

Abstract
Here we report on a highly sensitive potentiometric detection of DNA hybridization. The new assay uses a low-volume solid-contact silver ion-selective electrode (Ag+-ISE) to monitor the depletion of silver ions induced by the biocatalytic reaction of the alkaline-phosphatase enzyme tag. The resultant potential change of the Ag+-ISE thus serves as the hybridization signal. Factors affecting the potentiometric hybridization response have been optimized to offer a detection limit of 50 fM (0.2 amol) DNA target. The new potentiometric assay was applied successfully to the monitoring of the 16S rRNA of E. coli pathogenic bacteria to achieve a low detection limit of 10 CFU in the 4 μL sample. Such potentiometric transduction of biocatalytically-induced metallization processess holds great promise for monitoring various bioaffinity assays involving common enzyme tags.
Introduction
Electrochemical devices have received considerable attention recently in the development of sequence-specific DNA hybridization biosensors.1-3 Such devices offer elegant and effective routes for interfacing the DNA-recognition and signal-transduction elements, and are uniquely qualified for meeting the size, cost, and power requirements of point-of-care DNA diagnostics. Enzyme labels, such as alkaline phosphatase (ALP) or horseradish peroxidase (HRP) have been widely used for such bioelectronic detection of DNA hybridization in connection to amperometric monitoring of the biocatalytic reaction product.4,5
The present article describes the use of an ion-selective electrode (ISE) transducer for highly sensitive potentiometric detection of enzyme-linked DNA hybridization connected to a biocatalytic metallization. Recent advances in direct potentiometry6,7 have led to powerful solid-contact ISE sensing devices, with remarkable selectivity and sensitivity (down to the sub-nanomolar level). Since the signal or detection limit of potentiometric transducers are not expected to deteriorate upon reducing the sample volume it is possible to use miniaturized ISEs for detecting femtomole amounts of ions in microvolume samples. Despite of these latest advances, the low-cost and simplicity of potentiometric ISEs and the inherent miniaturization and portability of the supporting instrumentation, such devices have rarely been applied for bioelectronic detection of DNA hybridization, with the exception of a recent work involving cadmium-sulfide nanocrystal labels.8 Such procedure involved the use of a Cd2+-ISE for detecting the hybridization event and required the dissolution of the captured nanoparticle tracer prior to the potentiometric detection, in a manner similar to analogous to the stripping-voltammetric measurements of nanoparticle tags.9,10
The new potentiometric DNA hybridization protocol, illustrated in Figure 1, involves the formation of the nucleic-acid sandwich, capturing of an ALP tracer and reduction of silver ions by the p-aminophenol (p-AP) product of the enzymatic reaction. The silver ISE transducer (shown in Figure 2B) offers a highly sensitive and rapid measurement of the depleted silver ion, with the decreased potential response serving as the hybridization signal. An analogous stripping-voltammteric detection of ALP-induced biometallization process was described recently by the groups of Kwak11 and Costa Garcia.12 Potentiometric microsensors were shown recently to be very attractive for probing in real-time biocatalytic metallization processes, as was illustrated for the NADH/ADH-mediated reduction of copper.13 Such direct and sensitive monitoring of biocatalytic metallization processes indicates great promise for potentiometric transduction of bioaffinity interactions in connection to common enzyme tags generating a reducing agent. The new silver-based biometallization potentiometric strategy represents an attractive alternative to the use of metal nanoparticle tags for potentiometric measurement of DNA hybridization,8 as it obviates the need of dissolving the nanocrystal tracer. Separating the hybridization and detection cells (Figure 1) prevents also the common problem of silver deposition onto the DNA and minimizes related background contributions. Such procedure thus holds great promise for detecting DNA hybridization, as illustrated below for highly sensitive potentiometric measurements of the E. coli bacterial pathogen.
Figure 1.
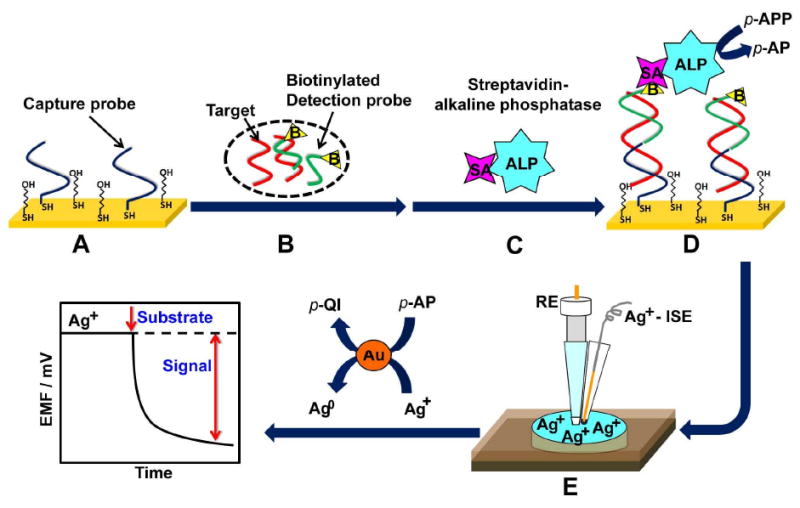
Representation of the potentiometric detection of DNA hybridization. (A) Formation of the mixed thiol monolayer (thiolated DNA capture probe and MCH) on the gold substrate; (B) hybridization of the target DNA/biotinylated detection-probe mixture with the surface capture probe; (C) binding of the SA-ALP enzyme; (D) addition of the ALP substrate to initiate the enzymatic reaction and (E) potentiometric detection of changes in the level of the silver ion upon adding an aliquot of the enzymatic reaction mixture to the Ag+-ISE cell.
Figure 2.

Photographs of the (A) 16-sensor electrochemical gold array for the DNA hybridization and of the (B) potentiometric detection setup. The latter involves the micropipette-based Ag+-ISE and the Ag/AgCl reference electrode (RE) along with a home-made double junction compartment, immersed in a microcell containing the silver ion solution, Au NPs and a magnetic stirring bar.
Experimental Section
Materials
6-mercapto-1-hexanol (MCH), Trizma hydrochloride (Tris-HCl), ethylenediaminetetraacetic acid, sodium chloride, sodium hydroxide, sodium phosphate monobasic, sodium phosphate dibasic, potassium chloride, potassium phosphate dibasic, potassium phosphate monobasic, bovine serum albumin, streptavidin-alkaline phosphatase (SA-ALP), p-aminophenol (p-AP), glycine, gold nanoparticles (Au NPs) (10 nm diam.), and silver nitrate were purchased from Sigma-Aldrich (St. Louis, MO). The blocking agent casein was obtained from Pierce (Rockford). p-aminophenyl phosphate monosodium salt hydrate (p-APP) was purchased from Biosynth International Inc. (Switzerland). Magnesium sulfate was received from EMD Chemicals Inc., while potassium nitrate was obtained from Fisher Scientific (Fair Lawn, NJ).
The buffer solutions used in this study were as follows: The DNA immobilization buffer (IB) was 10 mM Tris-HCl, 1 mM ethylenediaminetetraacetic acid, and 0.3 M NaCl (pH 8.0). The hybridization buffer (HB) was a 1M phosphate buffer solution containing 0.78 M K2HPO4, 0.27 M NaH2PO4 and 2.5% bovine serum albumin (pH 7.2). The binding buffer (BB) for associating with SA-ALP consisted of 0.14 M NaCl, 0.003 M KCl, 0.002 M KH2PO4, 0.01 M Na2HPO4 and 0.5% casein (pH 7.2). Glycine (50 mM, pH 9.0) was used as the potentiometric detection buffer (DB). Solutions of 0.5 μM p-AP and 0.2 mM p-APP were prepared daily in the DB containing 10 mM KNO3 and 5 mM MgSO4 and were stored at 4 °C.
All synthetic oligonucleotides were purchased from Thermo Fisher Scientific (Ulm, Germany). The sequences of the thiolated capture probe, biotinylated detection probe, and of the complementary target are listed in Table 1. The E. coli pathogen isolates were obtained from the University of California-Los Angeles (UCLA) medicine department. One milliliter of bacteria in Luria broth (containing 2.25×107 CFU, estimated from optical density measurements at 600 nm) was centrifuged at 10,000g for 5 min. The supernatant was discarded, and the bacteria left in the centrifuge tube were stored at -80 °C to prepare the bacterial isolate pellets.
Table 1. Sequence of Oligonucleotides Employed in This Work.
| Oligonucleotide | Sequence |
|---|---|
| Thiolated capture probe | 5′-Thiol-TAT TAA CTT TAC TCC-3′ |
| Biotinylated detection probe | 5′-CTT CCT CCC CGC TGA-Biotin-3′ |
| Target probe | 5′-TCA GCG GGG AGG AAG GGA GTA AAG TTA ATA-3′ |
The following chemicals were used for preparing the Ag+-ISE: The ionophores o-xylylenebis(N,N-diisobutyldithiocarbamate) (copper(II) ionophore(I)), sodium tetrakis[3,5-bis-(trifluoromethyl)phenyl]borate (Na-TFPB) and lipophilic salt tetradodecylammonium tetrakis(4-chlorophenyl)-borate (ETH 500) were purchased in Selectophore® or puriss grade from Fluka (Buchs, Switzerland). The methylene-chloride solvent was obtained from Fisher (Pittsburgh, PA), poly(3-octylthiophene) (POT) was synthesized following the procedure of Jarvinen et. al14 and was purified according to the patent application.15 The synthesis of methyl methacrylate-decyl methacrylate (MMA-DMA) copolymer matrix was described previously.16 All stock and buffer solutions were prepared using double-deionized water (18.2 MΩ cm).
Preparation of Ag+-ISE
Membranes
The membrane for the Ag+-ISE was prepared by dissolving a total weight of 50 mg components including copper(II) ionophore (I) (17 mmol kg-1), Na-TFPB (7 mmol kg-1), ETH 500 (12 mmol kg-1), and copolymer MMA-DMA (∼98%) in 0.8 mL of methylene chloride, in accordance to earlier studies17,18 with slight modifications. The membrane ‘cocktail’ was degassed by sonication for 1 min prior to coating the microelectrodes.
Potentiometric Microelectrode
The solid-contact Ag+-ISE was prepared by using a 2 cm long gold wire (200 μm diam.) that was soldered to a copper wire for electrical contact. Before use, the gold wires were thoroughly cleaned with diluted sulfuric acid, rinsed with water, then acetone and were left in chloroform for 10 min. The POT solution was applied by dip coating along the length of the gold wire at least three times until the color of the wire turned black. After the gold wires were fully covered with POT they were allowed to dry. Subsequently, the wires were inserted into a 10 μL polypropylene pipette tip, reaching the end of the micropipette tip. Finally, the membrane ‘cocktail’ was applied to the tip of the POT-coated wire for three times at 15 min intervals and was allowed to dry for at least 1 hour. The resulting microelectrodes were conditioned prior to use for one day in a 10-3 M AgNO3 solution, and for another day in a 10-9 M AgNO3.18
Formation of Oligonucleotide Probe at the Gold Surface
DNA hybridization was performed on an array of 16 gold electrodes (2.5 mm diam.; GeneFluidics Inc. Monterey Park, CA; Figure 2A). The Au sensors were incubated with a 6 μL aliquot of the 0.05 mM thiolated capture probe in the IB overnight at 4 °C in a humidified surrounding. Subsequently, the probe-modified Au sensors were treated with 6 μL of the 1 mM MCH aqueous solution for 40 min to obtain a mixed self-assembled monolayer. Finally, the sensors were thoroughly rinsed with water and dried under nitrogen.
DNA Hybridization Assay
The DNA detection strategy is illustrated in Figure 1. Different concentrations of the DNA target were mixed with the biotinylated detection probe (0.25 μM) in the HB and were incubated for 10 min at room temperature to allow the target-probe hybridization. Aliquots (4 μL) of this target/detection-probe hybrid solution were cast on each of the oligonucleotide-modified gold sensors and were incubated for 15 min. After the sensors were washed and dried, a 4 μL aliquot of 2.33 U mL-1 SA-ALP (in the BB) was cast on each sensor and was incubated for 15 min to allow the enzyme binding, followed by washing with water and drying with nitrogen. An 8 μL aliquot of the 0.2 mM p-APP substrate solution was subsequently dropped on each sensor, and the enzymatic reaction proceeded for 30 min (under dark). Subsequently, the supernatant was immediately transferred to the ISE detection cell for the potentiometric measurement.
E. coli 16S rRNA Hybridization Assay
The applicability of the present method was tested with E. coli 16S rRNA. To produce the E. coli 16S rRNA, the bacteria were lysed by resuspending a pellet containing 2.25×107 CFU bacteria (as determined by serial plating) in 10 μL of 1 M NaOH and waiting for 5 min. A 50 μL aliquot of the biotinylated detection-probe (0.25 μM) in HB was added to the bacterial lysate, leading to 2.25×107 CFU per 60 μL. Different concentrations of the pathogen were obtained by serial dilutions of this bacterial-lysate/detection-probe mixture with the 0.25 μM biotinylated detection probe solution to give E. coli solutions ranging from 288 to 4.5×106 CFU per 60 μL. A 10 min incubation was used for hybridizing the detection probe to the target. Aliquots (4 μL) of this bacterial-target/detection-probe hybrid were cast on each capture-probe modified sensor and incubated for 15 min, followed by capture of the ALP enzyme and the enzymatic reaction step (described earlier for the DNA hybridization assay). All procedures were carried out at room temperature.
Potentiometric Measurements
Potentiometric measurements were performed with a multifunctional data acquisition board (779026-01 USB-6009 14 Bit, National Instruments, Austin, TX) connected to a six-channel high Z interface (WPI Inc., Sarasota, FL). A conventional two-electrode configuration was employed all through the experiment, involving an Ag+-ISE and an Ag/AgCl reference electrode along with a home-made double junction compartment (Figure 2B). The detection process was carried out in a microcell containing 100 μL 10-6 M AgNO3 in DB in the presence of 2.5 μL Au NPs and using a small magnetic stirring bar (Figure 2B). Changes in the potential of the Ag+-ISE were monitored after adding the enzymatic-reaction solution (containing the p-AP product); the potential change after 2 min served as the hybridization signal. All measurements were carried out at room temperature and under stirring. Activity coefficients of silver were calculated according to the Debye-Huckel approximation and the potential values were corrected for liquid-junction potentials with the Henderson equation.
Results and Discussion
The goal of this work was to develop a highly sensitive potentiometric detection of enzyme-linked DNA hybridization connected to a biocatalytic metallization based on a highly sensitive Ag ISE transducer that delivers nM detection limits. The resulting potentiometric hybridization assay relies on a thiolated DNA probe, confined to the gold surface through a mixed monolayer (Figure 1 and Figure 2A). Hybridization of the surface-confined capture probe with the target/biotinylated-detection-probe conjugate results in a sandwich structure on the modified gold surface. Subsequent ALP binding and addition of the p-APP substrate leads to the biocatalytic generation of the p-AP reducing agent. The resulting solution is then transferred to the detection cell containing the Ag+-ISE, the silver ion and the gold nanoparticle seeds (Figure 2B). The change in the ISE potentiometric response, due to the decreased activity of the silver ion (by the p-AP reduction), thus reflects the level of target DNA in the sample.
Recently, solid-contact Ag+-ISE with a very high selectivity toward silver ions and femtomole detection limits have been successfully prepared and characterized.18,19 we used a micropipette-based Ag+-ISE and an Ag/AgCl reference electrode (along with a home-made double junction compartment) for measuring changes in the Ag+ concentration in the 100 μL microwell detection cell (Figure 2B). The attractive analytical performance of the new ISE transducer was examined first from a calibration experiment over a broad Ag+ concentration range, 1 nM – 0.1 mM, in a 100 μL glycine (pH 9.0) solution (Figure 3). The ISE responds rapidly to these nM and μM changes in the Ag+ concentration, and displays a nearly Nernstian response with a slope of 55.1 and a detection limit of 10-8 M (inset, Figure 3). Such detection limit corresponds to 1 pmol Ag+ in the 100 μL sample. Such low detection limit makes the micropipette-based Ag+-ISE attractive to the potentiometric DNA measurements described below. Note that unlike analogous stripping-voltammetric measurements, such nanomolar detection limits are achieved without a preceding accumulation step.
Figure 3.
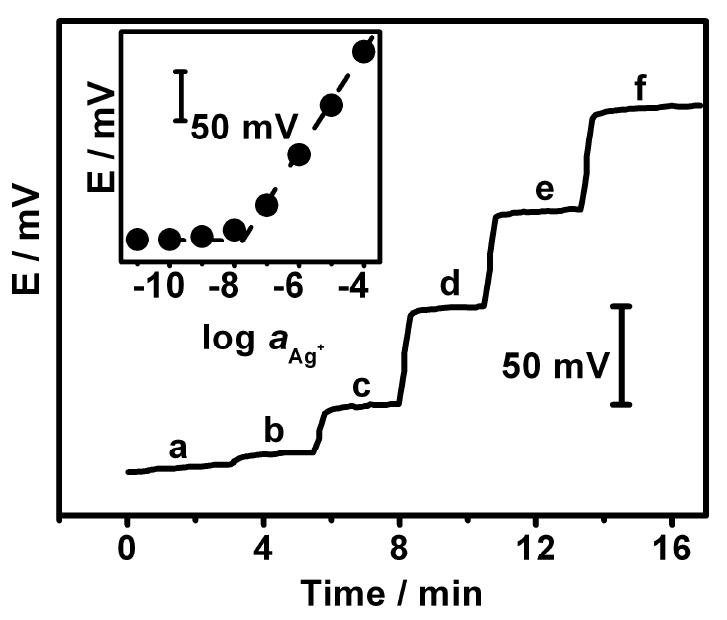
Potential-time potentiometric response of the Ag+-ISE to increasing levels of Ag+: (a) 10-9; (b) 10-8; (c) 10-7; (d) 10-6; (e) 10-5 and (f) 10-4 M in a 100 μL glycine (pH 9.0) solution. Inset displays the corresponding calibration plot.
Factors affecting the potentiometric transduction of the p-AP induced biometallization were evaluated and optimized. The amount of Au NPs was examined first owing to their pronounced effect upon the silver deposition process. While the p-AP enzymatic product can spontaneously reduce silver ions to metallic silver this reduction process is too slow without the Au NP nucleation sites.20 Figure 4A displays the influence of the level of Au NPs on the silver deposition at a fixed (10-6 M) Ag+ concentration. The potential-change signal (ΔE) increases linearly with the volume of the Au NPs solution up to 2.5μL (in 100 μL of the DB) and levels off for higher volumes of Au NPs.
Figure 4.
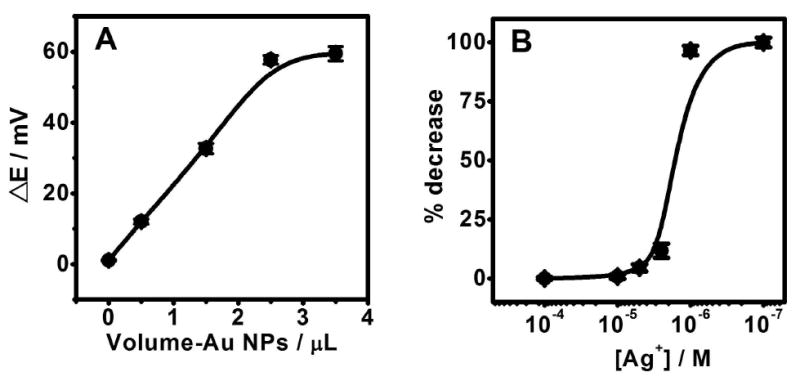
(A) Influence of the amount of Au NPs on the silver deposition caused by 0.5 μM p-AP at Ag+ concentration of 10-6 M. (B) Percentage decrease of the potential response at different Ag+ concentrations from 10-7 to 10-4 M in glycine buffer (pH 9.0) containing 0.5 μM p-AP.
Other experimental variables, including the levels of Ag+ and of the p-APP enzyme substrate, have also been optimized. Figure 4B displays the dependence of the percentage potential change upon the level of Ag+ in the presence of 0.5 μM p-AP. No apparent potential change was observed for Ag+ concentrations higher than 10-5 M. Such negligible change reflects the very low (0.5 μM) level of the p-AP reducing agent compared to the large excess of Ag+. Yet, the relative response increases rapidly between 10-5 and 10-6M Ag+ and levels off at approximately 100% at lower (submicromolar) silver concentrations. An Ag+ concentration of 10-6 M was selected for all subsequent measurements.
Other parameters, such as the concentration of the thiolated capture probe, the concentration of the enzyme (SA-ALP) and substrate (p-APP), the pH of the DB were also evaluated. Optimum signals were obtained for a 0.05 mM thiolated capture probe, 2.33 U mL-1 SA-ALP, 0.2 mM p-APP and a pH of 9.0 of DB (data not shown).
The analytical performance of the new DNA hybridization potentometric assay was characterized under the optimal experimental conditions and using microliter (4 μL) samples. Figure 5A displays the potential-time potentiometric response obtained at the Ag+-ISE (in the presence of Au NPs and 10-6 M Ag+) for different concentrations of the DNA target: 0(a), 100 fM (b), 1 pM (c), 10 pM (d), 100 pM (e), 1 nM (f), and 10 nM (g). Well-defined potentiometric signals are obtained for each target concentration. The potential decreases rapidly within the first 120 sec and then slowly, reaching its steady-state value. Compared to common optical or voltammetric detection the ISE allows real-time monitoring and can be used for probing the kinetics of such biometallization processes, as it responds continuously and instantaneously to the changes in the Ag+ concentration. The small contribution observed without the DNA target (a) reflects the contribution of p-AP generated from the non-specifically adsorbed ALP. The resulting calibration plot, shown in Figure 5B, indicates a linear relationship between the potential response (ΔE, corresponding to the Ag+ depletion) and the logarithm of the target DNA concentration over the entire 100 fM to 10 nM range, i.e., over 5 orders of magnitude (correlation coefficient, 0.986). A detection limit of 50 fM DNA can thus be estimated from the signal-to-noise characteristics of these data. Such detection limit corresponds to 0.2 amol in the 4 μL sample, and represents a 175-fold improvement in the detectability compared to analogous stripping-voltammetric detection of ALP-induced silver biometallization processes.12 The fM detectability of the new ISE based DNA detection compares favorably with common ALP-based amperometric DNA assays that offer nM – sub-pM detection limits.21-24 This was confirmed in analogous amperometric measurements of the p-AP product, carried out under identical (immobilization, hybridization) conditions as the new ISE protocol, that yielded a detection limit of 1.5 nM DNA (not shown). The new potentiometric protocol is also comparable with advanced fluorescence-based enzyme-linked DNA assays that yield detection limits in the pM – fM range,25,26 but requires bulky and expensive instrumentation. Further improvements in the detection limits are expected through modern amplification schemes (used in amperometric DNA assays), involving nanoscale ‘carriers’ to maximize the number of enzyme tags per binding event.27
Figure 5.
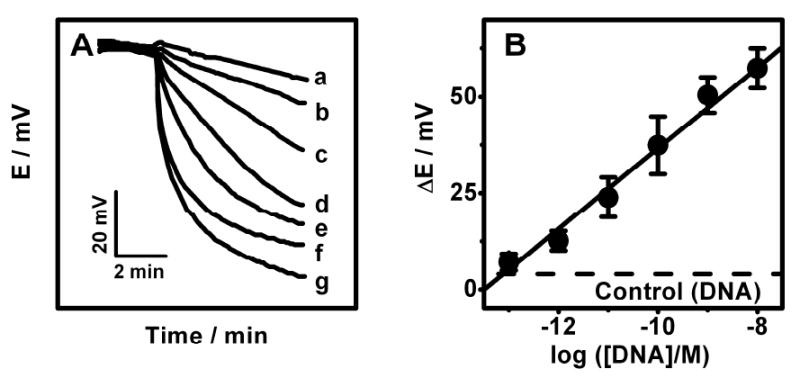
Potential-time potentiometric hybridization response (A) and calibration plot (B) for increasing concentrations of the DNA target: (a) 0, (b) 100 fM, (c) 1 pM, (d) 10 pM, (e) 100 pM, (f) 1 nM, (g) 10 nM. (B) The corresponding calibration plot obtained using the potential change after 2 min as the hybridization signal. See Experimental Section for details.
The real-life utility of the new potentiometric DNA assay was illustrated using the detection of 16S rRNA of E. coli pathogenic bacteria. Figure 6 displays potential-time recordings obtained for samples containing increasing levels of the E. coli bacteria from 288 to 4.5×106 CFU per 60μL (b-e). Well-defined potentiometric signals are observed for these low levels of E. coli. The corresponding semi-log calibration plot (Figure 6B) exhibits a well defined linear dependence between the potential change and the log[E. coli] (correlation coefficient, 0.988). The data of Figure 6A indicate a detection limit of around 150 CFU per 60μL. Considering the 4 μL sample volume such detection limit corresponds to a total of 10 CFU per sensor. This value is significantly lower than the 2600 CFU per sensor value reported previously for amperometric measurements of 16S rRNA of E. coli based on a HRP tag.28
Figure 6.
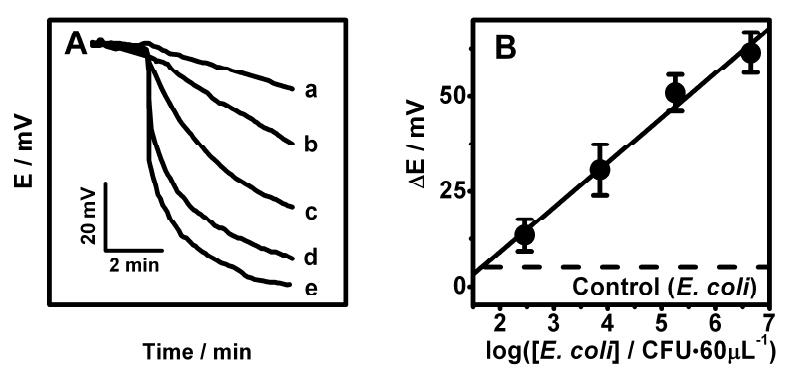
Potential-time potentiometric hybridization response (A) and calibration plot (B) for increasing levels of E. coli: (a) 0, (b) 288, (c) 7200, (d) 180000, (e) 4500000 CFU per 60μL. (B) The corresponding calibration plot using the potential change after 2 min as the hybridization signal. See experimental Section for details of the E. coli sample preparation.
The reproducibility of the potentiometric response for E. coli bacteria was also examined. Figure 7 displays 10 simultaneous measurements of E. coli, at 12000 CFU per sensor, using different sensors of the same array. Well defined and reproducible potential signals are observed. A favorable relative standard deviation (RSD) of 3.83% was thus obtained for the E. coli potential signal (after 2 min). The accuracy was estimated from a recovery experiment (that yielded a 6.7% error at the 36000 cfu/60μL level) and from correlation to the established HRP-based amperometric method.28 Orthogonal regression analysis of the resulting correlation plot (present method vs amperometric method, for the pathogen bacteria) yielded the equation of y = 1.02× − 0.08937 (R2 = 0.9864; not shown). These data reflect the good accuracy of the new method.
Figure 7.
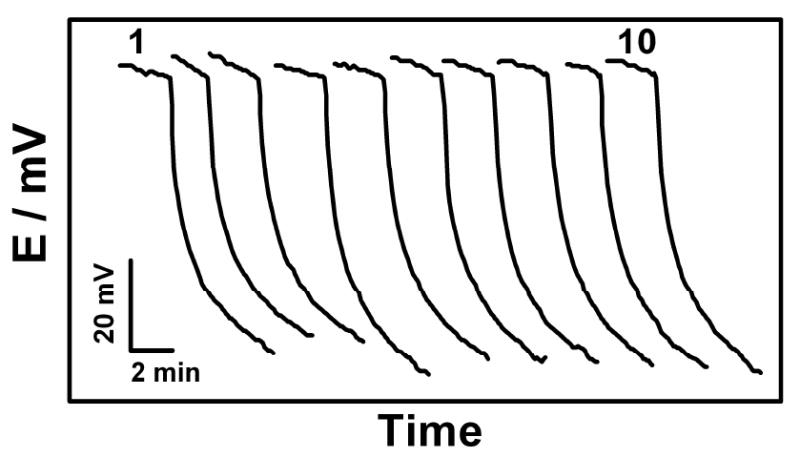
Reproducibility study involving 10 parallel measurements of 12000 CFU E. coli per sensor at the different sensors of the same array chip (of Figure 2A); 4 μL sample droplets.
Conclusions
We described an attractive electrochemical method for detecting DNA hybridization based on the potentiometric monitoring of the ALP-induced depletion of silver ions in the presence of Au NPs. This represents the first example of using ISEs for probing hybridization-induced biometallization processes. Such effective monitoring of enzyme-linked DNA hybridization events reflects the rapid, direct, continuous and sensitive potential response of modern ISEs. The present potentiometric technique exhibits an improved performance compared to other electrochemical techniques that use nanoparticle tags, as it obviates the need for particle-dissolution or electrodeposition/preconcenration steps and problems associated with metallization of the DNA itself. Advanced amplification schemes involving nanoscale ‘carriers’ are expected to push the detectability of potentiometric DNA bioassays to the sub-fM level. The new protocol thus holds great promise for various bioaffinity assays (including immunoassays) involving common enzyme tags, and for related diagnostic testing, in general. The compact and portable supporting instrumentation makes such ISE transduction mode particularly attractive for decentralized and point-of-care DNA diagnostics.
Acknowledgments
Financial support from the National Institutes of Health (RO1 EB002189 and U01 AI075565) and National Science Foundation (CHE 0506529) are gratefully acknowledged.
References
- 1.Mikkelsen SR. Electroanalysis. 1996;8:15–19. [Google Scholar]
- 2.Palecek E, Fojta M. Anal Chem. 2001;73:74A–83A. doi: 10.1021/ac0123936. [DOI] [PubMed] [Google Scholar]
- 3.Wang J. Anal Chim Acta. 2002;469:63–71. [Google Scholar]
- 4.Hernandez-Santos D, Diaz-Gonzalez M, Gonzalez-Garcia MB, Costa-Garcia A. Anal Chem. 2004;76:6887–6893. doi: 10.1021/ac048892z. [DOI] [PubMed] [Google Scholar]
- 5.Roth KM, Peyvan K, Schwarzkopf KR, Ghindilis A. Electroanalysis. 2006;18:1982–1988. [Google Scholar]
- 6.Bakker E, Pretsch E. Anal Chem. 2002;74:420A–426A. doi: 10.1016/j.trac.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radu A, Shane P, Bakker E, Diamond D. Electroanalysis. 2007;19:144–154. [Google Scholar]
- 8.Numnuam A, Chumbimuni-Torres KY, Xiang Y, Bash R, Thavarungkul P, Kanatharana P, Pretsch E, Wang J, Bakker E. J Am Chem Soc. 2008;130:410–411. doi: 10.1021/ja0775467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Liu G, Polsky R, Merkoci A. Electrochem Comm. 2002;4:722–726. [Google Scholar]
- 10.Wang J, Xu D, Kawde N, Polsky R. Anal Chem. 2001;73:5576–5581. doi: 10.1021/ac0107148. [DOI] [PubMed] [Google Scholar]
- 11.Hwang S, Kim E, Kwak J. Anal Chem. 2005;77:579–584. doi: 10.1021/ac048778g. [DOI] [PubMed] [Google Scholar]
- 12.Fanjul-Bolado P, Hernandez-Santos D, Gonzalez-Garcia MB, Costa-Garcia A. Anal Chem. 2007;79:5272–5277. doi: 10.1021/ac070624o. [DOI] [PubMed] [Google Scholar]
- 13.Chumbimuni-Torres KY, Wang J. Analyst. 2009;134:1614–1617. doi: 10.1039/b902171j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarvinen H, Lahtinen L, Nasman J, Hormi OAL. Synthetic Metals. 1995;69:299–300. [Google Scholar]
- 15.U.S. Patent Application 20040254336
- 16.Qin Y, Peper S, Bakker E. Electroanalysis. 2002;14:1375–1381. [Google Scholar]
- 17.Chumbimuni-Torres KY, Rubinova N, Radu A, Kubota LT, Bakker E. Anal Chem. 2006;78:1318–1322. doi: 10.1021/ac050749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubinova N, Chumbimuni-Torres KY, Bakker E. Sens Actuators B. 2007;121:135–141. doi: 10.1016/j.snb.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szigeti Z, Malon A, Vigassy T, Csokai V, Grun A, Wygladacz K, Ye N, Xu C, Chebny V, Bitter I, Rathore R, Bakker E, Pretsch E. Anal Chim Acta. 2006;572:1–10. doi: 10.1016/j.aca.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willner I, Baron R, Willner B. Adv Materials. 2006;18:1109–1120. [Google Scholar]
- 21.Nebling E, Grunwald T, Albers J, Schafer P, Hintsche R. Anal Chem. 2004;76:689–696. doi: 10.1021/ac0348773. [DOI] [PubMed] [Google Scholar]
- 22.Miranda-Castro R, De-los-Santos-Alvarez P, Loba-Castanon MJ, Miranda-Ordieres AJ, Tunon-Blanco P. Anal Chem. 2007;79:4050–4055. doi: 10.1021/ac062260q. [DOI] [PubMed] [Google Scholar]
- 23.Lucarelli F, Marrazza G, Mascini M. Langmuir. 2006;22:4305–4309. doi: 10.1021/la053187m. [DOI] [PubMed] [Google Scholar]
- 24.Kim E, Kim K, Yang H, Kim YT, Kwak J. Anal Chem. 2003;75:5665–5672. doi: 10.1021/ac034253x. [DOI] [PubMed] [Google Scholar]
- 25.Niu SY, Li QY, Ren R, Zhang SS. Biosens Bioelectron. 2009;24:2943–2946. doi: 10.1016/j.bios.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 26.Li ZH, Hayman RB, Walt DR. J Am Chem Soc. 2008;130:12622–12623. doi: 10.1021/ja8053018. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Liu GD, Jan MR. J Am Chem Soc. 2004;126:3010–3011. doi: 10.1021/ja031723w. [DOI] [PubMed] [Google Scholar]
- 28.Liao JC, Mastali M, Gau V, Suchard MA, Moller AK, Bruckner DA, Babbitt J, Li Y, Gornbein J, Landaw EM, McCabe ERB, Churchill BM, Haake DA. J Clin Microbiol. 2006;44:561–570. doi: 10.1128/JCM.44.2.561-570.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]