs. 3) MT turnovers between shortening and elongation phases can push and pull small granules to form larger ones. We finally explored experimentally the validity of our hypotheses and especially investigated the implication of MTs in SG assembly in HeLa Cells. Using YB-1 as marker of SGs, we found that dynamic MTs promote the coalescence of small cytoplasmic granules into larger ones via pushing or pulling. In addition, sliding of mRNP particles along MTs most probably acts in conjunction with the mechanism of SG pushing or pulling.
MATERIALS AND METHODS
Tubulin and MTs Preparation
Tubulin was purified from sheep brain crude extracts as described previously (23). Aliquots were stored at −80 °C in 50 mm MES-KOH, pH 6.8, 0.5 mm dithiothreitol, 0.5 mm EGTA, 0.25 mm MgCl2, 0.5 mm EDTA, 0.1 mm GTP, 30% glycerol (v/v), for long term storage. Taxol-stabilized MTs were prepared using 40 μm tubulin in 50 mm MES-KOH, pH 6.8, 0.5 mm dithiothreitol, 0.5 mm EGTA, 6 mm MgCl2, 0.5 mm EDTA, 0.6 mm GTP, 20% glycerol, and 10 μm taxol at 37 °C for 30 min. MTs were sedimented by centrifugation (52,000 × g, 30 min at 37 °C) at the end of which the MT pellet was resuspended in 25 mm MES-KOH, pH 6.8, 0.5 mm EGTA, 1 mm dithiothreitol, 5 μm taxol.
YB-1 Purification
Recombinant YB-1 was expressed in Escherichia coli and purified as previously described (24). Purified proteins were dialyzed against 200 mm NaCl, 20 mm HEPES-KOH, pH 7.6, 1 mm dithiothreitol, and stored at −80 °C.
Synthesis of Fluorescent mRNA
Plasmid pSP72-2Luc was used as a template for the synthesis by T7 polymerase 2Luc mRNAs (3000 nucleotides) (25). For mRNA synthesis, we used a solution containing a mixture of UTP and aminoallyl-UTP (Sigma, aminoallyl-UTP:UTP ratio was 1:5). After transcription, unincorporated NTPs were removed by gel filtration through an NAP-5 column (Amersham Biosciences), and mRNAs were further isolated with RNAble (Eurobio) following the manufacturer's recommendation. mRNA was then labeled with Cy2 using the FluoroLink-Ab Cy2 Labeling Kit (Amersham Biosciences).
Plasmid Construction and Transfection
The cDNA encoding the full-length YB-1 was amplified by PCR and cloned into the XhoI and BamH1 sites of vector pEGFP-C3 (Clontech). PCR-amplified products were then sequenced. HeLa cells were transfected with DNA plasmids by using Lipofectamine 2000 (Invitrogen). The efficacy of transfection and the integrity of the encoded protein (GFP-YB-1) were demonstrated by immunoblotting (supplemental Fig. S3A).
Cell Culture
HeLa cervical carcinoma and Chinese hamster ovary cells were maintained in Dulbecco's modified Eagle's medium supplemented with 5% fetal calf serum, 2 mm l-glutamine, and antibiotics (penicillin and streptomycin) in a humidified 5% CO2 atmosphere at 37 °C.
Immunofluorescence
HeLa cells grown on coverslips were washed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min at 37 °C. After fixation, cells were permeabilized with 0.5% Triton X-100 for 15 min then washed and incubated for 1 h with a rabbit anti-YB-1 antibody (produced as described in Ref. 26) and either a mouse monoclonal anti-tubulin antibody (clone tub 2.1, 1:5000), a mouse anti-HuR antibody (Molecular Probes, 10 μg/ml), or a mouse anti-pericentrin (Abcam, 1:1000 dilution) in blocking solution.
Cells were then washed extensively in PBS and incubated for 1 h with fluorochrome-coupled secondary antibodies (AlexaFluor488 and AlexaFluor555, 1:2500 dilution) in blocking solution. After final washes with PBS, coverslips were prepared for fluorescence microscopy.
The measurements of the radius and integrated intensity of SGs were performed using National Institutes of Health ImageJ. Because SGs are not round-shaped, the apparent radius, r, was estimated using the measured areas of SGs and stating that areas were equal to πr2. Integrated intensities were defined as the immunofluorescence signal intensity integrated over the granule structure minus the integrated intensity in the surrounding cytoplasmic background over the same area.
Immunoblotting
HeLa cells were washed once with PBS, and then lysed in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, protease inhibitor. Lysates were centrifuged at 14,000 × g for 15 min at 4 °C, and supernatants were collected. Proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Invitrogen). The membranes were blocked in 5% nonfat dry milk/PBS for 30 min at room temperature and incubated for 1 h at room temperature with anti-YB-1 (supplemental Fig. S3A) or anti-Phospho-eIF2α antibodies (Cell Signaling, 1:1000 dilution (supplemental Fig. S2C)). Bound antibodies were detected using anti-rabbit-IRDye 800 and anti-mouse-IRDye 680 secondary antibodies (Odyssey, 1:2000 dilution) with an Odyssey system (LI-COR Biosciences).
Analysis of Free and Polymerized Tubulin Fractions from HeLa Cells
We followed the method described by Gundersen et al. with minor modifications (27). HeLa Cells were rinsed in MSB buffer (85 mm Pipes, 1 mm EGTA, 1 mm MgCl2, 2 m glycerol, and protease inhibitors, pH 6.9) and then extracted with 400 μl of MSB containing 0.4% Triton X-100. After 3 min, this fraction, which contains free tubulin, was gently removed to a graduated tube. One-fourth volume of 5× SDS-PAGE buffer was then added, and the sample was boiled for 5 min. The polymerized tubulin fraction, corresponding to microtubules that remained in the cells, was solubilized in the same final volume of 1× SDS-PAGE buffer and boiled for 5 min. Finally, tubulin bands were separated by SDS-PAGE electrophoresis and quantified using the Odyssey system.
Videomicroscopy of SGs
We monitored the assembly of SGs of HeLa cells in real-time. Using DNA constructs encoding GFP alone and GFP-YB-1, HeLa cells were transiently transfected with GFP-YB-1 and cultured for 24 h (supplemental Fig. S3A).
Fluorescence videomicroscopy was implemented on an inverted microscope (Axiovert 220, Carl Zeiss MicroImaging, Inc.) equipped with phase-contrast and optic filters. GFP emission was detected with a 65×/0.5 numerical aperture objective. Time-lapse images were captured at 1- or 2-min intervals using a cooled charge-coupled device camera (Zeiss).
For quantitative analysis, image series were first corrected for translational or rotational movements. The distances covered by individual SGs and the direction of their displacements were measured by analyzing sequential images with NIH ImageJ software. Ambiguous movements due to coalescence between granules and collective movements of SGs in the same direction due to cell shrinkage upon stress were discarded. To measure the orientation of SG movements, we selected SGs that were farther than 15 μm from the cell nucleus so that the solid angle of view from the granule to the nucleus was restricted.
RESULTS AND DISCUSSION
Theory
In this section, we develop a simple analytical model to study the consequences of hindered diffusion in the cytoplasm on the formation of SGs. We then describe the potential mechanisms, which could overcome the limitation due to hindered diffusion and allow the formation of micrometric SGs.
Small SGs (<100 nm) but Not Larger Ones Can Be Rapidly Assembled by Bulk Diffusion in the Cytoplasm
Let us assume that the mechanism of mRNP aggregation during stress results from diffusion in the bulk cytoplasm. In the ideal case, we consider an irreversible association between two mRNP particles after collision via thermal diffusion. The time, tg, which is required to form an aggregate of radius R is therefore diffusion-limited (28):
where td ≈ η/(KBTCRNP) ≈ 1/(4πDrCRNP) represents the time lapse between mRNP collision, η is the cytoplasm viscosity, r and CRNP are the radius and the concentration of the initial RNP particles, respectively, D is the bulk diffusion constant, KBT is the thermal energy, and df is the fractal dimension (∼1.8 for colloidal particles (29, 30)). Because the cytoplasm of mammals contains ∼15,000–150,000 mRNA molecules per cell (31) and the volume of typical cells is ∼2,000 μm3, the typical cytoplasm concentration of mRNA is thus ∼12–120 nm. In mammalian cells, in situ staining showed that a large portion of all mRNA could be recruited to SGs (50% of all poly(A)+ mRNA (12)), although this point is still discussed (11). For numerical applications, we arbitrary considered that the concentration of stalled pre-initiation complexes, CRNP, is roughly equal to 20 nm. The other parameters are: r ∼10 nm (6), η ∼ 10−2 pascals(Pa)/s as measured for inert dextran particles of 10 nm radii (30), df = 1.8, and T = 37 °C. With these values, td ∼ 0.2 s. It indicates that the time, spent to form SGs with 100 nm and 1-μm radii, tg, are ∼12 s and 12 min, respectively. In this ideal model of free diffusion, the rate of mRNP aggregation in a viscous cytoplasm is then sufficient to explain the appearance of mRNP granules of radius larger than 1 μm after ∼10 min. However, in the cytoplasm, the diffusion of macromolecules is significantly hindered, especially due to actin filaments (17). With a typical distance between nearest actin filaments of 100 nm, such a thin mesh immobilizes granules larger than ∼50–100 nm (18). Consequently, the aggregation process should end up with small and immobile SGs. Interestingly, the ultrastructure analysis of SGs reveals the presence of ∼30–100 nm aggregates (6, 32), which are assembled in large structures, i.e. SGs. In summary, although small SGs can potentially be formed via free cytoplasmic diffusion, other mechanisms must intervene for the assembly of large SGs (R ≫ 100 nm).
How Do Cells Form Large SGs?
We propose three hypotheses, which are not mutually exclusive, to explain how the limitation of SG size due to hindered diffusion can be overcome. These hypotheses are here ordered according to their potential importance, the least probable is presented first.
Shuttling of mRNP Particles from Small to Large Granules
In the Ostwald ripening model (33, 34), self-aggregating particles on the surface of smaller granules are more loosely bound and thus more easily released into the solution than from the surface of larger granules. Owing to their small size (∼10–20 nm), stalled pre-initiation complexes can rapidly shuttle from the surface of smaller granules to the surface of larger granules. The consequence is that large granules should increase in size at the expense of smaller granules, which then tend to disappear.
Sliding of mRNP Along MTs via Thermal Agitation or Active Transport Mediated by Molecular Motors
We assume here that MTs can serve as tracks on which small SGs or mRNP particles may slide via thermal diffusion or ATP-driven active transport. In the present discussion, we consider thermal sliding, but minor modifications would be required to incorporate an active transport, which increases the benefit of mRNP sliding for rapid SG assembly. The common prerequisite is that mRNP granules interact with the MT surface via cationic mRNA binding partners like poly(A)-binding protein, YB-1 (35), and (or) adapter proteins like kinesin or dynein (36).
The advantage provided by mRNP sliding along MTs is not to increase the rates of collision between granules larger than 100 nm because the diffusion of large granules is also hindered in the vicinity of MT. The point is, rather, that small mRNP particles have a higher probability to encounter long MTs than another mRNP in the bulk. Therefore, as rapid diffusion of small bodies is allowed in the cytoplasm, remaining particles or small granules (<100 nm) can collide with long MTs and slide to support the growth of large granules on MTs. To decipher whether this mechanism can take place, we first need to estimate the time required for a small particle to find MTs, tm, and then the time to collide, via thermal sliding, with the nearest particle on MTs, ts. After some algebra (see supplemental Text 1A), we found that tm and ts scale like,
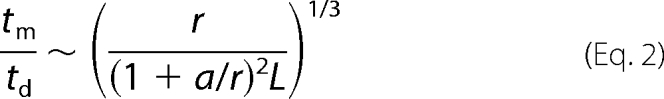 |
 |
where a is the MT radius (∼12 nm), L is the microtubule length, D and Ds are the particle diffusion constants in the bulk cytoplasm and on the MT surface, respectively, td, r, and CRNP are as defined in Equation 1.
tm is generally shorter than td because L ≫ r. Concerning ts, it is shorter that td provided that surface diffusion is not significantly slowed down by a strong mRNP attraction on MT. With r = 10 nm, a = 12 nm, L = 20 μm, and CRNP = 20 nm, the time lapse between two collisions on MTs, tm+ ts, is then ∼0.01 + 0.022 D/Ds, which is shorter than td ∼ 0.2 s, unless D/Ds > 9.
At the end of this process, there should be, in an ideal case, one large granule per MT. In HeLa cells, there are ∼100 MTs per cell, and their mean length is ∼20 μm. With ∼20,000 mRNP particles per cell, 200 particles could be assembled per MT to form small granules of radius R ∼ r(200)1/df ∼ 180 nm. A dense microtubule network may thus limit the size of SGs. When there are only 20 MTs to participate in granule assembly, R is much larger (∼1 μm).
Pushing and Pulling Granules via MT Turnovers Can Promote the Formation of Large Granules
Dynamical MTs can push or pull SGs during growing or shortening phases. Pushing or pulling could promote large granule formation by coalescence of granules larger than 100 nm, which otherwise are immobile due to hindered diffusion. The benefit is therefore an increased mobility of granules larger than 50–100 nm. To analyze the possibility of such mechanism, we note that the velocity of SG for a radius R is v = f/(6πηR). Because the maximum pushing force, f, generated by a growing MT can be higher than a few piconewtons (pN) (37, 38), we obtain v ∼ 2.8 μm/min for f ∼ 2 pN, R ∼ 750 nm, and an effective cytoplasmic viscosity of 3 pa.s, as measured with vesicles of radius R ∼ 750 nm (39). The granule speed mediated by pushing is therefore higher than the speed expected for bulk diffusion (the mean displacement is ∼95 nm for 1 min using the same viscosity). MT-mediated pulling forces can also displace granules during shortening phases of the plus ends, thus dragging granules toward the centrosome. As reported previously, pulling forces could be comparable to pushing forces but require an attraction force between MTs and SGs (38) to grasp MTs during the shortening phase.
Numerical Simulations
To further evaluate the role of free diffusion in SG formation and the relationships between SGs and the MT network, we performed numerical simulations. In the simulation model, mRNP particles were allowed to diffuse in a cubic volume with a size-dependent dampening mimicking the cytoplasm environment (Fig. 1, see supplemental Text 1B). As shown in Fig. 1B, hindered diffusion leads to the formation of small granules with a narrow distribution of sizes that do not exceed a critical limit defined by the actin mesh (radius smaller than 100 nm). To analyze the effect of MTs, we consider that, after their collision with a MT, mRNPs are able to randomly slide with a coefficient of diffusion, Ds. Fig. 1, C and D, shows the assembly of mRNP particles in the presence of different number of MTs. When there are just a few MTs, the results indicate that MTs favor the formation of very large granules (Fig. 1C), as described in Equations 2 and 3. In this case, small particles participate in the growth of mRNP aggregates on MTs by first binding to MTs and then by sliding on their surfaces. On the other hand, increasing the density of MT over a threshold results in the dispersion of mRNP particles onto the dense MT network, and this favors the formation of small aggregates immobilized on MTs that cannot grow larger due to hindered diffusion (Fig. 1D). In addition, we note the formation of even smaller granules when mRNP sliding on MTs is slowed down due to a strong surface friction on MT wall (Fig. 1E).
FIGURE 1.

Numerical simulation showing the effects of mRNP sliding on MTs and MT-mediated stirring on SG assembly. 400 mRNP particles were allowed to self-aggregate in a cubic volume, 6 × 6 × 6 μm3, for 10 min. Parameters: r = 20 nm; viscosity, η = 0.01 pa.s; df = 1.8; rC = 60 nm (see supplemental Text 1B). For a better visualization of the aggregation mechanism, graphs show the surface projection on the plane z = 0. In addition, the radii of the circles represented in the graphs show the number of mRNP particles on each granule not the physical radii. A, t = 0. B, hindered diffusion leads to the appearance of small and immobile granules, which inhibits the formation of large granules. t = 10 min. C, in the presence of two stable MTs, large granules appear on the MTs. t = 10 min, D = Ds. D, in the presence of ten stable MTs, mRNPs are dispersed along MTs, which results in the appearance of smaller aggregates (compare with C). t = 10 min. E, if the thermal diffusion of mRNPs on MTs is slowed down (Ds/D = 0.03), the aggregation mechanism is impaired and many smaller granules are formed on MTs. t = 10 min. F, same as E with dynamic MTs (five turnovers during 10 min). Granules or mRNP particles on MTs are pushed or pulled over a distance equal to 1.5 μm, leading to the formation of large granules.
Finally, to analyze the potential effect of MT dynamics on SG formation, we incorporate in the model one MT turnover per 2 min. During MT turnover, the mRNP granules located along a MT trajectory are either pushed (elongation) or pulled (shortening). The stalled force of MTs being high enough to move SGs (see previous section), coalescence could then occur if two granules are situated in an MT trajectory. The simulation shows that MT dynamics can considerably improve the formation of large SGs even if the mRNP sliding along MTs is dampened (Fig. 1F). Another point is that an attraction force increases the concentration of granules along MTs and thus promotes granule coalescence via MT-mediated pushing or pulling (Fig. 1F).
Experiments
In the following experiments, the formation of SGs was triggered by arsenite-mediated oxidative stress (40), the most widely used and best-known model of SG formation. All in vivo experiments presented here were conducted on HeLa cells. However, the results may also be extended to other cell lines as similar results were obtained with Chinese hamster ovary cells (see supplemental Fig. S1). The formation and movements of granules were analyzed using YB-1, one of the major core mRNA-binding proteins with multiple functions (24) and a reliable marker of SGs (1, 41). The very function of YB-1 in SGs is still under debate (41), and, in the present study, we will not address this issue. We only consider YB-1 as an interesting marker of SGs. Indeed the tracking of YB-1 should allow us to observe the early stage of granule formation due to its association with cytoplasmic mRNA before and after stress. In contrast, nuclear T cell-restricted intracellular antigen, a common marker of SGs, is redirected from the nucleus to the cytoplasm under stress. To first assess that YB-1 is an actual SG marker in our system, we show that YB-1 distributes homogeneously throughout the cytoplasm in the absence of stress, whereas it relocates rapidly in granules of increasing size under stress conditions (Fig. 2A). In these granules, YB-1 co-localizes with HuR (42), another SG marker (supplemental Fig. S1). A striking feature of arsenite-treated cells is that SGs are unevenly distributed in the cytoplasm with an apparent exclusion from the centrosome array (Fig. 2B). The range of this exclusion is of several microns, which suggests an influence of MTs on the spatial distribution of SGs. In this section, we attempt to evaluate experimentally whether hindered diffusion in the cytoplasm limits the size of SGs and then examine the three hypotheses advanced in the previous section to explain micrometric SG formation.
FIGURE 2.

YB-1 is a reliable cytoplasmic marker of mRNA to investigate SG formation. A, HeLa cells were grown in control conditions or stressed by exposure to 0.5 mm arsenite for 45 min. Although YB-1 is homogenously distributed in the cytoplasm of control cells, it localizes in large granules after arsenite treatment. B, pericentrin labeling (red) of arsenite-treated cells reveals the exclusion of SGs from the centrosomal region, which is also clearly visible on panel A after arsenite treatment. Cells were fixed and immunostained for YB-1 and α-tubulin as described under “Materials and Methods.”
Passive Diffusion in the Absence of MT Leads to the Formation of Submicrometric SGs
Due to hindered diffusion in the cell cytoplasm, passive diffusion of self-attracting mRNP particles should lead to the formation of small granules (≪1 μm). In agreement with this, we observed that, in the presence of arsenite and vinblastine to disrupt MTs, smaller YB-1 granules were formed compared with control cells (Fig. 3A). These small granules have a typical size smaller than 1 μm and are actual SGs as indicated by a clear co-localization between HuR and YB-1 (supplemental Fig. S2, A and B, and Fig. 4 for quantification). We also checked that the appearance of these small granules during MT disruption was not, indirectly, the result of an alteration of the post-translational machinery leading to the arsenite-induced translational arrest. We indeed observed that MT-destabilizing drugs do not affect the phosphorylation of the initiation factor eIF2 (supplemental Fig. S2C), which should then efficiently trigger the formation of stalled pre-initiation complexes. To complete our observations, we investigated whether MT-disrupted cells can resume the formation of micrometric granules after MT regrowth. When vinblastine was removed after 45-min exposure to arsenite, MTs rapidly repolymerized leading once again to the formation of micrometric granules after 45 min (see Fig. 3B). In agreement with the results of Kobolova et al. (21), the data indicate that MTs are not required for the formation of small SGs, whereas they are necessary for the formation of large SGs. To extend our results to more relevant in vivo conditions, we also explored the effect of MT disruption on SG formation under levels of stress lower than that generally used to obtain well defined and large SGs. It turned out that, even at very low concentrations of arsenite (down to 100 μm), smaller mRNP granules than in control cells were formed in the presence of vinblastine (Fig. 3A).
FIGURE 3.

Microtubule disruption leads to the formation of small SGs after arsenite exposure. A, MT disruption leads to the formation of small SGs at high and low level of stress. HeLa cells were treated with 0.1% DMSO (Control) or with 3 μm vinblastine (VLB). These treatments were applied 2 h prior to and during 45-min exposure to arsenite at varying concentrations. At all concentrations of arsenite, large SGs are formed in control cells, even though they appear less contrasted at the lowest arsenite concentration (100 μm). In the presence of vinblastine, SGs are smaller and homogenously distributed in the cytoplasm whatever the arsenite concentration. Cells were stained with anti-YB-1 antibody. B, cells resume the formation of large SGs after MT repolymerization. i, HeLa cells after exposure to 0.5 mm arsenite for 90 min; ii, vinblastine-treated HeLa cells exposed to 0.5 mm arsenite for 90 min; iii, same as ii except that vinblastine was removed during the last 45 min of exposure to arsenite; iv, HeLa cells were first exposed to arsenite for 45 min to form large granules and then treated with vinblastine for the last 45 min of arsenite exposure. We note that, upon removal of vinblastine to allow MT regrowth, cells are again able to form large SGs (iii). However, the dissociation of large SGs into small SGs upon MT disruption is not possible, meaning that, after their formation, large SGs are rather stable when MTs are disrupted (iv).
FIGURE 4.

Statistical analysis of SG size in cells treated with microtubule-targeting drugs. A, immunofluorescence of HeLa cells treated with 0.1% DMSO (arsenite), 0.3 and 3 μm vinblastine (VLB), 3 μm nocodazole, 3 μm taxol or 1 mm erythro-9-(2-hydroxy-3-onyl)adenine (EHNA), 2 h prior to and during exposure to 0.5 mm arsenite for 45 min. MT stabilization by taxol or disruption by high concentrations of vinblastine or nocodazole (3 μm) affect spatial distribution, size, and shape of SGs. Note that the larger granules are localized at the cell periphery. EHNA, an ATPase inhibitor that targets motor activity, has no obvious influence on SG distribution and size. B, apparent radii and integrated intensities of SGs. We note that, in the presence of taxol or vinblastine (3 μm), SGs are significantly smaller and have lower integrated intensities.
To quantify the effect of MTs on granule sizes, we measured the apparent radii and the integrated intensities of SGs immunostained for YB-1 after arsenite treatment. The integrated intensity reflects the number of YB-1 proteins in granules and is therefore a good indicator of the number of mRNP particles per granule. We found that the number of smaller granules increased in the absence of MTs compared with control cells (3 μm vinblastine (Fig. 4, A and B)). A similar pattern was obtained for cells treated with 3 μm nocodazole (see Fig. 4A; quantification not shown). However, at lower concentrations of MT-destabilizing drugs, at which few MTs remain in the cytoplasm (0.3 μm vinblastine (Fig. 4, A and B) and 0.3 μm nocodazole, data not shown), the population of micrometric granules increased (Fig. 4B). Interestingly, in the absence of MTs, larger granules were preferentially located near the cell margin rather than close to the nucleus, an effect that we attributed to large membrane movements at the cell periphery (Fig. 4A).
Using GFP-YB-1 and fluorescence videomicroscopy, the kinetics of SG formation was analyzed in the absence or presence of 3 μm vinblastine (Fig. 5A). Like endogenous YB-1, GFP-YB-1 was evenly distributed in the cytoplasm without arsenite. This control indicates that GFP-YB-1 by itself did not trigger the formation of SGs. In the absence of vinblastine, the formation of the first GFP-YB-1 granules appeared between 10 and 14 min exposure to 0.5 mm arsenite (Fig. 5A). A few minutes (∼8 min) after the appearance of these small structures, GFP-YB-1 accumulated into large granules of micrometric size (t = 18 min, Fig. 5A). Such a rapid aggregation is in agreement with the first report of Kedersha and co-workers (14). In the presence of 3 μm vinblastine, the aggregation kinetics was severely impaired (Fig. 5A). Small granules indeed appeared between 10- and 14-min exposure to arsenite, like in control cells, but larger SGs remained rare even 16 min after the appearance of the first small granular structures (t = 26 min).
FIGURE 5.

Kinetics of SG assembly in cells treated with microtubule-targeting drugs. A, kinetics of SG assembly in transfected HeLa cells using GFP-YB-1. Cells were treated with 0.1% DMSO (Control), 3 μm vinblastine, 5 μm taxol, 1 mm EHNA for 1 h prior to arsenite stress (0.5 mm arsenite). A high concentration of taxol (5 μm) was used to ascertain that microtubules were fully stabilized during experiments. Images of GFP fluorescence were captured using digital capture at various times after addition of arsenite, as indicated in each panel. During the first 10 min of stress, the cytoplasmic distribution GFP-YB-1 remained unaffected. B, analyzes of the displacements of SGs by videomicroscopy. HeLa cells were transfected with GFP-YB-1 to analyze the motion of SGs. Cells were treated with 0.1% DMSO (Control), 3 μm vinblastine, 5 μm taxol, or 1 μm cytochalasin D for 1 h prior to arsenite addition. Sequential fluorescence images were recorded after 40-min arsenite treatment when SGs were assembled. SG displacements were classified into three groups: short (1), middle (2), and long (3) range displacements (Δ×) per minute. Graphs show the mean ± S.D. values derived from 10 different cells. C, directions of SG movements. In control cells, SG displacements showed two preferential orientations, pointing toward the nucleus and in the opposite direction. In D: i, differential images representing the granule movements obtained by subtracting two consecutive images in a time-lapse series (first image, black granules; second image after 2 min, bright granules). As indicated by the low contrast of the differential image, the SGs of taxol-treated cells are nearly immobile compared with control; ii, two differential images reveal coalescence between two large SGs in control cells. Time-lapse videos showing the displacements of SGs are available in the supplemental files.
In summary, these results indicate that, in the absence of MTs, mRNP particles can diffusive freely in the cytoplasm, which leads to the formation of small submicrometric granules due to hindered diffusion (see the “Theory” section). However, the second step leading to the formation of larger micrometric granules is inhibited and can be reactivated after MT repolymerization, thus indicating the need of microtubules for the formation of micrometric stress granules.
Mechanisms of Large SG Assembly in the Cytoplasm
mRNP Shuttling from Small to Large Granules
In favor of this hypothesis, it has been shown that SGs are highly dynamic structures, and detachment of mRNPs from SGs is possible (11, 14). However, the requirement of MTs to trigger the formation of micrometric SGs is not in agreement with the ripening model, because the mRNP shuttling from small to large SGs should not rely on MTs. To further address this point, we analyzed whether smaller granules (<1 μm) disappear without collision in parallel to the increase in larger granule radii. Subtractions of sequential fluorescence images of GFP-YB-1 during the formation of SGs revealed that granules grow by coalescence (Fig. 5D). However, the disappearance of granules independently of collision was not observed and should rarely occur.
Microtubule Dynamic Instability Promotes Coalescence of Small Granules into Larger Ones
To explore the idea that MT turnovers may favor the formation of micrometric SGs by pushing or pulling SGs, the influence of taxol, a drug that suppresses MT dynamics, was considered. Compared with control, there were many more submicrometric granules in taxol-treated cells (Fig. 6A). To decipher whether the effects of taxol on SGs indeed result from MT stabilization, we analyzed its effects at varying concentrations (100 nm to 3 μm). We remarked that 100 nm taxol was sufficient to increase the proportion of small SGs, and this pattern was accentuated at 300 nm taxol (Fig. 6, A and B). At higher taxol concentrations, the sizes of granules were similar to that observed at 300 nm taxol. This concentration threshold matches the taxol concentration required for a complete stabilization of MTs, as observed by using free tubulin as an indicator of microtubule instability (Fig. 6B). The effect of taxol on SGs is then most probably due to MT stabilization.
FIGURE 6.

Both MT dynamics and the number of MTs per cell modulate SG size. A, effect of taxol concentration on SG assembly. HeLa cells were treated with 0.1% DMSO (control) or varying concentrations of taxol. These treatments were applied 1 h prior to and during exposure to 0.5 mm arsenite for 45 min. At low concentration (<0.3 μm), taxol gradually decreases SG size, leading to the formation of small SGs with a homogenously distribution in the cytoplasm. Further increases of taxol concentration (0.3 μm to 3 μm) do not accentuate the influence of taxol on SG formation. B, Western blotting showing the effect of taxol on free and polymerized tubulin in HeLa cells. After 1-h incubation with taxol at various concentrations, cells were treated to quantify the free and polymerized tubulin pools. At low concentrations (<300 nm), taxol gradually decreases the concentration of free tubulin due to progressive MT stabilization. Above 300 nm, taxol fully stabilized MTs. In the graph representing the ratio of free to total tubulin and the mean value of SG intensity over 500 granules for various taxol concentrations, we clearly observed a correlation between the number of mRNP per SG and the ratio of free to total tubulin. Error bars represent the ± S.D. C, effect of the presence of small and numerous stable MTs on SG formation. i, control cells. HeLa cells were incubated on ice for 1 h and then returned to 37 °C to 30 min and then were exposed to 0.5 mm to arsenite for 45 min. ii, same as i except that cells were treated with 3 μm taxol during the whole process. Because taxol-stabilized MTs are resistant to cold depolymerization, long stable MTs were not disrupted. iii, same as ii but taxol was added 30 min after MT depolymerization on ice. In these conditions, taxol promotes the polymerization of stable but small MTs from depolymerized tubulin. SGs formed under such conditions are smaller than that formed in the presence of long and stable MTs. The presence of many small MTs thus inhibits large SG formation. D, influence of taxol and EHNA on SG exclusion from the centrosomal region. We note that there are fewer granules near the centrosome of arsenite-stressed cells (control). This heterogenous distribution remains unchanged when cells are treated with EHNA, an ATPase inhibitor of molecular motors. However, in the presence of taxol, cells display a homogenous distribution of SGs, with SGs also localized near the centrosome. Immunofluorescence was performed using anti-pericentrin antibodies to label the centrosome (red) or anti-YB-1 to label SGs (green).
Videomicroscopy analyses of the kinetics of SG formation in taxol-treated cells also revealed the formation of SGs smaller than in control cells (Fig. 5). In addition, after MT disruption, the first stage leading to the formation of small granules is not affected by taxol, whereas the second step leading to micrometric granule formation is inhibited. In agreement with a slow kinetics of assembly, the analysis of the movements of isolated granules in the presence of taxol shows that granules were less mobile compared with control, with a nearly complete disappearance of the long ranged movements (Fig. 5, B and D, and supplemental video S3). Additionally, SG movements showed preferential orientations toward the nucleus as well as in the opposite direction, which reflects the expected directions of MT forces (Fig. 5C). This result also indicates that not only pushing but pulling forces could act on SGs, most probably due to the presence of an attraction force between granules and MTs. In summary, MT turnovers generate long ranged movements of SGs and thus promote SG coalescence. This experimental result is in agreement with our theoretical estimation, which indicates that the forces generated by MT shortening and elongation are sufficient to displace large SGs. We observed a direct consequence of this mechanism in the spatial distribution SGs. Indeed, dynamic MT plus-ends are not homogenous distributed throughout the cytoplasm (43); active plus-ends are mostly localized away from the centrosomal region. Coalescence of large SGs should then occur with a higher probability away from the centrosome, thus resulting in apparent SG avoidance of the centrosomal region (Fig. 6D).
Actin filaments are also dynamical structures and thus can potentially participate in the pushing or pulling mechanisms to favor SG coalescence. However, even though the stall force of a single actin filament (1 pN (44, 45)) is sufficient to displace SGs, the involvement of actin in granule formation is less clear. The separation distance between actin filaments is ∼100 nm, and large SGs are thus probably embedded in the thin actin mesh (supplemental Fig. S3) so that the resulting force of actin dynamics on granules should be weak. This explains why, as previously described, arsenite-mediated stress continues to trigger the formation of SGs in cells treated with actin-disrupting drugs (here cytochalasin D, an inhibitor of actin polymerization (Fig. 5B) (20)). Actin filaments could, however, participate to cytoplasmic movements of SGs via long ranged redistribution of actin filaments, as reported during cell motility (46) or blebbing (47). In addition, the peripheral retrograde flow of actin may be involved as, in the absence of MTs, SGs are larger in the cell periphery than close to the nucleus (Fig. 4A).
Is mRNP Sliding or Active Transport Along MT Implicated in SG Formation?
We first attempted to address this question by a series of in vitro experiments. As recently reported by our group, mRNA can be attracted in vitro onto stable MTs in the presence of mRNA cationic partners, like YB-1 and poly(A)-binding protein. MTs can thus serve as tracks to promote mRNP aggregation (35). This interaction is of electrostatic origin and takes place because the cationic domains of proteins interacting with mRNA can be shared between anionic mRNA and MTs. The promotion of mRNP aggregation on stable MTs results from the sliding of mRNP particles on MT surface due to thermal agitation. We extend here these findings using mRNPs made with YB-1 and fluorescent mRNA and observe the formation of mRNP aggregates on taxol-stabilized MTs in vitro. YB-1·mRNA complexes were first preformed with a YB-1/mRNA ratio sufficiently high to allow the formation of small mRNP aggregates (for details see Ref. 35). In the absence of MTs, micron-size mRNPs were homogenously distributed on the coverslip (Fig. 7) showing that an excess of cationic proteins like YB-1 promotes mRNP self-aggregation. In the presence of few MTs, we noted the formation of larger aggregates along MTs (Fig. 7). In contrast, when the concentration of MTs was increased, the formation of large granules was inhibited and mRNP particles appeared dispersed along many MTs. This most probably results from the dispersion of mRNP particles along a dense MT network, which limits the number of mRNP per MT and thus granule size nucleated on MTs. These results are in good agreement with our theoretical prediction (see Fig. 1).
FIGURE 7.

Formation of large mRNP granules on stable MTs in vitro. Fluorescence imaging of Cy2-labeled mRNA (2 LUC mRNA, 3000 nucleotides). Small mRNP granules were formed in the presence of 30 μm YB-1 and 30 μg/ml fluorescent mRNA. Non-fluorescent taxol-stabilized MTs were added at varying concentrations, as indicated in each panel. At low concentrations (0.3 μm tubulin), MTs favor the formation of large mRNP granules, whereas the increase of MT mass (10 μm tubulin) leads to a progressive dispersion of mRNP on their surface (see higher magnification images).
We then extended our investigations to living cells. Because the tracking of mRNP particles sliding on MTs during stress is not trivial, we rather studied whether or not the number of stable MTs per cell modulates the size of granules as observed in vitro. To vary the number of stable MTs per cell, HeLa cells were incubated on ice to depolymerize MTs and then exposed to taxol to promote the formation of many short and stable MTs. After incubation at 37 °C for 30 min, cells were finally exposed to arsenite. Under such conditions, SGs were smaller than in cells treated with taxol prior to incubation on ice to stabilize long MTs (Fig. 6C). We also noted that, with low concentrations of vinblastine (0.3 μm) to reduce the number of stable MTs (48, 49), SGs were bigger than in taxol-treated cells (Fig. 4). These experimental results suggest that the number of stable MTs influences the formation of large SGs and indirectly indicate that mRNP particles use MTs as tracks for the nucleation and growth of SGs.
Because a mechanism of mRNP sliding along MTs probably participates in the assembly of SGs, a question arises about the implication of active mRNP transport mediated by molecular motors such as kinesin or dynein. To the best of our knowledge and in agreement with our results, a direct observation by videomicroscopy showing SGs translocations along MTs is still lacking, whereas directional movement of p-bodies was already reported (50). A model of active transport for SG formation is hard to reconcile with the immobilization of SG movements that we observed in taxol-treated cells by videomicroscopy (Fig. 5, B and D). Indeed, taxol is not known to inhibit active transport. In addition, we observed that two ATPase inhibitors known to affect active transport (51–54), vanadate and erythro-9-(2-hydroxy-3-onyl)adenine (EHNA), failed to inhibit the formation of micrometric SGs after arsenite treatment (Figs. 5 and 6 and supplemental Fig. S4C). The avoidance of the centrosomal region was again observed in the presence of EHNA (Fig. 6D). However, in favor of an active transport, ATP depletion during stress leads to the formation of smaller SGs than in control cells (supplemental Fig. S4), but it is known that ATP depletion also affects MT dynamics and MT organization (55). At this point, we are no able to demonstrate the implication of an ATP-driven transport of mRNP in the formation of micrometric granules, even though this hypothesis is reasonable.
Summary
We studied three possible mechanisms to account for the rapid assembly of mRNA and associated proteins into micrometric SGs in the viscous cytoplasm despite the limit of hindered diffusion. We then attempted to evaluate their pertinence experimentally as follows: The “ripening” model suggests that mRNP particles shuttle from small to large granules. We found no experimental evidence for this mechanism. The “sliding” model implies that mRNP particles slide along MTs via thermal agitation and (or) active transport. MTs then act as substrate for granule nucleation and growth. This model seems to be in agreement with in vitro and in vivo experimental results, even though the dense MT network of living cells reduces its effectiveness. The potential participation of ATP-driven transport of mRNP mediated by kinesin or dynein remains to be explored. In the “pushing or pulling” model, we propose that cells take advantage of the dynamicity of its cytoskeleton to overcome hindered diffusion in the viscous cytoplasm. Indeed MTs can pull and push immobilized granules to form larger granules by coalescence. Both theoretical predictions and experimental results suggest that MT dynamical instability indeed participates to the formation of SGs and acts in coordination with the mechanism of mRNP sliding on MTs.
As the mechanism of SG assembly is critical for cell survival, these findings could be helpful to design new therapeutic strategies for targeting the mechanism of SG formation. This is particularly relevant, because stress conditions like hypoxia, hyperthermia, or UV irradiation are realistic means to contribute to the treatment of cancer.
Acknowledgments
We gratefully acknowledge the Genopole Evry for constant support of the laboratory. We thank Julie Lameth for her help in immunostaining and microscopy.
This work was supported by INSERM.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4, text, equations, and Videos 1–3.
- mRNP
- messenger ribonucleoprotein
- SG
- stress granule
- MT
- microtubule
- MES
- 4-morpholineethanesulfonic acid
- PBS
- phosphate-buffered saline
- GFP
- green fluorescent protein
- EHNA
- erythro-9-(2-hydroxy-3-onyl)adenine.
REFERENCES
- 1.Kedersha N., Anderson P. (2007) Methods Enzymol. 431, 61–81 [DOI] [PubMed] [Google Scholar]
- 2.Hua Y., Zhou J. (2004) FEBS Lett. 572, 69–74 [DOI] [PubMed] [Google Scholar]
- 3.Kim W. J., Back S. H., Kim V., Ryu I., Jang S. K. (2005) Mol. Cell Biol. 25, 2450–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moeller B. J., Cao Y., Li C. Y., Dewhirst M. W. (2004) Cancer Cell 5, 429–441 [DOI] [PubMed] [Google Scholar]
- 5.Yamasaki S., Anderson P. (2008) Curr. Opin. Cell Biol. 20, 222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nover L., Scharf K. D., Neumann D. (1989) Mol. Cell Biol. 9, 1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore M. J. (2005) Science 309, 1514–1518 [DOI] [PubMed] [Google Scholar]
- 8.Anderson P., Kedersha N. (2006) J. Cell Biol. 172, 803–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kedersha N. L., Gupta M., Li W., Miller I., Anderson P. (1999) J. Cell Biol. 147, 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazroui R., Sukarieh R., Bordeleau M. E., Kaufman R. J., Northcote P., Tanaka J., Gallouzi I., Pelletier J. (2006) Mol. Biol. Cell 17, 4212–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mollet S., Cougot N., Wilczynska A., Dautry F., Kress M., Bertrand E., Weil D. (2008) Mol. Biol. Cell 19, 4469–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kedersha N., Anderson P. (2002) Biochem. Soc. Trans. 30, 963–969 [DOI] [PubMed] [Google Scholar]
- 13.Tourrière H., Chebli K., Zekri L., Courselaud B., Blanchard J. M., Bertrand E., Tazi J. (2003) J. Cell Biol. 160, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Kedersha N., Cho M. R., Li W., Yacono P. W., Chen S., Gilks N., Golan D. E., Anderson P. (2000) J. Cell Biol. 151, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson P., Kedersha N. (2008) Trends Biochem. Sci. 33, 141–150 [DOI] [PubMed] [Google Scholar]
- 16.Luby-Phelps K. (2000) Int. Rev. Cytol. 192, 189–221 [DOI] [PubMed] [Google Scholar]
- 17.Luby-Phelps K., Castle P. E., Taylor D. L., Lanni F. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 4910–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dauty E., Verkman A. S. (2005) J. Biol. Chem. 280, 7823–7828 [DOI] [PubMed] [Google Scholar]
- 19.Lukacs G. L., Haggie P., Seksek O., Lechardeur D., Freedman N., Verkman A. S. (2000) J. Biol. Chem. 275, 1625–1629 [DOI] [PubMed] [Google Scholar]
- 20.Ivanov P. A., Chudinova E. M., Nadezhdina E. S. (2003) Exp. Cell Res. 290, 227–233 [DOI] [PubMed] [Google Scholar]
- 21.Kolobova E., Efimov A., Kaverina I., Rishi A. K., Schrader J. W., Ham A. J., Larocca M. C., Goldenring J. R. (2009) Exp. Cell Res. 315, 542–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon S., Zhang Y., Matthias P. (2007) Genes Dev. 21, 3381–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castoldi M., Popov A. V. (2003) Protein Expr. Purif. 32, 83–88 [DOI] [PubMed] [Google Scholar]
- 24.Evdokimova V., Ruzanov P., Imataka H., Raught B., Svitkin Y., Ovchinnikov L. P., Sonenberg N. (2001) EMBO J. 20, 5491–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skabkin M. A., Kiselyova O. I., Chernov K. G., Sorokin A. V., Dubrovin E. V., Yaminsky I. V., Vasiliev V. D., Ovchinnikov L. P. (2004) Nucleic Acids Res. 32, 5621–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davydova E. K., Evdokimova V. M., Ovchinnikov L. P., Hershey J. W. (1997) Nucleic Acids Res. 25, 2911–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gundersen G. G., Khawaja S., Bulinski J. C. (1987) J. Cell Biol. 105, 251–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen T. T., Shklovskii B. I. (2002) Phys. Rev. E. Stat. Nonlin Soft Matter Phys. 65:031409 [DOI] [PubMed] [Google Scholar]
- 29.Weitz D. A., Lin M. Y. (1986) Phys. Rev. Lett. 57, 2037–2040 [DOI] [PubMed] [Google Scholar]
- 30.Arrio-Dupont M., Cribier S., Foucault G., Devaux P. F., d'Albis A. (1996) Biophys. J. 70, 2327–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halbeisen R. E., Galgano A., Scherrer T., Gerber A. P. (2008) Cell Mol. Life Sci. 65, 798–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilks N., Kedersha N., Ayodele M., Shen L., Stoecklin G., Dember L. M., Anderson P. (2004) Mol. Biol. Cell 15, 5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finsy R. (2004) Langmuir 20, 2975–2976 [DOI] [PubMed] [Google Scholar]
- 34.Yao J. H., Elder K. R., Guo H., Grant M. (1993) Phys. Rev. B Condens. Matter 47, 14110–14125 [DOI] [PubMed] [Google Scholar]
- 35.Chernov K. G., Curmi P. A., Hamon L., Mechulam A., Ovchinnikov L. P., Pastré D. (2008) FEBS Lett. 582, 2875–2881 [DOI] [PubMed] [Google Scholar]
- 36.Tsai N. P., Tsui Y. C., Wei L. N. (2009) Neuroscience 159, 647–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoué S., Salmon E. D. (1995) Mol. Biol. Cell 6, 1619–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dogterom M., Kerssemakers J. W., Romet-Lemonne G., Janson M. E. (2005) Curr. Opin. Cell Biol. 17, 67–74 [DOI] [PubMed] [Google Scholar]
- 39.Mogilner A., Oster G. (1996) Biophys. J. 71, 3030–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ercal N., Gurer-Orhan H., Aykin-Burns N. (2001) Curr. Top. Med. Chem. 1, 529–539 [DOI] [PubMed] [Google Scholar]
- 41.Yang W. H., Bloch D. B. (2007) RNA 13, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai N. P., Ho P. C., Wei L. N. (2008) EMBO J. 27, 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vorobjev I. A., Rodionov V. I., Maly I. V., Borisy G. G. (1999) J. Cell Sci. 112, 2277–2289 [DOI] [PubMed] [Google Scholar]
- 44.Footer M. J., Kerssemakers J. W., Theriot J. A., Dogterom M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 2181–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mogilner A., Oster G. (2003) Biophys. J. 84, 1591–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollard T. D., Borisy G. G. (2003) Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 47.Charras G. T., Hu C. K., Coughlin M., Mitchison T. J. (2006) J. Cell Biol. 175, 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan M. A., Thrower D., Wilson L. (1992) J. Cell Sci. 102, 401–416 [DOI] [PubMed] [Google Scholar]
- 49.Pourroy B., Carré M., Honoré S., Bourgarel-Rey V., Kruczynski A., Briand C., Braguer D. (2004) Mol. Pharmacol. 66, 580–591 [DOI] [PubMed] [Google Scholar]
- 50.Aizer A., Brody Y., Ler L. W., Sonenberg N., Singer R. H., Shav-Tal Y. (2008) Mol. Biol. Cell 19, 4154–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clark T. G., Rosenbaum J. L. (1982) Proc. Natl. Acad. Sci. U.S.A. 79, 4655–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shah J. V., Flanagan L. A., Janmey P. A., Leterrier J. F. (2000) Mol. Biol. Cell 11, 3495–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouchard P., Penningroth S. M., Cheung A., Gagnon C., Bardin C. W. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 1033–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lalli G., Gschmeissner S., Schiavo G. (2003) J. Cell Sci. 116, 4639–4650 [DOI] [PubMed] [Google Scholar]
- 55.Bershadsky A. D., Gelfand V. I. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 3610–3613 [DOI] [PMC free article] [PubMed] [Google Scholar]