

Abstract
Background
The role of a non-synonymous A118G polymorphism of the human mu-opioid receptor gene (OPRM1) for alcohol reward and therapeutic efficacy of naltrexone remains controversial. A functionally equivalent OPRM1 C77G polymorphism in rhesus macaques allows this to be addressed under controlled experimental conditions.
Methods
Twenty one rhesus macaques (13 females, 8 males) were genotyped for OPRM1 C77G, and studied during 1h sessions for preference between an aspartame sweetened alcohol solution (8.4% v/v) and a non-alcoholic control fluid, in a baseline session followed by naltrexone (1 mg/kg) and vehicle treatment in a counterbalanced within-subject design.
Results
Mixed-model ANOVA controlling for baseline and sex showed a highly significant (p=0.003) interaction between genotype and treatment. Post-hoc analysis showed that vehicle treated 77G carriers had markedly higher alcohol preference than 77C homozygous subjects (p=0.001). Following naltrexone administration, 77G carriers decreased their preference (p=0.002), and no longer differed from 77C homozygous subjects. In contrast, the latter group was unaffected by treatment, and in fact showed a trend-level increase of preference following naltrexone.
Conclusions
These results support a critical pharmacogenetic role of OPRM1 variation for therapeutic efficacy of naltrexone.
Keywords: alcoholism, naltrexone, family history, opioids
The opioid receptor antagonist naltrexone (NTX) was discovered as an alcoholism treatment almost two decades ago (1,2), based on observations in non-human primates (3). Today, meta-analyses of more than 30 randomized controlled trials support NTX efficacy, but the average effect size is modest (4), and clinical use of NTX has not become widespread. The limited overall effect size of NTX may reflect heterogeneity in responsiveness among patients. Secondary analyses of clinical trials suggest that family history of alcoholism is predictive of NTX response (5), and direct support for this notion has been provided under laboratory conditions, both with regard to subjective alcohol effects (6) and alcohol self-administration (7). Together, these findings point to the clinically important possibility that the effect size of NTX in treatment of alcoholism could be considerably improved in appropriately selected patient populations.
In agreement with the human findings, blockade of opioid transmission results in suppression of alcohol consumption in a range of animal models. Rodent studies show that among the three cloned opioid receptors, the μ-subtype is critical for alcohol reinforcement and therefore presumably for the therapeutic efficacy of NTX. For instance, genetic disruption of the μ-opioid receptor gene (OPRM1) results in loss of alcohol self-administration in mice, while pharmacological blockade of this subtype decreases ethanol drinking both in non-selected Wistar rats and in rat lines bred for high alcohol preference [reviewed in (8)].
The role of family history as a factor predictive of therapeutic NTX response in alcoholism points to genetic factors as mediators of response heterogeneity. Taken together with the role of the μ-opioid receptor as a mediator of alcohol reward and as the target for NTX actions, this points to OPRM1 gene variation as a potential pharmacogenetic determinant of NTX responses. In this context, a nonsynonymous A118G OPRM1 SNP discovered over a decade ago (9), is of interest. This polymorphism remains controversial as a genetic susceptibility factor for alcoholism (10), but is more consistently found to modulate responses to alcohol and to μ-opioid receptor blockade (11,12). Some secondary analyses of published clinical trials suggest that OPRM1 118G carriers are particularly responsive to NTX (13,14), while others have not found this to be the case (15).
An OPRM1 SNP that is functionally equivalent to the human A118G polymorphism (C77G) has been identified in rhesus macaques (16) and offers an attractive opportunity to study the role of OPRM1 variation for alcohol and NTX responses in a closely controlled experimental system. Using this model, we recently reported increased psychomotor stimulation in response to alcohol, increased alcohol preference, and increased frequency of alcohol consumption to intoxication in carriers of the rhesus (rh) OPRM1 77G variant (17). These findings directly predict that 77G carriers should be preferentially sensitive to suppression of alcohol preference by NTX, a hypothesis that was examined in the present study.
Methods
Procedures were approved by the NIAAA Institutional Animal Care and Use Committee. Genotyping and assessment of alcohol preference were as described (17). Briefly, at approximately 4 years of age, 21 socially housed rhesus macaques (n=21; 13 females, 8 males; 13 C/C, 8 C/G) were trained to consume alcohol (8.4 %v/v) in an aspartame sweetened solution and the vehicle, during 1h sessions carried out 5 days a week in their home enclosure. Tracking of individual drinking was accomplished using a transponder chip attached to the collar. Alcohol preference was chosen as outcome, because under these conditions, it most directly reflects choice between alcohol and non-alcoholic control solution, while total alcohol consumption is influenced by additional factors, e.g. rank, that determine whether the animal at all enters the drinking station. Following completion of training, drug testing was carried out over three weeks. During week 1, baseline preference was assessed. On three consecutive days, an intramuscular injection of saline (0.1 ml/kg) was given 30 min prior to a 1h sessions, during which access to both the alcohol solution and vehicle was provided. Baseline preference was calculated as the mean of these three sessions. The same procedure was repeated week 2 and 3, with half of the subjects randomly assigned to saline, while the other half received NTX (1.0 mg/kg), in a counterbalanced, within-subjects design. Data were analyzed using mixed model ANOVA (STATISTICA 8.0, Statsoft, Tulsa, OK). Baseline preference, sex and order were evaluated as co-variates and retained in the model because their inclusion reduced residual variance. Post-hoc comparisons were carried out using the Newman-Keul test.
Results
There were no main effects of sex (F[1,12]=1.3, p=0.27) or order of treatment (F[1,12]=0.6, p=0.81) on alcohol preference. Baseline preference was a significant co-variate (F[1,12]=22.2, p=0.0005), and a significant main effect of treatment was found (F[1,12]=5.5, p=0.04). Most importantly, treatment differentially affected subjects as a function of genotype, shown by a highly significant interaction between OPRM1 genotype and treatment (F[1,12]= 15.9, p=0.002; Figure 1). This interaction accounted for 57% of the variance in preference, measured as partial eta2. Following vehicle treatment, 77G carriers had markedly higher alcohol preference than 77C homozygous subjects (p=0.001). After NTX administration, 77G carriers decreased their preference compared to vehicle (p=0.002), and no longer differed from 77C homozygous subjects. In contrast, the latter group was unaffected by treatment, and in fact showed a trend-level increase of preference following NTX. A similar interaction was present when analyzing females only (F[1,16]=8.1; p=0.01). The analysis for males was underpowered, because only two male 77G carriers were available, but the trend was the same. Finally, total intake of aspartame-sweetened solution, a control for taste, nausea and other non-specific factors, had baseline consumption as a significant co-variate (F[1,16]=16.3; p=0.0001), but was unaffected by treatment (F[1,16]=0.21; p=0.66).
Figure 1.
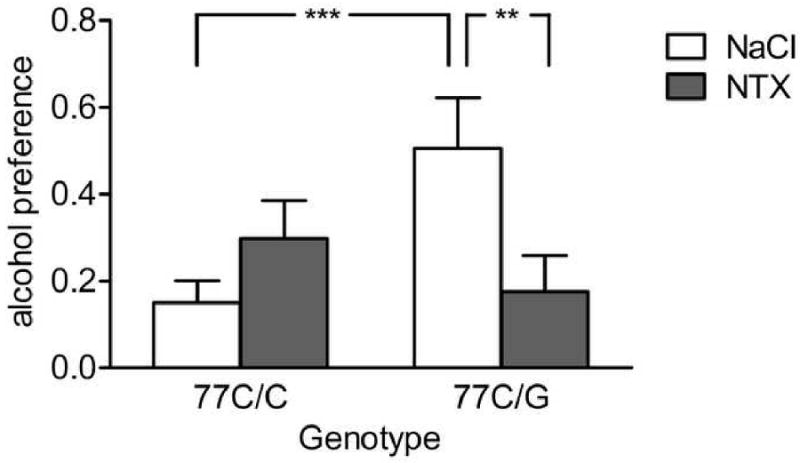
Selective suppression of alcohol preference by naltrexone (0.1 mg/kg) in rhesus macaques carrying the 77G allele of the μ-opioid receptor gene. Genotype strongly interacted with treatment (p=0.002). Post-hoc analysis showed that following saline treatment, 77G carriers had significantly higher preference than 77A homozygous subjects (***p=0.001), and that naltrexone treatment markedly reduced alcohol preference compared to saline within the 77G carrier group (**p=0.002). In contrast, no naltrexone effect was found in the 77A homozygous group. Subjects were socially housed in their regular enclosures, and allowed access to drinking stations during 1h sessions 5 days a week. During drinking sessions, they were offered choice between an 8.4% v/v alcohol containing, aspartame sweetened solution, and the aspartame sweetened vehicle without alcohol. Individual baseline preference was assessed during an initial week, during which all subjects received saline injections. All subjects subsequently received naltrexone or saline, in counterbalanced order, during the second and third week of testing. Baseline preference was controlled for in the analysis. For detailed statistics, see Results.
Discussion
This study replicates our previous finding that rhOPRM1 77G carriers have higher basal preference for alcohol (17). In the original report, this effect of OPRM1 genotype was most pronounced in males, although a similar trend was also seen in females. Presently, the effect was observed in a predominantly female cohort and remained when analysis was restricted to females. It is therefore likely that rhOPRM1 77G, in fact, confers increased alcohol preference irrespectively of sex. We originally also found increased psychomotor stimulation by alcohol in male but not female 77G carriers. This is not inconsistent with the present findings, because psychomotor stimulation was examined following administration of a high alcohol dose (2.0-2.1 g/kg), at which sedative alcohol effects in females may have obscured differences in psychomotor stimulation.
The key finding of the present study is that NTX only suppressed alcohol preference in 77G carriers. Human data that address the ability of NTX to reduce alcohol drinking are presently limited to retrospective secondary analyses of published clinical trials, and results are mixed. Two studies support a role of 118G as a predictor of treatment efficacy (13,14), while one does not (15). Furthermore, human studies cannot easily isolate the influence of A118G from that of other polymorphisms in linkage disequilibrium (LD) with it. One study found that other polymorphisms within the same haplotype block, but not A118G, were associated with diagnoses of substance dependence (18). In contrast, a haplotype based re-analysis of the COMBINE study found NTX response to be specifically attributable to 118G (19). The situation is further complicated by the recent finding that alternative isoforms of the μ-opioid receptor are encoded by transcripts that originate from different initiation sites, and that genotype may serve as a proxy for isoform identity (20). Combined, however, the human and rhesus findings strongly suggest that the rhOPRM1 C77G and the hOPRM1 A118G SNPs, respectively, are functional with regard to NTX response in the respective species. Opioid signaling mediates in part alcohol reward (8), and these data therefore suggest that the respective variant may confer a higher degree of opioid response to alcohol, for example through elevated β-endorphin release.
The statistical interaction between genotype and treatment in our study reflects opposite directionality of the NTX effect in 77G carriers and subjects homozygous for the major 77C allele. This pattern parallels a recent study that examined family history of alcoholism as a moderator of NTX response under laboratory conditions, and found suppression of self-administration in family history positive subjects, but significantly increased self-administration following NTX treatment in family history negative participants (7). These parallel findings are consistent with OPRM1 A118G variation fully or in part carrying the role of family history for NTX responses.
In summary, we show here that carriers of rhOPRM1 77G, a functional equivalent of hOPRM1 118G, are selectively sensitive to suppression of alcohol preference by NTX treatment. A possible limitation of the findings is that they were obtained following brief duration of treatment in non-dependent animals that do not achieve high blood alcohol levels. This is unlikely to be a major limitation, because human laboratory studies under similar conditions have shown good predictive validity for clinical NTX effects [reviewed in (8)]. If the non-human primate laboratory model used here can be generalized to the same extent as the corresponding human paradigm, these data suggest that genotype at the corresponding human locus should be considered a key matching variable for NTX treatment.
Acknowledgments
Supported by the intramural research programs of the NIAAA and NICHD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Volpicelli JR, Alterman AI, Hayashida M, O'Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 2.O'Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence. A controlled study. Arch Gen Psychiatry. 1992;49:881–887. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- 3.Altshuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- 4.Bouza C, Angeles M, Munoz A, Amate JM. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction. 2004;99:811–828. doi: 10.1111/j.1360-0443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 5.Rubio G, Ponce G, Rodriguez-Jimenez R, Jimenez-Arriero MA, Hoenicka J, Palomo T. Clinical predictors of response to naltrexone in alcoholic patients: who benefits most from treatment with naltrexone? Alcohol Alcohol. 2005;40:227–233. doi: 10.1093/alcalc/agh151. [DOI] [PubMed] [Google Scholar]
- 6.King AC, Volpicelli JR, Frazer A, O'Brien CP. Effect of naltrexone on subjective alcohol response in subjects at high and low risk for future alcohol dependence. Psychopharmacology (Berl) 1997;129:15–22. doi: 10.1007/s002130050156. [DOI] [PubMed] [Google Scholar]
- 7.Krishnan-Sarin S, Krystal JH, Shi J, Pittman B, O'Malley SS. Family history of alcoholism influences naltrexone-induced reduction in alcohol drinking. Biol Psychiatry. 2007;62:694–697. doi: 10.1016/j.biopsych.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 8.Heilig M, Egli M. Pharmacological treatment of alcohol dependence: Target symptoms and target mechanisms. Pharm Therap. 2006;111:855–876. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, Gong J, Schluger J, Strong JA, Leal SM, Tischfield JA, Kreek MJ, Yu L. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci U S A. 1998;95:9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arias A, Feinn R, Kranzler HR. Association of an Asn40Asp (A118G) polymorphism in the mu-opioid receptor gene with substance dependence: A meta-analysis. Drug Alcohol Depend. 2005 doi: 10.1016/j.drugalcdep.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 11.Wand GS, McCaul M, Yang X, Reynolds J, Gotjen D, Lee S, Ali A. The mu-opioid receptor gene polymorphism (A118G) alters HPA axis activation induced by opioid receptor blockade. Neuropsychopharmacology. 2002;26:106–114. doi: 10.1016/S0893-133X(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 12.Ray LA, Hutchison KE. Effects of naltrexone on alcohol sensitivity and genetic moderators of medication response - A double-blind placebo-controlled study. Arch Gen Psychiatry. 2007;64:1069–1077. doi: 10.1001/archpsyc.64.9.1069. [DOI] [PubMed] [Google Scholar]
- 13.Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, O'Brien CP. A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology. 2003;28:1546–1552. doi: 10.1038/sj.npp.1300219. [DOI] [PubMed] [Google Scholar]
- 14.Anton RF, Oroszi G, O'Malley S, Couper D, Swift R, Pettinati H, Goldman D. An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch Gen Psychiatry. 2008;65:135–144. doi: 10.1001/archpsyc.65.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelernter J, Gueorguieva R, Kranzler HR, Zhang HP, Cramer J, Rosenheck R, Krystal JH. Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: Results from the VA cooperative study. Alcoholism-Clinical and Experimental Research. 2007;31:555–563. doi: 10.1111/j.1530-0277.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 16.Miller GM, Bendor J, Tiefenbacher S, Yang H, Novak MA, Madras BK. A mu-opioid receptor single nucleotide polymorphism in rhesus monkey: association with stress response and aggression. Mol Psychiatry. 2004;9:99–108. doi: 10.1038/sj.mp.4001378. [DOI] [PubMed] [Google Scholar]
- 17.Barr CS, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ, Higley JD, Heilig M. Association of a functional polymorphism in the mu-opioid receptor gene with alcohol response and consumption in male rhesus macaques. Arch Gen Psychiatry. 2007;64:369–376. doi: 10.1001/archpsyc.64.3.369. [DOI] [PubMed] [Google Scholar]
- 18.Zhang HP, Luo XG, Kranzler HR, Lappalainen J, Yang BZ, Krupitsky E, Zvartau E, Gelernter J. Association between two mu-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum Mol Genet. 2006;15:807–819. doi: 10.1093/hmg/ddl024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oroszi G, Anton RF, O'Malley S, Swift R, Pettinati H, Couper D, Yuan QP, Goldman D. OPRM1 Asn40Asp Predicts Response to Naltrexone Treatment: A Haplotype-Based Approach. Alcoholism-Clinical and Experimental Research. 2009;33:383–393. doi: 10.1111/j.1530-0277.2008.00846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shabalina SA, Zaykin DV, Gris P, Ogurtsov AY, Gauthier J, Shibata K, Tchivileva IE, Belfer I, Mishra B, Kiselycznyk C, Wallace MR, Staud R, Spiridonov NA, Max MB, Goldman D, Fillingim RB, Maixner W, Diatchenko L. Expansion of the human mu-opioid receptor gene architecture: novel functional variants. Hum Mol Genet. 2009;18:1037–1051. doi: 10.1093/hmg/ddn439. [DOI] [PMC free article] [PubMed] [Google Scholar]