

Abstract
Protein ubiquitylation is a complex enzymatic process that results in the covalent attachment of ubiquitin, via Gly-76 of ubiquitin, to an ε-NH2-group of an internal lysine residue in a given substrate. While E3 ligases frequently utilize lysines adjacent to the degron within the substrate, many substrates can be targeted to the proteasome via polyubiquitylation of any lysine. We have assessed the role of lysine residues proximal to the cyclin D1 phosphodegron for ubiquitylation by the SCFFbx4/αB-crystallin ubiquitin ligase and subsequent proteasome-dependent degradation of cyclin D1. The work described herein reveals a requisite role for Lys-269 (K269) for the rapid, poly-ubiquitin mediated degradation of cyclin D1. Mutation of lysine 269, which is proximal to the phosphodegron sequence surrounding Thr-286 in cyclin D1, not only stabilizes cyclin D1, but also triggers cyclin D1 accumulation within the nucleus thereby promoting cell transformation. In addition, D1-K269R is resistant to genotoxic stress induced degradation, similar to non-phosphorylatable D1-T286A, supporting the critical role for the post-translational regulation of cyclin D1 in the response to DNA damaging agents. Strikingly, while mutation of lysine 269 to arginine inhibits cyclin D1 degradation, it does not inhibit cyclin D1 ubiquitylation in vivo demonstrating that ubiquitylation of a specific lysine can influence substrate targeting to the 26S proteasome.
Introduction
Cell cycle progression is positively regulated by cyclin/cyclin-dependent kinase (cdk) complexes and counteracted through the activity of cyclin-dependent kinase inhibitors (CKIs). D-type cyclins are the first cyclins induced as cells enter the cell cycle from G0 and together with their catalytic partners cdk4/cdk6 promote G1/S transition by relieving Rb-dependent repression of E2F transcription factors (Matsushime et al., 1994; Kato et al., 1993). At the G1/S boundary, cyclin D1 is phosphorylated at Thr-286 by GSK-3β, triggering cyclin D1 nuclear export and subsequent ubiquitin-dependent proteolysis (Diehl et al., 1998). Cytoplasmic ubiquitylation of phosphorylated cyclin D1 is catalyzed by SCFFbx4/αB-crystallin ligase (Lin et al., 2006).
The importance of maintaining threshold levels of cyclin D1 is emphasized by the frequent cyclin D1 overexpression in human cancer. Cyclin D1 upregulation occurs in 50% of human breast, esophageal, lung and liver tumors (Lin and Beerm, 2004; Sato et al., 1999). While approximately 15% of cases can be attributed to chromosomal translocations and gene amplification (Worsley et al., 1996), inhibition of cyclin D1 proteolysis is either implicated or has been directly demonstrated to be a contributing factor in the majority of such tumors (Benzeno et al., 2006; Moreno-Bueno et al., 2003). In human esophageal cancer cyclin D1 overexpression occurs through the mutational inactivation of its ubiquitin ligase, Fbx4 (Barbash et al., 2008). Additionally, mutations in the cyclin D1 phosphodegron have been observed in human tumors (Benzeno et al., 2006; Moreno-Bueno et al., 2003). Importantly, these findings underline the importance of cyclin D1 proteolysis for normal cell homeostasis, which is often attenuated in human cancer.
Ubiquitin is covalently linked to substrates through the concerted activities of an enzymatic pathway containing E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme) and E3 (substrate specificity factor) enzymes via the C-terminal Gly-76 of ubiquitin and the εNH2-group of substrate's internal lysine. The molecular basis for the specificity of E3 ligases to particular lysines in substrates is well accepted but is poorly understood (Hershko et al., 1984). Structural studies suggest that the proximity of the substrate lysine chains and E2 enzymes plays a pivotal role in the efficiency of substrate ubiquitylation, suggesting that the specificity to the particular lysine can be explained by the relative spatial relationship of these lysines to the E3 ligase (Zheng et al., 2002; Wu et al., 2003). Indeed, some substrates are ubiquitylated only at specific lysines, proximal to the sites of E3 binding, including IκB (Scherer et al., 1995) and p27 (Shirane et al., 1999).
Previous work suggested that mutation of all cyclin D1 lysines to arginine confers protection from proteasome-dependent degradation, while the mutations of single lysines only lead to a modest increase in the stability of cyclin D1(Feng et al., 2007). This work implicated Lys-112 and Lys-114 as essential ubiquitin acceptors; complicating the interpretation of this work is the fact that these lysine residues also mediate the interaction of cyclin D1 with cdk4 (Feng et al., 2007), and their mutation disrupts the functionality of cyclin D1. In addition, previous analysis did not address the specificity of lysines as essential acceptors for the E3 ligase, SCFFbx4/αB-crystallin. We now demonstrate that the mutation of Lys-269 (D1-K269R) renders cyclin D1 protein resistant to proteasomal degradation. D1-K269R is resistant to the ubiquitylation by SCFFbx4/αB-crytsallin in vitro, consistent with increased stability in vivo. Critically, cyclin D1-K269R is ubiquitylated in vivo and the ubiquitin conjugation of both wild type cyclin D1 and D1-K269R in vivo occurs through Lys-48 linked polyubiquitin chains. Despite being ubiquitylated in vivo, D1-K269R exhibits diminished association with 19S proteasomal subunit, suggesting that the ubiquitylation of cyclin D1 at Lys-269 is essential for the targeting of cyclin D1 to proteasome and subsequent degradation.
Materials and Methods
Cell culture, plasmids and transfections
NIH-3T3 cells were maintained in DMEM medium containing 10% of fetal bovine serum, glutamine and antibiotics. Where indicated, 24 hours before transfection, cells were plated at optimal density and the following day transfected using Lipofectamine Plus reagent (Invitrogen, Carlsbad, CA). Stable cell lines expressing cyclin D1 were generated by puromycin (Sigma-Aldrich, St. Lois, MO) selection (5μg/ml) for 21 days and subsequently cultured in medium containing 2.5μg/ml puromycin. Insect Sf9 cells were maintained as described elsewhere. Flag-tagged cyclin D1 mutants were constructed using QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) using pFlex-cyclin D1 plasmid as template. PCR reactions were performed accordingly to manufacturer instructions. Clones were sequenced in their entirety to confirm the presence of mutations.
Immunoprecipitation and immunoblot analysis
Cells were lysed in buffer containing: 50 mM HEPES (pH 8.0), 150mM NaCl, 2.5mM EGTA, 1mM EDTA, 0.1% Tween 20, protease, and phosphatase inhibitors (1mM PMSF, 20U/ml aprotinin, 5μg/ml leupeptin, 1mM DTT, 0.4mM NaF, 10mM β-glycerophosphate, and 100nM okadaic acid). The protein concentration was determined by BCA assay (Pierce, Rockford, IL). Where indicated cyclin D1 was precipitated using either M2 agarose (Sigma-Aldrich, St. Lois, MO) or a cyclin D1–mouse monoclonal antibody, D1-72-13G. Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membrane and analyzed by immunoblot. Antibodies used were as follows: Fbx4 rabbit polyclonal antibody (Rockland, Gilbertsville, PA), cyclin D1 mouse monoclonal D1-72-13G, β-actin, Flag rabbit polyclonal (Sigma-Aldrich, St. Louis, MO), p27 mouse monoclonal antibody, CDK4 (BD Pharmingen, San Diego, CA), ubiquitin mouse monoclonal (Covance, Emerville, CA), cyclin E rabbit polyclonal, cyclin A rabbit polyclonal (Santa Cruz, Santa Cruz, CA).
Cycloheximide chase analysis
For cycloheximide chase experiments, NIH-3T3 cells were plated at equal densities and the next day asynchronous cells were treated with cycloheximide (100μg/ml, Sigma-Aldrich, St. Lois, MO) for indicated periods of times. Cells were lysed in buffer (as described above) and processed for immunoblot analysis.
In vitro kinase assay
Cyclin D1/cdk4 complexes were expressed in SF9 cells. Cells were lysed in Tween-20 buffer and immunoprecipitated using M2-agarose (sigma-Aldrich, St. Lois, MO). Purified complexes were mixed with recombinant GST-Rb as described previously described (Matsushime et al., 1994) and the phosphorylation of Rb was determined by autoradiography.
In vitro ubiquitylation
SCFFbx4/αB-crystallin complexes were purified from Sf9 cells using M2 agarose, mixed with purified substrate as indicated that had been phosphorylated in vitro with recombinant GSK3β (5U), E1, E2 (UbcH5A), ATP, and ubiquitin for indicated times at 37°C. Proteins were resolved on 10% SDS-PAGE and visualized by Western blotting with the D1-72-13G11 antibody.
Immunofluorescence
NIH3T3 derived cell lines were plated at optimal density on glass coverslips. 24 hours after splitting cells were permeabilized with Methanol:Acetone (1:1), washed with PBS and incubated in primary antibody (cyclin D1 17-13G11) for 2 hours. After washing and incubation with secondary anti-mouse FITC conjugated antibody (GE Healthcare, slides were mounted using ProLong Gold anti-fade DAPI reagent (Invitrogen, Carlsbad, CA) and analyzed by fluorescent microscopy using Nikon Eclipse E800 microscope. For quantification of immunofluorescence data we counted 100 cells for each cell line in three independent experiments.
Transformation assays
Anchorage-independent growth was determined by analyzing cellular growth in semisolid medium. Cells (5×103) were seeded in complete Iscove's media containing 0.65% noble agar/10% FCS. Cells were grown for 21 days in 8% CO2. For foci formation cells were plated at 1.5 × 105 cells/well of a 6-well plates. Cells were cultured in medium containing 5% FBS. Foci were visualized after 21days with Wright Giemsa stain (Sigma-Aldrich, St. Lois, MO).
γIR treatment
Asynchronously proliferating NIH-3T3 cells stably expressing WT cyclin D1, D1-T286A, or D1-K269R were subjected to 10Gy γIR, followed by recovery at 37°C. Cell lysates were prepared in Tween-20 buffer, followed by SDS-PAGE and immunoblot with the following antibodies: cyclin D1, cyclin E, cyclin A and β-Actin.
Cell cycle analysis
NIH 3T3 cells stably expressing cyclin D1 or D1-K269R were harvested and washed with phosphate-buffered saline, fixed with ethanol, and stained with propidium iodide for 1 hour, prior to FACS analysis. Cell cycle profiles based on DNA content were established by using FlowJo software.
Results
Mutation of Lys-269 to arginine stabilizes cyclin D1 protein with no attenuation of its functions
Cyclin D1 degradation is triggered by GSK-3β-dependent phosphorylation and subsequent ubiquitylation by SCFFbx4/αB-crystallin complex (Diehl et al., 1998; Lin et al., 2006). To determine whether lysines proximal to Thr-286 of cyclin D1 are essential for the proteasome-dependent degradation of cyclin D1, we generated mutant cyclin D1 alleles where lysine residues were changed to arginines (D1-K238R and D1-K269R). While both wild-type cyclin D1 and D1-K238R exhibited similar kinetics for proteasomal degradation, the half-life of D1-K269R was extended to 3-4 fold (from 20 to 90 minutes; Fig. 1A).
Figure 1. Mutation of Lys-269 in cyclin D1 leads to increased cyclin D1 protein stability without the attenuation of cyclin D1 functions.
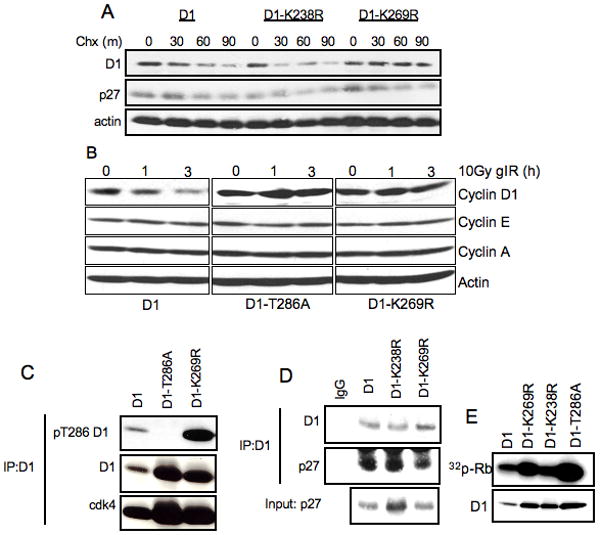
A. NIH-3T3 cells stably expressing wild-type cyclin D1, D1-K238R and D1-K269R were treated with 100μg/ml cyclohexemide for indicated periods of time. Cells were harvested and cyclin D1 protein levels were determined by immunoblot with D1-72-13G antibody. B. NIH 3T3 cells expressing wild type cyclin D1, D1-T286A, or D1-K269R were irradiated and harvested as indicated. Cyclin D1 levels were assessed by immunoblot. C. Cell lysates from NIH-3T3 cell lines stably expressing WT cyclin D1, D1-T286A and D1-K269R were used for immunoprecipitation of cyclin D1 with M2 agarose followed by immunoblot with phospho-Thr-286, D1-72-13G and cdk4 antibodies. D. Same as B, immunoblot with p27 antibody. E. Cyclin D1/cdk4 complexes were assembled in SF9 cells and used to phosphorylate recombinant GST-Rb, followed by autoradiography.
Previous work demonstrated that cyclin D1 proteolysis induced by γIR is SCFFbx4/αB-crystallin –dependent (Pontano et al., 2008). Thus, we tested whether D1-K269R is refractory to γIR-dependent proteasomal degradation. NIH-3T3 cells stably expressing wild-type cyclin D1, D1-K269R and D1-T286A were exposed to 10Gy of γIR (Fig 1B), and accumulation of cyclin D1, A and E were assessed by immunoblot at various intervals following γIR exposure. While wild-type cyclin D1 was degraded in response to γIR, D1-K269R and D1-T286A mutants were stable following γIR exposure. Thus, the K269R mutation renders cyclin D1 resistant to ubiquitin-dependent proteolysis in proliferating cells and following genotoxic stress.
Since the phosphorylation of cyclin D1 at Thr-286 is absolutely required for its degradation, we determined whether the K269R mutation might perturb normal phosphorylation of Thr-286 and thereby indirectly impact cyclin D1 degradation. Wild-type and mutant cyclin D1 proteins were immunoprecipitated from asynchronous NIH-3T3 cells that stably express Flag-tagged D1 alleles and phosphorylation was assessed with a phospho-specific Thr-286 antibody. We noted increased Thr-286 phosphorylation of D1-K269R relative to wild type cyclin D1. The increase in phosphorylation is expected given the reduced rate of degradation of cyclin D1-K269R (Fig1C, panel 1 and 2).
We next determined whether the cyclin D1-K269R mutant retains normal catalytic function and the capacity to bind to p27Kip1. Indeed, co-precipitation revealed that associated with p27Kip1 cyclin D1-K269R (Fig. 1D). We subsequently evaluated the ability of cyclin D1-K269R to associate with and induce CDK4 catalytic activity toward Rb. Immunoprecipitation/western analysis revealed that wild-type and mutant cyclin D1 both associate with CDK4 (Fig 1C, lower panel) and efficiently catalyzed CDK4-dependent phosphorylation of recombinant Rb (Fig 1E). In summary, the mutation of Lys-269 to arginine increases the stability of cyclin D1 without disrupting either the established functional activities of cyclin D1 or the phosphorylation signals critical for cyclin D1 degradation. Collectively, these results are consistent with the interpretation that D1-K269R stabilization occurs due to loss of an essential ubiquitin acceptor residue required for cyclin D1 proteolysis.
Lys- 269 of cyclin D1 is essential for SCFFbx4/αB-crystallin –dependent ubiquitylation in vitro
Given that the Lys-269 to Arg mutation resulted in reduced cyclin D1 degradation, we hypothesized that this mutation would inhibit ubiquitylation of cyclin D1 by SCFFbx4/αB-crystallin. We therefore determined the ability of SCFFbx4/αB-crystallin to directly catalyze ubiquitylation of wild-type D1 and D1-K269R in vitro. For this experiment, we utilized GST-fusion proteins wherein the C-terminal 41 residues of cyclin D1 (or K269R) are fused in frame with GST. In this fusion, the only available lysine is K269, as lysines within GST are not ubiquitylated. Consistent with in vivo stabilization, purified, recombinant SCFFbx4/αB-crystaliin could not catalyze ubiquitylation of cyclin D1-K269R, although it effectively ubiquitylated wild type cyclin D1 in vitro (Fig 2A). To ensure that Lys-269 is the ubiquitin acceptor in full-length cyclin D1 we performed in vitro ubiquitylation assay using full-length wild type and K269R cyclin D1 purified from Sf9 cells. Again, wild-type cyclin D1, but not D1-K269R, was effectively ubiquitylated (Fig 2B), suggesting that Lys-269 is an acceptor of ubiquitin from SCFFbx4/αB-crystallin in vitro. Importantly, wild-type and mutant cyclin D1 retained the ability to bind to SCFFbx4/αB-crystallin in vitro, demonstrating that the disruption of the ubiquitylation was not due to the inability of D1-K269R to bind to the SCF complex (Fig 2C). Thus, Lys-269 is required for the efficient cyclin D1 ubiquitylation by SCFFbx4/αB-crystallin complex in vitro.
Figure 2. D1-K269R is resistant to SCFFbx4/αB-crystallin ubiquitylation in vitro.
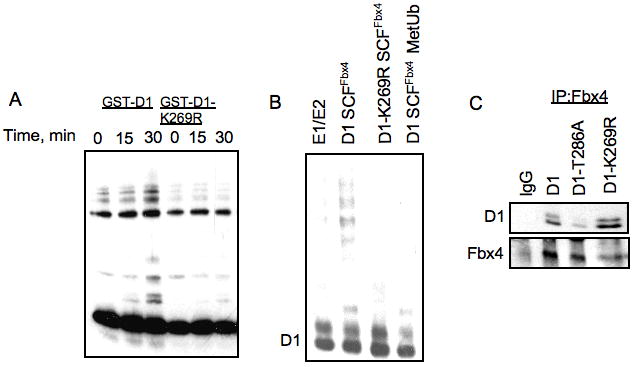
A. SCFFbx4/αB-crystallin complexes were assembled in SF9 cells, purified with M2 agarose and used to ubiquitylate recombinant GST-D1 or GST-D1-K269R. Reaction mixtures were separated by SDS-PAGE, followed by immunoblot with cyclin D1 antibody. B. D1/K4 and D1-K269R/K4 complexes were purified form SF9 cells using M2 agarose and used for in vitro ubiquitylation reaction as described in A. C. SCFFbx4/αB-crystallin complexes were expressed in SF9 cells, purified with M2 agarose and mixed with GST-WT cyclin D1, D1-T286A or D1-K269R for binding analysis. Complexes were separated by SDS-PAGE, followed by immunoblot with cyclin D1 and Fbx4 antibodies.
Lys-269 of cyclin D1 is not essential for ubiquitylation in vivo but is necessary for rapid proteasome-dependent degradation
Because Lys-269 cannot be ubiquitylated in vitro, and is required for proteolytic turnover in vivo, we determined whether Lys-269 was necessary for cyclin D1 ubiquitylation in vivo. NIH-3T3 cells stably expressing wild-type cyclin D1 and D1-K269R were treated with the proteasome inhibitor, MG132 and following lysis of cells under denaturing conditions, ubiquitylated proteins were purified by precipitation using a ubiquitin specific antibody and cyclin D1 ubiquitylation was assessed by immunoblot with a Flag reactive antibody (Fig 3A). D1-K269R ubiquitylation was readily apparent in vivo, indicating that lysines besides Lys-269 of cyclin D1 can function as ubiquitin acceptors in vivo.
Figure 3. In vivo ubiquitylation of D1-K269R.
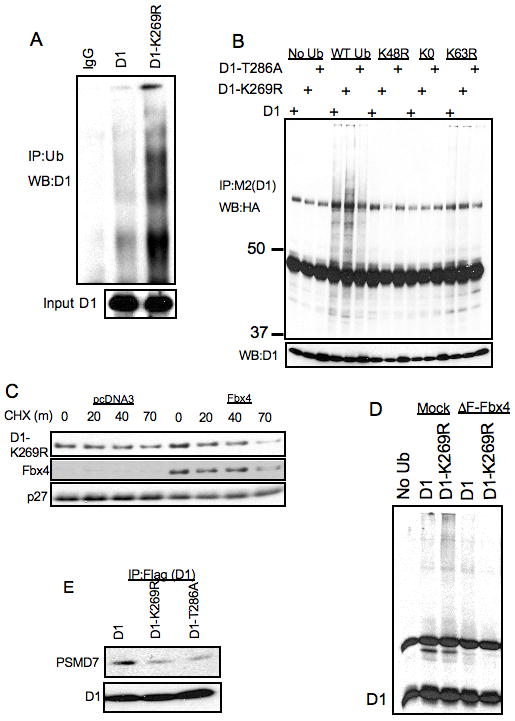
A. NIH-3T3 cells stably expressing WT and K269R cyclin D1 were treated with 10μM MG132 for 6 hours, proteins were lysed under denaturing conditions (50mM Tris-HCl(pH 7.4), 1% SDS and 5mM DTT). Proteins were precipitated with anti-ubiquitin antibody in the buffer containing 50mM Tris-HCL (pH 7.4), 250mM NaCl, 5mM EDTA and 0.5% NP-40 and separated by SDS-PAGE and detected by immunoblot with a cyclin D1 antibody. B. NIH-3T3 cells stably expressing WT cyclin D1, D1-T286A and D1-K269R cyclin D1 were transfected with WT, K48R, K0 and K63R ubiquitin constructs. 48 hours post-transfection cells were treated with 10 μM MG132 for 6 hours and cell lysates were immunoprecipitated with M2 agarose followed by SDS-PAGE of protein complexes and immunoblot with anti-HA, cyclin D1 and Fbx4 antibodies. C. NIH-3T3 stably expressing D1-K269R were transfected with an Fbx4 expression vector. 48 hours post-transfections cells were used for cycloheximide chase assay at indicated time intervals. Cell lysates were separated by SDS-PAGE, followed by immunoblot with cyclin D1, Fbx4 and p27 antibodies. D. 293T cells were transfected with ubiquitin, ΔF-Fbx4 and cyclin D1 constructs as indicated. 48 hours post-transfection cells were treated with 10 μM MG132 for 6 hours and cell lysates were immunoprecipitated with anti-cyclin D1 antibody followed by SDS-PAGE of protein complexes and immunoblot with the cyclin D1 antibody. E. Same as in B, followed by immunoblot with cyclin D1 and PSMD7 antibodies.
Since D1-K269R was ubiquitylated in vivo, we determined whether ubiquitin linkages formed on wild-type cyclin D1 were retained in D1-K269R. Normally, Lys-48 linked polyubiquitin chains provide signals for the targeting of the substrates to the proteasome. Since cyclin D1 is degraded in a ubiquitin-dependent manner, we hypothesized that it is ubiquitylated through Lys-48 linked polyubiquitin chains. We transfected NIH-3T3 lines stably expressing wild-type cyclin D1, D1-T286A and D1-K269R with HA-tagged wild-type, K48R, K0R and K63R ubiquitin expression vectors. Cells were treated with MG132 and cyclin D1 was immunoprecipitated with the M2 monoclonal antibody and ubiquitylated protein species were visualized by immunoblot with the HA-antibody (Fig 3B). Both, wild-type cyclin D1 and D1-K269R were ubiquitylated in vivo, while the ubiquitylation of D1-T286A was reduced. The expression of K48R ubiquitin significantly reduced the ubiquitylation of wild-type cyclin D1, D1-T286A and D1-K269R (Fig 3B), suggesting that the ubiquitin chains formed on cyclin D1 wild-type and mutants were primarily Lys-48-linked. The expression of K63R ubiquitin only slightly reduced the polyubiquitylation of cyclin D1 alleles, which suggests that K63-linked ubiquitin chains or mixed K48-linked and K63-linked chains might occasionally form on cyclin D1. In summary, K269R cyclin D1 is a stable mutant of cyclin D1 that is resistant to SCFFbx4/αB-crytsallin ubiquitylation in vitro, but is ubiquitylated in vivo primarily through Lys-48 polyubiquitin chains.
Based on this data, we suggest that Fbx4 can ubiquitylate additional lysines in cyclin D1, but with reduced processivity. If this is the case, overexpression of Fbx4 should increase potential cyclin D1-Fbx4 association frequency and potentially increase productive cyclin D1 poly-ubiquitylation and thereby increase cyclin D1 degradation. To determine whether D1-K269R degradation is sensitive to Fbx4 levels and thus presumably steady-state Fbx4 E3 ligase activity, Fbx4 was overexpressed in NIH-3T3 cells stably expressing D1-K269R (Fig 3C) and cyclin D1-K269R half-life was measured. D1-K269R turnover was modestly accelerated upon Fbx4 overexpression, suggesting that in the absence of Lys-269 SCFFbx4 could modify other lysines, providing alternative signals for cyclin D1 degradation. Consistent with ubiquitylation remaining Fbx4-dependent, expression of dominant-negative mutant of Fbx4 (ΔF) abolished in vivo ubiqutylation of wild-type and D1-K269R (Fig 3D).
We hypothesized that D1-K269R is ubiquitylated in vivo but the ubiquitylation of Lys-269 is critical for cyclin D1 targeting to the proteasome in vivo. To test this notion, NIH-3T3 cells stably expressing wild-type cyclin D1, D1-K269R and D1-T286A were treated with MG132, followed by immunoprecipitation with M2-agarose and immunoblot with the antibody to the 19S cap proteasome non-ATPase subunit PSMD7 (Fig 3D). D1-K269R and D1-T286A exhibited reduced association with PSMD7 compared to the wild-type cyclin D1, suggesting that D1-K269R targeting to proteasome is attenuated in vivo.
D1K269R exhibits increased nuclear localization and exhibits neoplastic potential
Attenuation of cyclin D1 proteolysis through the inhibition of SCFFbx4/αB-crystallin function leads to the accumulation of cyclin D1 in the nuclear compartment (Barbash et al., 2008). We therefore anticipated that mutation of a critical lysine acceptor in cyclin D1 might also result in nuclear accumulation of the mutant cyclin D1 allele. We analyzed the subcellular localization of D1-K269R in NIH-3T3 cells. Wild-type D1 localized to the nucleus in 10% of asynchronous cells and D1-K269R in 31%, while constitutively nuclear D1-T286A mutant had 100% nuclear localization (Fig 4A-B). Therefore, consistent with our previous observation, the disruption of cyclin D1 proteolysis leads to an increase in the fraction of nuclear cyclin D1. Additionally, expression of both, wild-type and K269R cyclin D1, only slightly changed the distribution of cells during cell cycle (Fig 4C), suggesting that overexpression of both wild-type cyclin D1 and D1-K269R similarly contribute to cellular proliferation.
Figure 4. D1-K269R has increased nuclear localization.
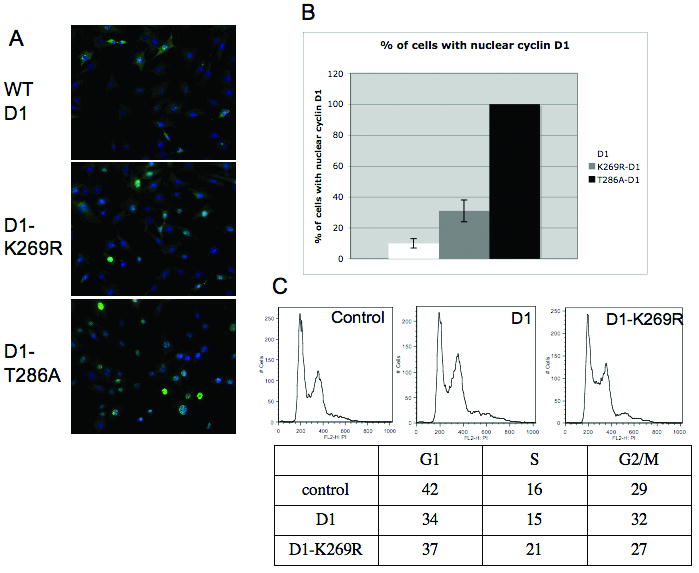
A. NIH-3T3 cells stably expressing WT cyclin D1, D1-K269R and D1-T286A were subjected to immunofluorescence analysis with cyclin D1 antibody (green), slides were counterstained with DAPI (blue). B. Quantification of A from three independent experiments (error bars represent standard deviation). C. Cell cycle analysis of asynchronous NIH-3T3 cell lines by PI/FACS.
Constitutively nuclear cyclin D1 mutant, D1-T286A, is a potent oncogene in in vitro and in vivo models. Additionally, we have previously demonstrated that disruption of cyclin D1 proteolysis through the stable knockdown of either Fbx4 or αB-crystallin in NIH-3T3 cells results in cellular transformation. Since D1-K269R is resistant to ubiquitin-dependent proteolysis and has increased nuclear localization, we investigated whether D1-K269R will transform NIH-3T3 cells. While wild-type cyclin D1 was weakly transforming, D1-K269R promoted the growth of cells in soft agar (Fig 5A-B) and resulted in foci formation in vitro (Fig 5C). In summary, the mutation of Lys-269 renders cyclin D1 resistant to ubiquitin-dependent proteolysis, leading to the accumulation of cyclin D1 in nuclear compartment and cellular transformation.
Figure 5. K269R cyclin D1 transforms NIH-3T3 cells.
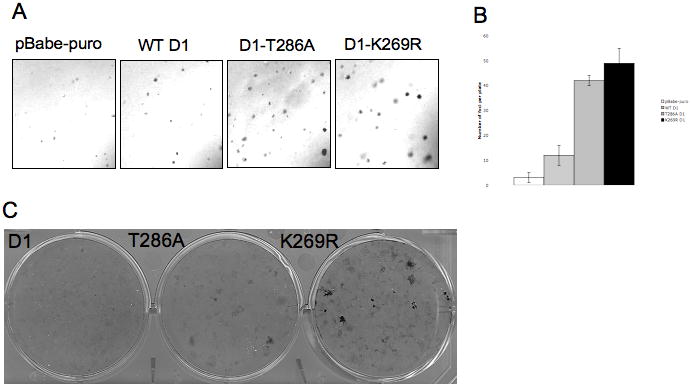
A. Anchorage –independent growth of NIH-3T3 cells stably expressing WT cyclin D1, D1-T286A, D1-K269R or pBabe-puro vector was analyzed by growth in soft agar. Colonies were visualized by microscopy. B. Quantification of triplicate samples shown in A. C. Foci formation ability of NIH-3T3 cells stably expressing WT cyclin D1, D1-T286A and D1-K269R was determined by Giemsa stain of foci grown for 21 days.
Discussion
Efficient degradation of proteins through the 26S proteasome requires that proteins are unfolded prior access to the catalytic core of the proteasome. Polyubiquitylation of substrates, which generally occurs on internal lysine residues within the substrate, is postulated to serve as the signal that targets a substrate to the proteasome cap where unfoldases and deubiquitinating enzymes unfold the substrate and recycle ubiquitin prior to its degradation. Previous studies have addressed the role of cyclin D1 lysines in protein turnover and revealed that the mutation of Lys-112 and Lys-114 extend cyclin D1 protein half-life; however these mutations also disrupt a functional interaction between cyclin D1 and its catalytic partner, cdk4. The degradation of free cyclin D1 is phosphorylation-independent (Germain et al., 2000) and thus is not SCFFbx4/αB-crystallin-dependent (Lin et al., 2006). Because cyclin D1 is rarely detected as a monomer (Parry et al., 1999), this mechanism likely contributes to the reduced pool of free cyclin D1. In the current work, we have identified a lysine residue within cyclin D1, near the GSK3β phosphorylation site, that targets cyclin D1 for polyubiquitylation by the SCFFbx4/αB-crystallin ligase. Our findings reveal that Lys-269 of cyclin D1 is essential for cyclin D1 proteolysis. Importantly, the K269R mutation disrupts cyclin D1 ubiquitylation by SCFFbx4/αB-crystallin complex in vitro, implicating the Lys-269 as a bona fide ubiquitin acceptor site for this E3 ligase.
One surprising observation that has stemmed from this series of experiments is that D1-K269R can still be ubiquitylated in vivo. However, while it is ubiquitylated, it remains as resistant to proteasome-dependent degradation as the non-phosphorylatable cyclin D1-T286A allele (Diehl et al., 1997). The potential of cyclin D1 to be ubiquitylated on alternative lysine residues is not unanticipated. In fact, there are instances wherein client proteins are ubiquitylated on the amino-terminal residue in the complete absence of internal lysines (Bloom et al., 2003; Breitschopf et al., 1998). However, the absence of efficient degradation of ubiquitylated D1-K269R demonstrates that it remains a poor substrate for the 26S proteasome. One possible explanation that we considered was alternative ubiquitin linkages, such as K63, which are not associated with degradation. However, ubiquitylation of D1-K269R is dependent upon K48 in ubiquitin. A second possibility is that the ubiquitin chains on D1-K269R are of insufficient length and thus poorly recognized by the proteasome. Consistent with this notion, binding of D1-K269R and PSMD7 (a subunit of the proteasome cap) was reduced relative to that observed with wild-type cyclin D1.
Our work demonstrates that efficient proteasome-dependent destruction of cyclin D1 requires ubiquitylation of a specific lysine acceptor. Similar mechanisms have been described for cyclin A, where the mutations in lysines proximal to D-box (Lys37, 54, 68) extend the half-life of cyclin A that is still ubiquitylated in vivo(Fung et al., 2005). Additionally, our experiments demonstrate that the degradation of D1-K269R is promoted by Fbx4 overexpression, indicating that in the absence of the Lys-269 acceptor site, Fbx4 can promote the degradation of cyclin D1, albeit with reduced efficacy, through alternative ubiquitylation sites. An alternative possibility is that the stability of cyclin D1 is controlled through the balance of the ubiquitylation and the modifications by other ubiquitin-like proteins, such as SUMO and ISG15 (Feng et al., 2008), where ubiquitin-like modifiers compete for the acceptor lysines and therefore K269R mutation might lead to the disruption of ubiquitylation of proximal lysines (such as Lys-238) allowing for their modification with ubiquitin-like proteins and, potentially by such means preventing cyclin D1 degradation.
D1-K269R accumulates in cells to higher levels than wild-type cyclin D1 and D1-K269R localization is shifted to the nuclear compartment. This is consistent with our previous observation that inhibition of Fbx4-dependent cyclin D1 ubiquitylation and proteolysis leads to the accumulation of cyclin D1 in the nuclei (Barbash et al., 2008). We demonstrate that D1-K269R is normally phosphorylated at Thr-286, the residue required for the nuclear export, suggesting that the increase in the nuclear fraction of D1-K269R is not a result of disrupted phosphorylation-dependent nuclear export. The increase in the nuclear fraction of D1-K269R is likely a result of the increased nuclear import of cyclin D1 upon the inhibition of its proteolysis, but the precise mechanism of relocalization requires additional studies.
Destruction of nuclear cyclin D1 is required to prevent cyclin D1-dependent cell transformation (Aggarwal et al., 2007; Gladden AB, 2006). Consistent with its increased nuclear accumulation, expression of D1-K269R triggered neoplastic transformation of murine fibroblasts, similar to overexpression of cyclin D1-T286A or downregulation of Fbx4 (Barbash et al., 2008). One of the mechanisms whereby nuclear cyclin D1 promotes tumorigenesis is through attenuation of Cullin-4 expression with subsequent accumulation of its substrate, replication factor, Cdt1 (Aggarwal et al., 2007). As a consequence, cells expressing constitutively nuclear cyclin D1 exhibit over-replication phenotype and chromosomal abnormalities. Indeed, cells expressing D1-K269R exhibit significant downregulation of Cul4A expression (data not shown), suggesting a common with T286A mechanisms of oncogenic transformation.
In summary, our work demonstrates that Lys-269 is ubiquitin acceptor site for SCFFbx4/αB-crystallin. The data presented underlines the importance of Lys-269 ubiquitylation for cyclin D1 degradation during cell cycle and in response to genotoxic stress. Our experiments suggests that despite the fact that multiple lysines in cyclin D1 can be ubiquitylated in vivo, the ubiquitylation of Lys-269 provides critical basis for cyclin D1 degradation.
Acknowledgments
The authors wish to thank Serge Fuchs for the plasmids encoding the various ubiquitin lysine-arginine mutants, Petia Zamfirova and Margarita Romero for technical assistance. This work was supported by CA93237 (NIH) and a Leukemia & Lymphoma Scholar award (JAD).
References
- Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21:2908–22. doi: 10.1101/gad.1586007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash O, Zamfirova P, Lin DI, Chen X, Yang K, Nakagawa H, et al. Mutations in Fbx4 Inhibit Dimerization of the SCF(Fbx4) Ligase and Contribute to Cyclin D1 Overexpression in Human Cancer. Cancer Cell. 2008;14:68–78. doi: 10.1016/j.ccr.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, et al. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene. 2006;25:6291–303. doi: 10.1038/sj.onc.1209644. [DOI] [PubMed] [Google Scholar]
- Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115:71–82. doi: 10.1016/s0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998;17:5964–73. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–72. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Feng Q, Sekula D, Guo Y, Liu X, Black CC, Galimberti F, et al. UBE1L causes lung cancer growth suppression by targeting cyclin D1. Mol Cancer Ther. 2008;7:3780–8. doi: 10.1158/1535-7163.MCT-08-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Sekula D, Muller R, Freemantle SJ, Dmitrovsky E. Uncovering residues that regulate cyclin D1 proteasomal degradation. Oncogene. 2007;26:5098–106. doi: 10.1038/sj.onc.1210309. [DOI] [PubMed] [Google Scholar]
- Fung TK, Yam CH, Poon RY. The N-terminal regulatory domain of cyclin A contains redundant ubiquitination targeting sequences and acceptor sites. Cell Cycle. 2005;4:1411–20. doi: 10.4161/cc.4.10.2046. [DOI] [PubMed] [Google Scholar]
- Germain D, Russell A, Thompson A, Hendley J. Ubiquitination of free cyclin D1 is independent of phosphorylation on threonine 286. J Biol Chem. 2000;275:12074–9. doi: 10.1074/jbc.275.16.12074. [DOI] [PubMed] [Google Scholar]
- Gladden AB, W R, Aggarwal P, Wasik MA, Diehl JA. Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene. 2006;25:998–1007. doi: 10.1038/sj.onc.1209147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Heller H, Eytan E, Kaklij G, Rose IA. Role of the alpha-amino group of protein in ubiquitin-mediated protein breakdown. Proc Natl Acad Sci U S A. 1984;81:7021–5. doi: 10.1073/pnas.81.22.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331–42. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–66. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Beerm DG. Molecular biology of upper gastrointestinal malignancies. Semin Oncol. 2004;31:476–86. doi: 10.1053/j.seminoncol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–76. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, Hardisson D, Sarrio D, Prat J, et al. Cyclin D1 gene (CCND1) mutations in endometrial cancer. Oncogene. 2003;22:6115–8. doi: 10.1038/sj.onc.1206868. [DOI] [PubMed] [Google Scholar]
- Parry D, Mahony D, Wills K, Lees E. Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol. 1999;19:1775–83. doi: 10.1128/mcb.19.3.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontano LL, Aggarwal P, Barbash O, Brown EJ, Bassing CH, Diehl JA. Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability. Mol Cell Biol. 2008;28:7245–58. doi: 10.1128/MCB.01085-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Itoh F, Hareyama M, Satoh M, Hinoda Y, Seto M, et al. Association of cyclin D1 expression with factors correlated with tumor progression in human hepatocellular carcinoma. J Gastroenterol. 1999;34:486–93. doi: 10.1007/s005350050301. [DOI] [PubMed] [Google Scholar]
- Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci U S A. 1995;92:11259–63. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane M, Harumiya Y, Ishida N, Hirai A, Miyamoto C, Hatakeyama S, et al. Down-regulation of p27(Kip1) by two mechanisms, ubiquitin-mediated degradation and proteolytic processing. J Biol Chem. 1999;274:13886–93. doi: 10.1074/jbc.274.20.13886. [DOI] [PubMed] [Google Scholar]
- Worsley SD, Jennings BA, Khalil KH, Mole M, Girling AC. Cyclin D1 amplification and expression in human breast carcinoma: correlation with histological prognostic markers and oestrogen receptor expression. Clin Mol Pathol. 1996;49:M46–M50. doi: 10.1136/mp.49.1.m46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–56. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]