

Abstract
Chronic exposure to ultraviolet radiation (UVR) is the major etiologic factor in the development of human skin cancers including squamous cell carcinoma (SCC). We have shown that PKCε transgenic mice on FVB/N background, which overexpress PKCε protein approximately 8-fold over endogenous levels in epidermis, exhibit about 3-fold more sensitivity than wild-type littermates to UVR-induced development of SCC (Cancer Research, 64, 7756, 2004). To determine whether it is PKCε and not the mouse genetic background, that determines susceptibility to UVR carcinogenesis, we cross-bred PKCε FVB/N transgenic mice with SKH-1 hairless mice to generate PKCε overexpressing SKH-1 hairless mice. To evaluate the susceptibility of PKCε SKH-1 hairless transgenic mice to UVR carcinogenesis, the mice were exposed to UVR (1–2 KJ/m2) three times weekly from a bank of six kodacel-filtered FS40 sunlamps. As compared to the wild-type hairless mice, PKCε overexpression in SKH-1 hairless mice decreased the latency (12 weeks) while increased the incidence (2-fold) and multiplicity (4-fold) of SCC. The SKH hairless transgenic mice were observed to be as sensitive as FVB/N transgenic mice to UVR-induced development of SCC and expression of proliferative markers (PCNA, Stat3 and ERK1/2). The results indicate that PKCε level dictates susceptibility, irrespective of genetic background, to UVR carcinogenesis.
Keywords: PKC, Stat3, SCC, Transgenic mice, Ultraviolet radiation
Introduction
Skin cancer accounts for half of all cancers diagnosed in the United States totaling over 1 million new cases every year (Anonymous, 2009). Squamous cell carcinoma (SCC) and basal cell carcinoma (BCC) are the most common forms of nonmelaoma skin cancer. SCC is the most aggressive form of nonmelanoma skin cancer and unlike basal cell carcinoma can metastasize (Goldman, 1998).
The most important risk factor for development of non-melanoma skin cancer is chronic exposure to UV radiation (UVR) in sunlight (Molho-Pessach et al. 2007, Cooper et al. 2007). The UV spectrum, part of the electromagnetic spectrum, lies between visible light and X-rays and is divided conventionally into three categories: UVA (315–400 nm), UVB (280–315 nm) and UVC (190–280 nm). Because stratospheric ozone absorbs most of the radiation below 310 nm, UVA and UVB components of sunlight are the most prominent and ubiquitous carcinogenic electromagnetic wavelengths in our natural environment (Wheeler et al., 2004).
UVR is a complete carcinogen, which both initiates and promotes carcinogenesis. UVB initiates photocarcinogenesis by directly damaging DNA (Marrot et al. 2008, Rass et al. 2008, Timares et al. 2008, de Gruijl et al. 2008) UVB-induced photoproducts include cyclobutane pyrimidine dimer (CPD), pyrimidine (6-4) pyrimidone dimer ([6-4]PD) and Dewar photoisomer of the (6-4)PD (Moriwaki et al. 2008). The CPD is the predominant photoproduct, accounting for 85% of the primary DNA lesions in UV-irradiated DNA (de Gruijl et al. 2001). The majority of the DNA lesions are removed by the nucleotide excision repair (Moiwaki et al. 2008, de Gruijl et al 2001). However, upon DNA replication, some cells acquire transition mutations (C → T) and tandem double mutations (CC → TT) arising at dipyrimidine sites (Berton et al. 1997, Brash et al. 1991). These mutations are frequently observed in UV-induced SCC in mice and humans (de Gruijl et al. 2008). Among a series of gene mutations (TP53, PITCH and oncogenes) that are associated with UV-induced skin cancer, C → T and CC → TT point mutations in the p53 gene are most frequent (Ziegler et al. 1994, Kanjilal et al. 1995) UVR can induce several types of epidermal injury including sunburn cell (apoptotic cell) formation (Ziegler et al. 1994, Kanjilal et al. 1995). The sunburn cells can be initiated by UV-induced DNA damage and subsequent induction of p53 protein. The p53-dependent apoptosis of UV-damaged normal cells (sunburn cells) is prevented due to p53 mutation. Thus, these mutated cells can clonally expand to form SCC following subsequent UVR exposures.
The tumor promotion component of UVR carcinogenesis, which involves clonal expansion of the initiated cells, is probably mediated by aberrant expression of genes altered during tumor initiation. UVR has been reported to alter the expression of genes regulating inflammation, cell growth and differentiation, and oncogenesis. Specific examples include upregulation of the expression of p21 (WAF1/C1P1) (Lu et al. 1999), p53 (Ziegler et al.1994), AP-1 activation (Cooper et al. 2007), ODC (Wheeler et al 2004), COX2 (Isoherranen et al 1999), TNFα, and a wide variety of cytokines and growth factors (Wheeler et al. 2004). UVR-induced initial signals linked to the development of skin cancer are not defined. We found that PKCε overexpression in epidermal cells of FVB/N mice sensitizes the skin to UVR-induced cutaneous damage and development of SCC.
Protein Kinase C (PKC), a family of phospholipid-dependent serine/threonine kinases, is not only the major intracellular receptor for the mouse skin tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) (Griner et al 2007, Mellor et al. 1998, Newton 2001, Mochly-Rosen et al 1998, Angel et al. 2003) but also is activated by a variety of stress factors including ultraviolet radiation (UVR) (Wheeler et al. 2004, Mellor et al. 1998, Mochly-Rosen et al. 1998) PKCε is among six isoforms (α, δ, ε, η, μ, ζ) expressed in the mouse skin(Mochly-Rosen et al. 1998). To determine the in vivo functional specificity of PKCε in mouse skin carcinogenesis, we generated PKCε transgenic mouse (FVB/N) lines 224 and 215 that overexpress approximately 8- and 18-fold respectively PKCε protein over endogenous levels in basal epidermal cells (Reddig et al. 2000, Jansen et al. 2001). PKCε transgenic mice were observed to be highly sensitive to the development of SCC elicited by the DMBA (100 nmol) - TPA (5 nmol) tumor promotion protocol (Reddig et al. 2000, Jansen et al. 2001). UVR exposure (1 kJ/m2 thrice weekly) induced irreparable skin damage in high PKCε overexpressing mouse line 215. However, the PKCε transgenic mouse line 224, when exposed to UVR (2 kJ/m2 thrice weekly), exhibited minimum cutaneous damage but increased SCC multiplicity by 3-fold and decreased tumor latency by 12 weeks (Wheeler et al. 2004). However it is unknown whether mouse genetic background contributes to the susceptibility of PKCε transgenic mice to UVR-induced development of SCC.
In this communication, we cross-bred PKCε FVB/N transgenic mice with SKH-1 hairless mice to generate PKCε overexpressing SKH-1 hairless mice to determine whether PKCε and not mouse genetic background, determines susceptibility to UVR carcinogenesis. We present that PKCε-overexpressing SKH-1 hairless mice are as sensitive as their FVB/N transgenic mice to UVR carcinogenesis.
Results
Generation of PKCε SKH-1 transgenic hairless mice
We have previously reported that PKCε transgenic FVB/N mice, which overexpress PKCε in the basal epidermal cells and the cells of the hair follicle, are highly sensitive to UVR-induced development of SCC (Wheeler et al., 2004). In the present study, we generated PKCε transgenic SKH-1 hairless mice to determine whether the mouse background contributes to the sensitivity of PKCε transgenic mice to UVR-induced carcinogenesis. In this experiment (Fig. 1), heterozygous (+/−) inbred FVB/N K14-T7PKCε mice were crossed for four generations to wildtype (−/−) out-crossed SKH1 hairless mice for transmission of the transgene and hairlessness. Four generations of crossing were sufficient as SKH-1 is an outcrossed line so genetic homogeneity within the mice is not necessary. PKCε transgenic SKH-1 hairless mice were phenotypically normal (Fig 1A). Histological examination of skin of untreated transgenic mice and wildtype mice revealed no histological differences (Fig. 1B). To determine epidermal PKCε expression level in transgenic and their wildtype mice, dorsal skin of untreated PKCε SKH-1 transgenic hairless mice and their wildtype littermates was excised and fixed in 10% formalin for the analysis of PKCε (Fig 1C). The dorsal skin of PKCε transgenic mice exhibited significantly more immunoreactivity throughout the epidermis compared to the wildtype dorsal skin with strong staining in the basal cells. The wild-type dorsal skin sample exhibited light immunoreactivity to the anti-PKCε antibody throughout the epidermis (Fig. 1C).
Fig. 1. Generation of K14-T7PKCε transgenic mice on SKH-1 background.
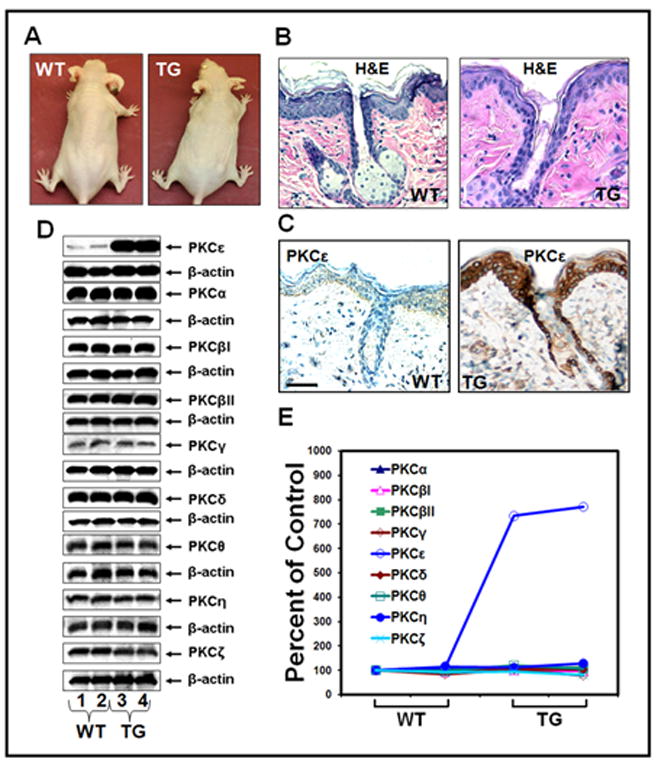
Heterozygous (+/−) FVB/N K14-T7PKCε mice were crossed for four generations to wildtype (−/−) outcrossed SKH-1 hairless mice for transmission of the transgene and hairlessness. a: Phenotype of female SKH-1K14-T7PKCε transgenic mice. Shown are the photographs of adult SKH-1 hairless PKCε transgenic mouse and its wildtype littermate at 11 weeks of age. b: Skin histology. The dorsal skin from a wildtype and PKCε SKH-1 hairless transgenic mouse was fixed in 10% neutral buffered formalin and embedded in paraffin, and sections (4μm) were stained with hematoxylin and eosin. c: Immunohistochemical staining for PKCε expression, scale bars = 50 μm. d: Epidermal PKC isoform expression profiles in PKCε SKH-1 hairless transgenic mice and their wildtype littermates; results representative of three replicates. e: The quantification of proteins (normalized to β-actin) was done by densitometry analysis using Total Lab Nonlinear Dynamics Image Analysis Software (Nonlinear, USA).
The possibility was explored that PKCε may cross-talk to other PKC isoforms by modulating their levels and their associated signals. In this experiment (Fig. 1D and E), we determined the levels of other PKC isoforms in the epidermis of PKCε transgenic SKH-1 hairless mice to examine whether the elevated PKCε levels in the epidermis altered the level of expression of other PKC isoforms. As compared to wild-type SKH-1 hairless mice, PKCε transgenic SKH-1 hairless mice elicited about 8-fold increase in PKCε expression level. However, there was no change in other PKC isoform expression levels between wildtype and transgenic mice (Fig. 1D and E).
Susceptibility of SKH-1 PKCε transgenic mice to UVR-induced development of SCC
PKCε transgenic SKH-1 hairless mice and their wildtype littermates (male and female) were treated three times (Monday, Wednesday, Friday) weekly with 2kJ/m2 UVR. After 7 weeks of UVR exposure, SKH-1 PKCε transgenic mice had severe cutaneous damage resulting in blistering, peeling, sloughing of the skin and the UVR dose was reduced to 1kJ/m2 for the remainder of the study. PKCε overexpression in SKH-1 hairless mice reduced the latency for SCC (Fig 2). The first carcinoma in PKCε transgenic SKH-1 hairless males and females mice appeared after 9 and 15 weeks of UVR exposure respectively as compared to 20 and 17 weeks in wildtype male and females littermates respectively. Carcinoma multiplicity in male PKCε transgenic mice was 1.18 after 23 weeks of exposure, compared to 1.0 in wildtype. Female PKCε transgenic mice had a carcinoma multiplicity of 2.57 compared to 1.6 in wildtype. Statistical analysis for determining the difference in multiplicities from male and female mice using a Poisson model for genotype and sex, found that males had a p-value of p=0.04 while females had a p-value of p=0.007. Carcinoma incidence in male PKCε SKH-1 transgenic and wildtype mice was 57.8% and 33.3% respectively (Fig 2B). Papilloma burden in male transgenic and wildtype mice was 1.0 and 2.0 respectively. Carcinoma incidence in female PKCε transgenic and wildtype mice was 53.8% and 20.0% respectively (Fig 2C). Papilloma burden in female transgenic and wildtype mice was 1.0 compared to 2.6 in respectively. Carcinoma incidence was first analyzed for statistical significance using the Cox proportional hazards model with pvalues for male mice being p=0.13 and female mice p=0.007. There was no significant gender difference with either the Poisson model (p=0.86) or Cox proportional hazards model (p=0.46). Transgenic mice are clearly more sensitive than wildtype littermates to UVR induced carcinogenesis. This sensitivity led to a massive decrease (~ 3 months) in latency and a significant increase in multiplicity.
Fig. 2. Susceptibility of PKCε transgenic SKH-1 hairless mice to the development of SCC by UVR.
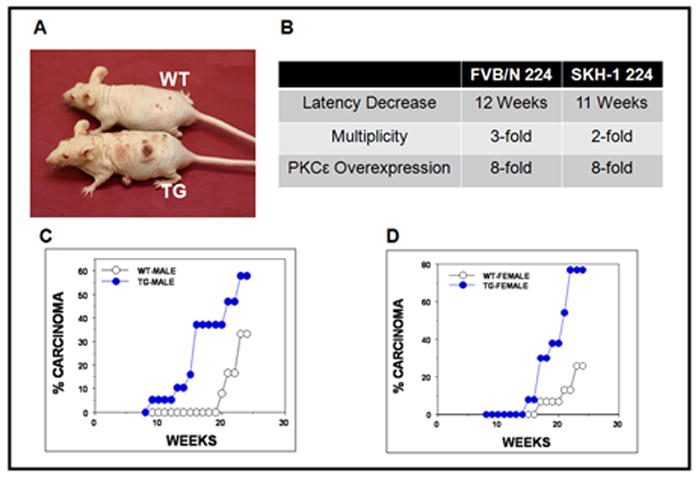
Male and female PKCε transgenic and wildtype mice at 11 weeks of age, mice were exposed to UVR (1–2kJ/m2) three times weekly from a bank of six Kodacel-filtered FS40 sunlamps. There were 15 mice per group. Carcinomas were recorded as downward invading lesions, which were confirmed histologically. The carcinoma data is expressed as the percentage of effectual total. Carcinoma incidence analyzed using Cox proportional hazards model. a: Representative photograph of a wildtype and PKCε transgenic SKH-1 mouse after 20 weeks of UVR exposure b: Similarity between FVB/N PKCε line 224 and SKH-1 PKCε line 224, comparison of decrease in carcinoma latency, increase in carcinoma multiplicity and PKCε overexpression. c: Comparison of male wildtype and PKCε transgenic mice carcinoma incidence. Carcinoma incidence in male mice was not found to be statistically different, p=0.13. d: Comparison of female wildtype and PKCε transgenic mice carcinoma incidence. Carcinoma incidence in female mice was found to be statistically different p=0.007.
Overexpression of PKCε in mouse epidermis increases sensitivity of skin to UVR-mediated phosphorylation of Stat3, ERK1/2 and PI3K
The effects of epidermal PKCε overexpression in SKH-1 hairless mice on UVR-induced activation of cell survival pathways (PI3K, MAPK and Stat3) were determined. In this experiment (Fig. 3), female PKCε transgenic SKH-1 hairless mice and their wild-type littermates were exposed to a single UVR (2 kJ/m2) dose. Mice were sacrificed at 1, 3, and 6h post UVR treatment. In wildtype mice, UVR treatment elicited rapid and transient increase in the expression level of PI3K (p110), PI3K (p85), pERK1/2 (Fig. 3A and C) and phosphorylated Stat3 at both Tyr705 and Ser727 (Fig. 3B and D). However, UVR treatment in PKCε transgenic SKH-1 hairless mice led to persistent activation of Stat3 phosphorylation at both Tyr705 and Ser727 (Fig. 3B and D), and pERK1/2 and PI3K (Fig. 3A and C).
Fig. 3. UVR-induced phosphorylation of PI3K, ERK1/2 and Stat3 in PKCε SKH-1 hairless transgenic mice and their wildtype littermates.
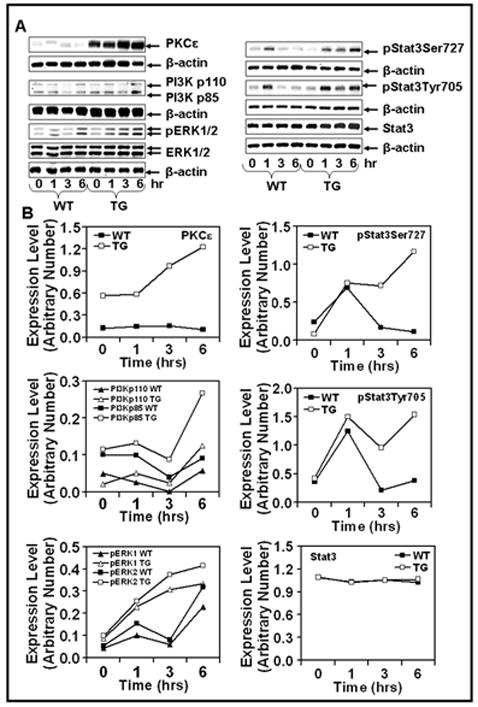
Female PKCε transgenic SKH-1 hairless mice (TG) and their wildtype littermates (WT) (3 mice per group) were exposed to a single UVR dose (1kJ/m2). The mice were sacrificed at 1, 3, and 6 hrs post UVR exposure. The mouse epidermis was scraped off and total lysate was prepared. Total epidermal lysate from three mice was pooled for the western blot analysis. a and b: The epidermal lysate (25μg protein) was subjected to SDS-PAGE followed by immunoblot analysis using indicated antibodies. Equal loading was confirmed by stripping and reprobing the blots with β-actin antibody. c and d: The quantification of proteins (normalized to β-actin) was done by densitometry analysis using Total Lab Nonlinear Dynamics Image Analysis Software (Nonlinear, USA).
SCC developed in PKCε transgenic SKH-1 hairless mice elicit elevated nuclear localization of PCNA and Stat3
In this experiment (Fig 4), SCC from PKCε transgenic SKH-1 hairless mice and their wild-type littermates (Fig 2) were excised and fixed in 10% neutral buffered formalin. Sections of SCC from PKCε transgenic SKH-1 hairless mice and their wild-type littermates were processed for immunohistochemistry of PKCε, Stat3 and PCNA (Fig 4B). SCC from PKCε SKH-1 transgenic mice showed a significant increase in PKCε, nuclear PCNA (Fig 4C) and nuclear Stat3 (Fig 4D) staining compared to wild-type mice (*=p<0.05).
Fig. 4. Immunohistochemistry of PKCε, Stat3, and PCNA in SCC.
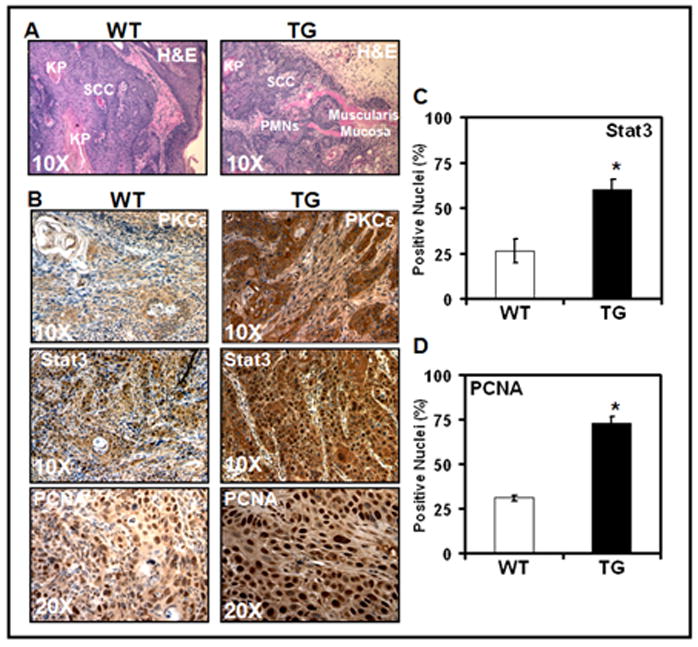
PKCε transgenic hairless mice (TG) and their wildtype littermates (WT) were exposed to UVR, as described in the legend to Fig 2 until development of SCC. SCC was excised promptly after euthanasia, placed immediately in 10% neutral buffered formalin, fixed for 1h in formalin, then transferred to PBS (pH7.4) and embedded in paraffin. Section of 4μm thickness was cut for H&E staining and immunohistochemical study. Each value is the mean +/− SE from an average of 10 fields from 3 different mice was used to calculate the students t-test. a: H & E Staining (Scale Bar = 50μm) b: Immunoreactivity of PKCε (Scale Bar = 50μm), Stat3 (Scale Bar = 50μm) and PCNA (Scale Bar = 25μm). c and d: Quantification of Stat3 and PCNA nuclear staining. Students t-test were performed to analyze nuclear staining differences (*p<.05).
Discussion
PKCε transgenic FVB/N mice, which overexpress PKCε protein 8-fold over endogenous levels in basal epidermal cells and cell of the hair follicle, are 3-fold more sensitive than wildtype to the development of SCC by repeated exposures to UVR (Wheeler et al., 2004). To determine whether it is PKCε and not the mouse genetic background, that determines susceptibility to UVR carcinogenesis, we cross-bred PKCε FVB/N transgenic mice with SKH-1 hairless mice to generate PKCε overexpressing SKH-1 hairless mice. We now present that PKCε overexpressing SKH-1 hairless mice are as sensitive as FVB/N PKCε transgenic mice to UVR-induced development of SCC (Fig 2B).
Genetic differences in susceptibility to two-stage skin carcinogenesis have been shown to be at the level of tumor promotion by TPA (Angel et al. 2003). PKCε is among the major intracellular receptor for the mouse skin tumor promoter TPA (Griner et al. 2007, Mellor et. al.1998) and is activated by a variety of stress factors including ultraviolet radiation (UVR) (Griner et al. 2003, Wheeler et al. 2004). Furthermore, genetic mouse background has been has been shown to be a major determinant of conversion of benign papillomas to malignant SCC (Woodworth et al 2004). FVB/N mice are moderately susceptible to papilloma formation but are highly prone to undergo malignant conversion (Reddig et al. 2000). PKCε overexpression in SKH-1 hairless mice, like FVB/N mice, suppressed papilloma formation but enhanced SCC development. Although, confirmation should await further experimentation involving crosses with inbred mouse strains (BALB/c, C57BL/6), the results presented imply that PKCε appears to be a highly penetrant susceptible gene.
We have previously reported generation and sensitivity of PKCε transgenic FVB/N mouse lines to UVR (Wheeler et al., 2004). PKCε transgenic line 215, which overexpresses about 18-fold PKCε protein over endogenous levels, elicited severe cutaneous damage after exposure to UVR (Wheeler et al., 2004). UVR-induced skin damage in PKCε transgenic line 215 after exposure to UVR (either 1 or 2 kJ/m2) was extensive and irreparable, and the experiment could not be continued until the appearance of carcinomas. However, the lower PKCε-overexpressing mice (line 224) tolerated three times weekly UVR exposures (2kJ/m2) for 38 weeks and, had 3-fold increased SCC multiplicity as well as decreased tumor latency by 12 weeks when compared to wildtype. PKCε overexpression in SKH-1 yielded similar results upon repeated UVR exposure.
UVR-induced activated PKCε mediates two potential signals which lead to inhibition of apoptosis and increased proliferation of preneoplastic cells. In this context, it is noteworthy that UVR-induced percentage of sunburn cells in PKCε transgenic mice was significantly lower than wildtype littermates (Wheeler et al., 2004). PKCε transgenic mice were more sensitive than their wildtype littermates to UVR-induced epidermal proliferation markers PCNA, ornithine decarboxylase, and constitutively activated Stat3 (Wheeler et al., 2004, Jansen et al., 2001, Aziz et al., 2007). Consistent with our previous report, PKCε SKH-1 transgenic mice elicited constitutive activation of Stat3 in both response to acute UVR exposure and in SCC developed during chronic UVR exposure. SCC from PKCε transgenic SKH-1 mice also had increased expression of nuclear PCNA (Fig 4).
PI3K/AKT and MAPK pathways are involved in regulation of survival pathways in tumor cells. Both PI3K/AKT and MAPK are activated during UVR carcinogenesis (Einspahr et al., 2008). However, the mechanism by which UVR treatment mediates activation of survival pathways (PI3K/AKT and MAPK) is unclear. PKCε overexpression in SKH-1 hairless mice accompanied increased expression of phosphorylated PI3K and MAPK (ERK1/2) implying that PKCε may impart sensitivity to UVR carcinogenesis via crosstalk to PI3K and MAPK pathways.
UVR treatment resulted in increased expression of PKCε (Fig 3B). This UVR-increased PKCε expression may be the result of the effect of UVR treatment on PKCε synthesis. It is noteworthy that PKCε levels are constant during chronic UVR treatment (data not shown) and in SCC developed by UVR treatment (Fig 4). These results (Fig 4) are in contrast to our previous findings with SCC developed in PKCε mice by DMBA-TPA protocol (Reddig et al., 2000). A possible explanation for this difference in PKCε expression level in SCC may be explained by a difference in antigen retrieval protocol for immunhistochemical localization.
In summary, PKCε overexpressing SKH-1 hairless mice, like FVB/N, are more sensitive than their wildtype littermates to induction of SCC by UVR treatment. UVR-induced activated PKCε cross-talks with survival pathways (PI3K, MAPK) and regulates constitutive activation of Stat3, critical factor involved in UVR carcinogenesis (Kim et al., 2009). PKCε transgenic SKH-1 hairless mice may provide a useful model to investigate UVR carcinogenesis.
Materials and Methods
Antibodies
The antibodies and sources of the antibodies used in this study were as follows: PKCε, Stat3, phosphorylated Stat3Tyr705 (pStat3Tyr705), phosphatidylinositol 3-kinase (PI3K; p85), PI3K (p110), β-actin from Santa Cruz Biotechnologies (Santa Cruz CA); and phosphorylated Stat3Ser727 (pStat3Ser727; BD Biosciences). Blocking peptides for PKCε and Stat3 were obtained from Santa Cruz Biotechnology.
The Generation of K14-T7PKCε Transgenic Mice on SKH-1 background
Transgenic mice overexpressing T7 epitope tagged Protein Kinase C epsilon (T7-PKCε), under the control of the human keratin 14 (K14) promoter, using the inbred FVB/N background were created as described previously (Reddig et al., 2000). Heterozygous (+/−) inbred FVB/N K14-T7PKCε mice were crossed for four generations to wildtype (−/−) out-crossed SKH1 hairless mice from Charles River Laboratories (Wilmington MA) for transmission of the transgene and hairlessness. Four generations of crossing were sufficient as SKH-1 is an outcrossed line so genetic homogeneity within the mice is not necessary. SKH-1 mice were maintained as an outcrossed line. Thus the difference between two “pure” SKH-1 mice is as much as the difference between a “pure” SKH-1 and a SKH-1//FVB/N cross. All animal experiments were performed in accordance with approval from IACUC at the University of Wisconsin-Madison.
UVR Treatment of Mice
Mice were housed in groups of two to three in plastic bottom cages in light-, humidity-, and temperature-controlled rooms; food and water were available ad libitum. The animals were kept in a normal rhythm of 12-h-light and 12-h-dark periods. The UVR source was Kodacel-filtered FS-40 sun lamps (approximately 60% UVB and 40% UVA). UVR dose was measured weekly using UVX-radiometer. Mice were used for experimentation at 11 to 14 weeks of age. Mice were exposed to UVR (1–2kJ/m2) three times weekly (Monday, Wednesday, and Friday). Tumor multiplicity was observed weekly and mouse weight was determined every other week. Carcinomas were recorded grossly as downward-invading lesions, which were confirmed histologically.
Histologic Analysis
Mouse skin was excised promptly after euthanasia and immediately placed in 10% neutral-buffered formalin, fixed for 24 hours in formalin, transferred to PBS (pH 7.4) and then embedded in paraffin. Four-μm sections were cut for hematoxylin, eosin staining and immunohistochemical study.
Immunohistochemistry of PKCε, Stat3 and PCNA
PKCε transgenic mice and their wildtype littermates were exposed to UVR (1–2 kJ/m2) three times weekly (Monday, Wednesday, and Friday), for 26 weeks. SCC specimens were fixed in 10% neutral-buffered formalin for 24 hours and embedded in paraffin. Four-μm-thick sections were cut for staining. Briefly, slides were first treated for antigen retrieval by incubating first in 1M citric acid (pH 6.0) at 95°C for 20 minutes and in 0.1M Tris-EDTA (pH 8.0) for 20 minutes. The slides were washed with PBS with 0.5% tween and incubated overnight at 4°C with the primary antibody. Subsequent incubation steps were performed in a moist chamber at room temperature. After intermediate washing steps in Tris-buffered saline (pH 7.4), the sections were incubated with biotin-labeled rabbit antimouse immunoglobulin G for 15 minutes at room temperature and then with streptavidin-peroxidase complexes for 15 minutes at room temperature. Visualization was performed using diaminobenzidine as a substrate for the peroxidase reaction. Slides were transferred into tap water and counterstained with hematoxylin for 4 minutes. Every experiment included a control that contained no primary antibody but preimmune rabbit serum. Antibody specificity was observed using blocking peptides. No immunoreactivity was observed using these controls. Specimens were analyzed using an Olympus BX 51 microscope.
For quantitation of PCNA and Stat3 positive staining cells, ten random fields were selected for each mouse. The number of cells demonstrating positive labeling and the total number of cells counted were recorded. An average percentage was then calculated based on the total number of cells and the number of positive staining cells from each set of 10 fields counted. Results are expressed as mean of percentages ± SE.
Western Blot Analysis
Mouseskin was excised and scraped to remove subcutaneous fat. The epidermis was scraped off on a ice-cold glass plate, homogenized in the lysis buffer [50 mmol/L HEPES, 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1.5 mmol/L MgCl2, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 200 μmol/L Na3VO4, 200 μmol/L NaF, and 1 mmol/L EGTA (final pH 7.5)]. The homogenate was centrifuged at 14,000 × g for 30 minutes at 4°C. Twenty micrograms of cell lysate were fractionated on 10% Criterion precast SDS-polyacrylamide gel (Bio-Rad Laboratories, Hercules CA). The protein was transferred to 0.45 μm Hybond-P polyvinylidene difluoride (PVDF)transfer membrane (Amersham Life Sciences, Piscataway NJ). The membrane was then incubated with the indicated antibody followed by a horseradish peroxidase secondary antibody (Santa Cruz Biotechnology), and the detection signal was developed with Amersham’s enhanced chemiluminescence reagent and autoradiography using BioMax film (Kodak, Memphis TN). The Western blots were quantitated by densitometric analysis using Totallab Nonlinear Dynamic Image analysis software (Nonlinear USA, Inc., Durham, NC).
Statistical Methods
The primary endpoint of interest was carcinoma. Those mice that did not develop carcinoma were censored at the last point that data was available for them. Differences in time to event, by gender and genotype and the interaction of gender and genotype, were examined visually using Kaplan-Meier method and tested with Cox proportional hazards models. Also of interest was tumor multiplicity. Differences in tumor multiplicity by gender, genotype, and interaction were tested with Poisson regression models. Computations were performed with R software. (R Development Core Team, 2007). A students ttest was also performed for the comparison of wildtype and transgenic nuclear localization of Stat3 and PCNA.
Acknowledgments
Grant Support: NIH grant number CA 35368 and Molecular and Environmental Toxicology Center predoctoral training grant T32ES007015, for Jordan M Sand. We would like to acknowledge Emily Siebers for breeding help.
Abbreviations
- PKC
Protein Kinase C
- UVR
Ultraviolet Radiation
- TPA
12-O-tetradecanoylphorbol-13-acetate
- DMBA
7, 12-Dimethylbenz[a]anthracene
- K14
human keratin 14 promoter
- SCC
squamous cell carcinoma
- STAT
signal transducers and activators of transcription
- PCNA
proliferating cell nuclear antigen
- ERK1/2
extracellular signal regulated kinase 1/2
- MAPK
mitogen activated protein kinase
- PI3K
phosphatidylinositol-3-kinase
Footnotes
Conflict of Interest
There is no conflict of interest for any authors.
References
- Angel JM, Caballero M, DiGiovanni J. Identification of novel genetic loci contributing to 12-O-tetradecanoylphorbol-13-acetate skin tumor promotion susceptibility in DBA/2 and C57BL/6 mice. Cancer Res. 2003;63:2747–51. [PubMed] [Google Scholar]
- Berton TR, Mitchell DL, Fischer SM, Locniskar MF. Epidermal proliferation but not quantity of DNA photodamage is correlated with UV-induced mouse skin carcinogenesis. J Invest Dermatol. 1997;109:340–7. doi: 10.1111/1523-1747.ep12335984. [DOI] [PubMed] [Google Scholar]
- Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, et al. A role for sunlight in skin cancer:UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88:10124–8. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ, Bowden GT. Ultraviolet B regulation of transcription factor families: roles of nuclear factor-kappa B (NF-kappaB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr Cancer Drug Targets. 2007;7:325–34. doi: 10.2174/156800907780809714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gruijl FR, Rebel H. Early events in UV carcinogenesis DNA damage, target cells and mutant p53 foci. Photochem Photobiol. 2008;84:382–7. doi: 10.1111/j.1751-1097.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- de Gruijl FR, van Kranen HJ, Mllenders LH. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J Photochem Photobiol B. 2001;63:19–27. doi: 10.1016/s1011-1344(01)00199-3. [DOI] [PubMed] [Google Scholar]
- Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–94. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- Isoherranen K, Punnonen K, Jansen C, Uotila P. Ultraviolet irradiation induces cyclooxygenase-2 expression in keratinocytes. Br J Dermatol. 1999;140:1017–22. doi: 10.1046/j.1365-2133.1999.02897.x. [DOI] [PubMed] [Google Scholar]
- Jansen AP, Verwiebe EG, Dreckschmidt NE, Wheeler DL, Oberley TD, Verma AK. Protein kinase C- transgenic mice: a unique model for metastatic squamous cell carcinoma. Cancer Res. 2001;61:808–12. [PubMed] [Google Scholar]
- Jansen AP, Ness KJ, Oberley TD, Verma AK. Relation of the induction of epidermal ornithine decarboxylase and hyperplasia to the different skin tumor promotion susceptibilities of protein kinase C –alpha, -delta, and – epsilon transgenic mice. Int J Cancer. 2001;93:635–43. doi: 10.1002/ijc.1395. [DOI] [PubMed] [Google Scholar]
- Kanjilal S, Strom SS, Clayman GL, Weber RS, el-Naggar AK, Kapur V, et al. p53 mutations in nonmelanoma skin cancer of the head and neck:molecular evidence for field cancerization. Cancer Res. 1995;55:3604–9. [PubMed] [Google Scholar]
- Lu YP, Lou YR, Yen P, Mitchell D, Huang MT, Conney AH. Time course for early adaptive responses to ultraviolet B light in the epidermis of SKH-1 mice. Cancer Res. 1999;59:4591–4602. [PubMed] [Google Scholar]
- Marrot L, Meunier JR. Skin DNA photodamage and its biological consequences. J Am Acad Dermatol. 2008;58:S139–48. doi: 10.1016/j.jaad.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochly-Rosen D, Kauvar LM. Modulating protein kinase C signal transduction. Adv Pharmacol. 1998;44:91–145. doi: 10.1016/s1054-3589(08)60126-x. [DOI] [PubMed] [Google Scholar]
- Molho-Pessach V, Lotem M. Ultraviolet radiation and cutaneous carcinogenesis. Curr Probl Dermatol. 2007;35:14–27. doi: 10.1159/000106407. [DOI] [PubMed] [Google Scholar]
- Moriwaki S, Takahashi Y. Photaging and DNA repair. J Dermatol Sci. 2008;50:169–76. doi: 10.1016/j.jdermsci.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–64. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- R. Core Development Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2007. URL http://www.R-project.org. [Google Scholar]
- Rass K, Reichrath J. UV damage and DNA repair in malignant and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:162–78. doi: 10.1007/978-0-387-77574-6_13. [DOI] [PubMed] [Google Scholar]
- Reddig PJ, Dreckschmidt NE, Zou J, Bourguignon SE, Oberley TD, Verma AK. Transgenic mice overexpressing protein kinase C epsilon in their epidermis exhibit reduced papilloma burden but enhanced carcinoma formation after tumor promotion. Cancer Res. 2000;60:595–602. [PubMed] [Google Scholar]
- Timares L, Katiyar SK, Elments CA. DNA damage, apoptosis and langerhans cell Activators of UV_induced immune tolerance. Photochem Photobiol. 2008;84:422–36. doi: 10.1111/j.1751-1097.2007.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Martin KE, Ness KJ, Dreckschmidt NE, Wartman M, Ananthaswamy HN. Protein kinase C is an endogenous photosensitizer that enhances ultraviolet radiation-induced cutaneous damage and development of squamous cell carcinomas. Cancer Res. 2004;64:7756–65. doi: 10.1158/0008-5472.CAN-04-1881. [DOI] [PubMed] [Google Scholar]
- Woodworth CD, Michael E, Smith L, Vijayachandra K, Glick A, Hennings H, et al. Strain-dependent differences in malignant conversion of mouse skin tumors in an inherent property of the epidermal keratinocyte. Carcinogenesis. 2004;25:1771–8. doi: 10.1093/carcin/bgh170. [DOI] [PubMed] [Google Scholar]
- Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–6. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]