

Abstract
The A/M2 proton channel of influenza A virus is a target for the anti-influenza drugs amantadine and rimantadine, whose effectiveness was diminished by the appearance of naturally occurring point mutants in the A/M2 channel pore, among which the most common are S31N, V27A and L26F. We have synthesized and characterized the properties of a series of compounds, originally derived from the A/M2 inhibitor BL-1743. A lead compound emerging from these investigations, spiro[5.5]undecan-3-amine, is an effective inhibitor of wild type A/M2 channels and L26F and V27A mutant ion channels in vitro, and also inhibits replication of recombinant mutant viruses bearing these mutations in plaque reduction assays. Differences in the inhibition kinetics between BL-1743, known to bind inside the A/M2 channel pore, and amantadine were exploited to demonstrate competition between these compounds; consistent with the conclusion that amantadine binds inside the channel pore. Inhibition by all of these compounds was shown to be voltage-independent, suggesting that their charged groups within the N-terminal half of the pore, prior to the selectivity filter that defines the region over which the transmembrane potential occurs. These findings not only help define the location and mechanism of binding of M2 channel-blocking drugs, but also demonstrate the feasibility of discovering new inhibitors that target this binding site in a number of amantadine-resistant mutants.
Keywords: Influenza A virus, M2 channel, proton channel, amantadine, spiro amine
Influenza A virus is a continuing cause of mortality and morbidity on an annual basis (1) and thus presents an important target for pharmaceutical investigation. Two classes of anti-influenza agents are available for human use: adamantane derivatives amantadine and rimantadine which target the virus A/M2 ion channel and oseltamivir and zanamivir, which targets the viral surface protein neuroaminidase (2). The use of amantadine and rimantadine is limited by the wide distribution of drug resistant virus variants, including the recently isolated influenza A (H1N1) virus (3–5).
The A/M2 protein of the influenza A virus forms a pH-gated proton channel that is essential for virus replication (6). The mature channel consists of four identical subunits of 96 amino acids, each subunit is a type III integral membrane protein (Nout, Cin) (7);. The highly conserved H37xxxW41 motif located in the single transmembrane domain of the protein is responsible for its channel activity and proton selectivity (7, 8). The activity of wild type (wt) A/M2 channels is known to be efficiently inhibited by amantadine, BL-1743 and its derivative azaspiro[5,5] undecane (spiro piperidine 20, Table 1) (Figure 1) (2, 9, 10). Naturally occurring point mutations of the pore lining residues located outside of the H37xxxW41 motif, such as L26F, V27A, A30T, S31N and G34E result in the formation of amantadine-insensitive influenza A virus phenotypes (3, 11–13). Extensive studies suggest that these residues are involved in the formation of the binding pocket for the drug (14–18). Amantadine-resistant phenotypes are prevalent in the currently circulating influenza A virus H3N2 and H1N1 2009 strains. Although the S31N mutation was observed in more then 90% of influenza A cases in certain years (19, 20), other amantadine insensitive phenotypes like L26F and V27A were isolated from influenza A patients with the emerging frequencies of 8–67% (19, 20). Other amantadine-resistant mutations have been found much less frequently (11, 20). Being resistant to amantadine, these naturally occurring mutants are also insensitive to all other known organic A/M2 channel inhibitors, including BL-1743 and spiro piperidine 20 (9, 10). Thus there is a great need for novel anti-influenza drugs that target the most common amantadine resistant phenotypes, S31N, V27A and L26F (19, 20).
Table 1.
Inhibition efficiency of the synthesized compounds on A/M2 channels.
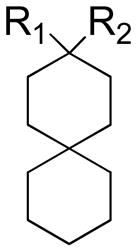 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | AM2 channel activity after 100μM compound inhibition | IC50 (μM) | Kd (μM)a |
| Amantadine | 6% | 15.76±1.24 | 15.17 | ||
| BL-1743 | 25% | 45.31±3.92 | 193.54 | ||
| 8 | H | NH3+Cl− | 11% | 12.59±1.11 | 9.16 |
| 9 | H | NH2+Cl−CH3 | 8% | 15.72±1.89 | 46.36 |
| 10 | H |  |
8% | 14.60±1.70 | 11.50 |
| 11 | H | 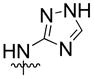 |
25% | n.d | |
| 12 | H | NH2+Cl−(CH3)2NH3+Cl− | 100% | n.d | |
| 13 | H | NH2+Cl−(CH3)3NH3+Cl− | 100% | n.d | >500 |
| 14 | H | 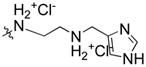 |
91% | n.d | |
| 15 | OH | 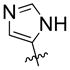 |
40% | n.d | |
| 16 | H |  |
13% | 12.54±1.24 | |
| 17 | F | 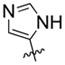 |
31% | 57.57±2.24 | |
| 18 | H | 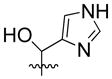 |
30% | n.d | |
| 19 | H | 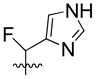 |
25% | 29.19±1.46 | |
| 20 | 5% | 0.92±0.11 | 12.87 | ||
Kd was obtained by global fitting of Circular Dichroism (CD) data of ligand titration to A/M2TM (22–46) using Igor Pro (wavemetrics). The variation of Kd values is ±25% based on different fitting values obtained from three repeats of amantadine titration and two repeats of compound 20 titration. See supplementary information for details.
Figure 1.

Chemical structures of A/M2 channel inhibitors.
In the current study we synthesized a family of spiro-[5,5]-undecane compounds based on the structure of the previously investigated A/M2 channel inhibitor BL-1743. We found a simple amino derivative of BL-1743, spiro[5.5]undecan-3-amine (spiran amine 8, Table 1) to be effective not only for inhibition of the wt A/M2 channel, but also for inhibition of two widely occurring amantadine resistant mutants, L26F and V27A. The efficiency of this compound was investigated on A/M2 channels heterologously expressed in Xenopus oocytes and confirmed by the in-vivo plaque reduction assay of recombinant influenza A virus.
The pharmacologically relevant binding site for amantadine has been found to lie either inside (15), or outside (21, 22) the pore, although the physiological relevance of the latter finding has not been confirmed with either electrophysiology in oocytes or plaque reduction assays with recombinant virus (23). However, BL-1743 was shown to inhibit channel activity by binding inside the channel pore (24). Previous findings have shown that the kinetics of A/M2 channel inhibition by BL-1743 are more rapid than those reported for amantadine (9, 25), making it possible to test for competition between these drugs to determine whether they compete for the same binding site inside the channel pore. Our results support the previously published structural and functional studies that showed that amantadine inhibits the A/M2 channel by coordinating with pore lining residues (12, 15, 16). We found that inhibition by amantadine, BL-1743, spiro piperidine 20 and spiran amine 8, all of which are positively charged at physiological pH, is independent of membrane voltage, consistent with binding in the N-terminal portion of the pore.
The current study shows that a novel compound, spiran amine 8, is a potent inhibitor of the L26F and V27A amantadine resistant mutants of the A/M2 protein. Additional evidence supports the conclusion that amantadine binds inside the N-terminal half of the channel pore. These findings show that novel anti-influenza drugs, capable of targeting wt and amantadine resistant virus phenotypes, can be identified and that the N-ternial part of the pore is a good target for such drugs.
MATERIALS AND METHODS
Spiran AM2 inhibitor library synthesis
The syntheses of the primary amine analog (8) of spiropiperidine-azaspiro[5,5]undecane and the methyl substituted secondary amine 9 are shown in Scheme 1. Intermediate spiro[5.5]undec-1-en-3-one 1 was prepared from both acid catalyzed one-pot Robinson annulation reaction and through Diels-Alder adduct followed by acid hydrolysis and aldol ring formation. The acid-catalyzed annulation often led to low yields (62% or lower) due to acid catalyzed polymerization of methyl vinyl ketone as evidenced by black oily substance formed in the reaction flask (26). While catalysis with proline derivatives might allow circumvention of these problems, we found the alternative Diels-Alder route provided better overall yields (75%) (27). Hydrogenesis of enone 1 with Pd/C with an H2 balloon gave spiro[5.5]undecan-3-one 2. Conversion of ketone 2 to amine 8 was achieved by treatment with hydroxylamine followed by LiAlH4 reduction. Methylamine 9 was prepared by reductive amination of 8 with formaldehyde as reported.
Scheme 1.

Synthesis of spiran amine 8, 9 and guanidine 10.
Syntheses of spiran triazole 11 and spiran amine 12–14 with extended linkers in scheme 2 were accomplished by reductive amination as described before.
Scheme 2.

Synthesis of spiran triazole 11 and spiran amine 12, 13 and 14 with extended linkers.
Compound 15, with an imidazole head group, was synthesized by nucleophilic attack of imidazol-4-yl anion (generated by treatment of N-trityl 4-iodoimidazole) onto ketone 2 (28), followed by deprotection in TFA/DCM as in scheme 3. The hydroxyl group in 15 was either reduced by Et3SiH/BF3*OEt2 to give 16 or fluorinated by DAST to give 17 after deprotection. Ketone 2 was converted to aldehyde 6 by the Wittig reaction, followed by acid hydrolysis. Comopunds 18 and 19 were then synthesized from compound 6 in the same manner as described for 15 and 17.
Scheme 3.

Synthesis of spiran with imidazole head group 15, 16, 17.
Molecular Biology, in vitro cRNA transcription
The cDNA encoding to the Influenza virus A/Udorn/72 A/M2 protein and to the A/M2 amantadine insensitive mutants were inserted into pGEMHJ (a gift from N. Dascal Tel-Aviv University, Israel) for the expression on Xenopus oocytes. cRNA was prepared as previously described (29).
Heterologous Expression and Electrophysiological Recordings
Stage V–VI Xenopus laevis oocytes were prepared as described previously (30). Oocytes injection and TEVC electrophysiological measurements were done as previously described (29). Amantadine (Sigma, St. Louis, MO) was applied to inhibit A/M2 induced currents. Data were analyzed using ORIGIN 8.0 software (OriginLab, Northampton, MA).
Cells, viruses and plasmids
293-T and Madin–Darby canine kidney (MDCK) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, Valencia, CA) supplemented with 10% FBS. Influenza A/Udorn/72 virus (wt) and mutant viruses were propagated in MDCK cells overlaid with serum-free DMEM containing 3.0 μg/ml N-acetyl trypsin (NAT; Sigma-Aldrich, St. Louis, MO) at 37°C. WT and mutant virus (V27A/L38F) were generated by using reverse genetics from cDNAs essentially as described previously (31). The eight genome- sense (pHH21) plasmids and four protein-expressing (pcDNA3.1) plasmids used to generate influenza virus by reverse genetics have been described previously (31). Mutation into the M2 gene in pHH21 vector was generated using Quick Change mutagenesis (Stratagene, La Jolla, CA). 293T cells were transfected using TransIT-LT1 (Mirus, Madison, WI) according to the manufacturer’s protocols. Virus stocks were propagated in MDCK cells, and the virus titers were determined by plaque assay on MDCK cells. For determination of viral genome sequences viral RNA was extracted by using the QIAamp viral RNA kit (Qiagen, Valencia, CA), followed by Super Reverse Transcriptase (Molecular Genetic Resources, Tampa, FL.) and using genome-specific primers, and amplified with AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA). The complete nucleotide sequences of the M genes were determined using a 3100-Avant genetic analyzer (Applied Biosystems).
Plaque reduction assays
Confluent monolayers of MDCK cells were incubated with the wt Udorn virus (100 plaque forming units (p.f.u.) per well) and V27A/L38F mutant virus (1000 and 100 p.f.u. per well) in DMEM-1% bovine serum albumin for 1 h at 37°C. The inoculums were removed, and the cells were washed with phosphate-buffered saline (PBS). The cells were then overlaid with DMEM-containing 0.6% Avicel microcrystalline cellulose (FMC BioPolymer, Philadelphia, PA) and NAT (2.0 μg/ml). To examine the effect of drugs (BL-1743, spiran amines and amantadine) on plaque formation, monolayers were preincubated with DMEM supplemented with indicated concentrations of the drugs at 37°C for 30 min, and virus samples were preincubated with DMEM-1% BSA with indicated concentrations of the drugs at 4°C for 30 min before infection. At 2 to 3 days after infection, the monolayers were fixed and stained with naphthalene black dye solution (0.1% naphthalene black, 6% glacial acetic acid, 1.36% anhydrous sodium acetate).
RESULTS
Structure-activity relationship (SAR) of 3-substituted spiro-[5,5]-undecanes
Our previous SAR study of 2-[3-azaspiro(5,5)undecanol]-2-imidazoline (BL-1743) revealed a very potent spiro-piperidine compound 20 with IC50 of 0.9 μM (10). However, compounds in this series failed to inhibit amantadine-resistant variants of A/M2, which prompted us to test alternative structures. In particular, a consideration of the overlay of the parent compound on derivatives of amantadine suggested that conversion of the piperidine in spiro piperidine 20 to a 3-amino-cyclohexyl amine while maintaining the second spiro-6-member ring would produce a more effective inhibitor. We therefore synthesized a family of spiro-[5,5]-undecanes, in which the 3-position was substituted with amines or other basic substituents (Table 1).
The compounds were tested on A/M2 channels expressed in Xenopus oocytes using TEVC technique. The inhibitory effect of the compounds was confirmed by in-vivo plaque reduction assays of influenza A virus (A/Udorn/72). The simple amino-derivative, 8, showed an activity on par with that of amantadine. However, further substitutions to the amine tended to cause a loss in activity. N-methylation (compound 9) led to a slightly less potent compound, while the guanidine derivative, 10, had similar potency as methylamine 9. Modifying the amine by addition of polar substituents and extended linkers (compounds 11 – 14) led to a marked decrease in activity. On the other hand, replacing the amine with an imidazole group caused a slight decrease in activity (compound 16). We also examined additional substitutions at the 3-methylene of 16, through the introduction of a hydroxyl and fluoro-substituent in 15 and 17, respectively. These substitutions gave rise to compounds with lower potency. Furthermore, the similarly substituted compounds 18 and 19 had decreased activity compared to the primary amine 8. In summary, these data show that the primary amino group is likely to be a nearly optimal substituent for the spiro-[5.5]-undecane scaffold. We therefore turned our attention to determining how the piperidine for cyclohexylamine substitution in 20 vs. 8 affected the ability of these compounds to inhibit amantadine-resistant forms of A/M2.
Inhibition effect of spiran amine compound 8 on wt and amantadine insensitive A/M2 channels
Amantadine resistant mutants carry naturally occurring point mutations of the pore lining residues of the A/M2 channel (3, 11, 12). Extensive structural, electrophysiological and in-silico investigations suggest that these residues form the binding pocket for the drug (14–18). We have tested the effect of spiran amine 8 on wt A/M2 channels and A/M2 channels with altered amantadine sensitivity and compared the inhibition by compound 8 to that of amantadine and BL-1743. We found that spiran amine 8 efficiently inhibits the activity of A/M2 wt channels and of A/M2-V27A mutants, with IC50 values of 12.6 μM and 84.9 μM respectively (Figure 2; Table 2). The inhibition of V27A mutants is particularly interesting, given that amantadine, BL-1743, and spiro piperidine 20 gave less than 10% inhibition of the mutant, when applied at 100 μM concentration. It is also interesting to note that the mutant V27G, which is also naturally occurring (32) is highly resistant to all compounds tested (Table 2).
Figure 2. Inhibition efficiency of amantadine, BL-1743 and spiran amine 8 on wt A/M2 channels and A/M2 L26F, V27A and S31N mutants.

Channel activity was assayed by TEVC for A/M2 channels expressed in Xenopus oocytes. Responses in the presence of various concentrations of an inhibitor (I) were normalized to the current evoked by application of the activating (pH 5.5) solution without inhibitor (I0). The experimental data are the average of three independent experiments. Each point is the mean (±SD) of 5–8 oocytes. IC50 values in μM: A/M2 with amantadine IC50=15.76±1.24; with BL-1743 IC50=46.25±3.56; with spiran amine 8 IC50=12.59±1.11. A/M2-L26F with amantadine IC50=164.46±14.40; with BL-1743 IC50>10 mM; with spiran amine 8 IC50=30.62±8.13. A/M2-V27A with amantadine IC50=1840; with BL-1743 IC50>10 mM; with spiran amine 8 IC50=84.92±13.61. A/M2- S31N with amantadine IC50=237.01±22.14; with BL-1743 IC50>10 mM; with spiran amine 8 IC50>10 mM.
Table 2. Inhibition effect of amantadine, BL-1743, spiran amine 8 and spiran amine 9 on A/M2 wt and amantadine insensitive mutant channels.
Data presented as a percent of remaining A/M2 activity after application of an inhibitor (100 μM) for 2 min. The experimental data are the average of three independent experiments. Each point is a mean (±SD) of 5–7 oocytes.
| BL-1743 | SPIRAN AMINE 8 | SPIRAN AMINE 9 | AMANTADINE | |
|---|---|---|---|---|
| % remaining activity after application of 100 μM compound | ||||
| A/M2 | 25.3±1.3 | 10.6±0.7 | 8.4±1.1 | 6.1±1.0 |
| A/M2-L26F | 89.2±4.8 | 32.4±3.3 | 72.6±8.4 | 51.8±3.5 |
| A/M2-V27A | 97.4±1.8 | 46.6±6.6 | 70.2±1.55 | 93.07±1.9 |
| A/M2-V27G | 82.9±9.3 | 87.9±6.1 | 89.6±4.1 | 95±4.9 |
| A/M2-A30T | 102±2.3 | 98.1±7.0 | 101. ±3.2 | 104.6±4.3 |
| A/M2-S31N | 100±0.67 | 99.1±0.9 | 100±4.1 | 65.3±2.7 |
| A/M2-G34E | 89.8±6.9 | 96. ±5.9 | 101.7±2.8 | 100.4±6.7 |
We next examined the ability of compound 8 to inhibit other pore-lining mutants. S31N is a highly frequent mutation, which gives rise to decreased sensitivity to amantadine (IC50 = 237.0 μM versus 15.8 μM for the wt A/M2 channel, Figure 2), and complete resistance to rimantadine (IC50 > 10mM). Similarly, compound 8 showed little inhibition of this mutant. Other mutations deeper inside the pore than V27 (A30T and G34E) completely eliminate the ability of all drugs tested to inhibit the channel (Table 2).
By contrast to the other mutations considered here, L26F does not involve a pore-lining residue, and instead involves a partially largely lipid-exposed residue that packs at the interface between the helices adjacent to the V27. Thus, this residue is expected to play a more subtle and less direct role in defining the steric properties of the drug-binding site. Indeed, amantadine inhibits this mutant with reduced affinity (IC50=164.5 μM), versus the much larger decreases seen for other variants. Compound 8 is an even more potent inhibitor of A/M2 L26F (IC50 = 30.6 μM, Figure 2).
The inhibitory effect of compounds 8 and 9 on the recombinantly expressed A/M2 wt and mutant channels was confirmed by in-vivo plaque reduction assays of influenza A viruses. Plaque formation of wt influenza virus (A/Udorn/72) was inhibited by amantadine, 8 and 9 at concentrations ranging from 0.5 to 5 μM (Figure 3A), with compound 8 showing slightly more potent activity than compound 9. BL- 1743 inhibited wt virus plaque formation only at high concentrations (50–100 μM) (Figure 3B). On the other hand, plaque count and size resulting from infection by an influenza virus A/M2-V27A/L38F, that contains the M2-V27A mutation was reduced by 50 μM spiran amine 8 (Figure 3C). These findings are consistent with the electrophysiological data (Figure 1). In comparison, amantadine and BL-1743 at the same concentration range failed to inhibit plaque formation of A/M2-V27A/L38F viruses (Figure 3C). The L38F mutation is naturally found in the Weybridge strain of the virus. This mutation is pharmacologically silent to the drugs used and does not affect M2 channel function in electrophysiological recordings (25, 33, 34). The activity of the double mutant A/M2-V27A/L38F was tested by TEVC and compared to that of a single mutant A/M2-V27A. We found that sensitivity of the double mutant channel to the tested compound was comparable to that of the single A/M2-V27A mutant. The A/M2-V27A/L38F mutant channels were not sensitive to either amantadine, or BL1743, while being efficiently inhibited by compound 8 (A/M2-V27A/L38F IC50=78.21±15.06; A/M2-V27A IC50= 84.92±13.61). Based on these findings we decided that the results of the plaque formation assay performed on the double mutant A/M2-V27A/L38F adequately represent the behavior of the single A/M2-V27A mutant.
Figure 3. In vivo plaque reduction assay.

Wt influenza virus (A/Udorn/72) and influenza virus containing mutations in the M2 TM domain (V27A/L38F) were recovered from cloned DNA and assayed for plaque formation on MDCK cells in the presence or absence of drugs as described in Materials and Methods. A. Effects of amandadine, BL-1743 and compounds 8 and 9 (0.5 and 5 μM) on Udorn plaque formation. B. Effect of BL-1743 (50 and 100 μM) on influenza virus plaque formation. C. Effects of amantadine, BL-1743 and spiran amine 8 (50 μM) on influenza mutant virus V27A/L38F plaque formation. C, upper panel: ~1000 p.f.u./well of mutant virus were used. C, bottom panel: ~100 p.f.u./well of mutant virus were used. Plaque count: no drug – 99 plaques/well; amantadine 50μM – 82 plaques/well; BL-1743 50 μM – 86 plaques/well; spiran amine 8 50 μM – 37 plaques/well.
Thus, spiran amine 8 is capable of efficiently inhibiting not only wt A/M2 channels and A/M2-L26F and A/M2-V27A mutant channels expressed in oocytes, but it also prevents replication of wt virus and these mutant recombinant viruses. As discussed below, we attribute to the greater potency of compound 8 in the plaque versus the electrophysiological studies to kinetic effects arising from the slow kinetics of binding of this class of inhibitors. Compounds are incubated with oocytes for brief periods in the electrophysiological experiments, but much longer periods in the plaque-binding studies.
We also measured the binding of selected drugs to the transmembrane domain of wt A/M2 protein (M2TM, residues 22–46), using a spectroscopic assay that relies upon changes in the CD spectrum of M2 induced by binding of drugs (Table 1 and described in supplementary materials). All drugs bound in a stoichiometry of approximately one drug/tetramer. At pH 7.4, potent compounds were found to bind to the transmembrane tetrameric form of M2TM with low μM binding constants. Compounds 8 and 10 were found to be approximately equipotent with amantadine, displaying dissociation constants in the range of 7 – 8 μM (Table 1). In agreement with the plaque assay, compound 9 was approximately four-fold less potent than amantadine or compound 8. These data support the expectation that the drugs inhibit the channel activity by binding directly to the A/M2 proton channel.
Competition among inhibitors
In 1999 Gandhi and co-workers concluded from competition experiments with Cu(II) that BL-1743 inhibits A/M2 channel activity by binding inside the channel pore (24). However, conflicting structural studies have shown that adamantane derivatives bind either inside (15), or outside (21) the channel pore, although the pharmacological relevance of the latter finding has not been confirmed (23). Furthermore, solid-state NMR experiments have shown that a member of the spiran series of compounds binds A/M2 in a manner similar to that of amantadine (10). Given that BL-1743 appears to bind within the pore, we were interested to determine whether amantadine binds competitively with respect to this compound, or whether it binds to an independent site.
The second-order rate constant (κon) for the association and inhibition of the wt A/M2 channel with BL- 1743 was calculated to be 720 M−1 s−1 (9), which is comparable with the κon of amantadine (600 to 900 M−1 s−1) (25). However, the dissociation rate constant (κoff) for BL-1743 is approximately an order of magnitude faster than that calculated for amantadine (10−3 s−1 and 10−4 s−1 respectively) (9, 25). We exploited this difference in κoff of BL-1743 and amantadine to investigate the mechanism of A/M2 channel inhibition by amantadine by testing whether this drug competes with BL-1743 for the inhibition of A/M2 channel activity. For these experiments A/M2 channels were expressed in Xenopus oocytes, and the channel activity was assayed by TEVC. A/M2 channel activity was evoked by application of the low pH activating solution (pH 5.5). In the control experiments the channel activity was inhibited by saturating concentrations of either BL-1743 (1000 μM) or amantadine (100 μM) alone (Figure 4A and B). As seen from Figure 4A and 4B, the inhibition of A/M2 was essentially fully reversible in the time frame of 5 min for BL-1743, but only about 10% complete for amantadine. This finding is consistent with the slower off-rate for amantadine than for BL-1743. Compound 8 also showed an off-rate similar to that of amantadine, as expected from their similar IC50 values (Figure 2, Figure 4C).
Figure 4. Inhibition of A/M2 channel activity by saturating concentrations of BL-1743, amantadine and spiran amine.

8. A–C; Representative traces of A/M2 channel activity inhibited by either BL-1743 (1000 μM, A), amantadine (100 μM, B), or spiran amine 8 (100 μM, C). A/M2 channel activity was induced by application of the activating solution (pH 5.5, red horizontal bar above the trace). The currents were inhibited by the application of the activating solution containing either BL-1743 (green horizontal bar, A), amantadine (yellow horizontal bar, B), or spiran amine 8 (orange horizontal bar, C) for 60–90 sec. After the maximal inhibition was achieved, oocytes were superfused with the non-activating solution (pH 8.5, second blue horizontal bar) for 5 min to allow current recovery and the channel activity was assayed again by the application of the activating solution (second red horizontal bar). Note that recovery was complete only for BL-1743.
We next examined the competition between BL-1743 and amantadine, chosen as a representative example of the slow-dissociating class of inhibitors. We subjected the oocytes to sequential treatment with drugs and drug-free recording solutions in a manner designed to probe both the kinetic and thermodynamic aspects of inhibition. A resting oocyte, incubated in the non-activating solution (pH 8.5), was treated as follows. 1) The oocyte was acidified by the application of the activating solution (pH 5.5) (red bar in Figure 5); 2) The oocyte was pulsed with the activation solution containing rapidly reversible BL-1743 at various concentrations ranging from 3 to 1000 μM for 60 sec (green bar); 3) BL- 1743 in the activating solution was replaced with the slowly reversible inhibitor amantadine (100 μM) for an additional 90 sec (yellow bar); 4) The pH was shifted to 8.5 to allow the system to re-equilibrate for 5 min (second blue bar); 5) The oocyte was re-acidified (second red bar) and the degree of recovered channel activity determined.
Figure 5. Inhibition of A/M2 channel activity by consecutive application of BL-1743 and amantadine.

A–D; Representative traces of the A/M2 channel activity modulated by consecutive application of BL- 1743 and amantadine. A/M2 channel activity was induced by the application of the activating solution (pH 5.5, red horizontal bar above the trace). The currents were first inhibited by the application of the activating solution (pH 5.5) containing various concentrations of BL-1743 (3–1000 μM, green horizontal bar) for 60 sec. After maximal inhibition of the channel activity was achieved, amantadine (100 μM) was substituted for BL-1743 (3–1000 μM) in the activating solution for 90 sec (yellow horizontal bar). The inhibitors were washed out for 5 min by the non-activating solution (pH 8.5, second blue horizontal bar) and the pH-induced activity of A/M2 channels was measured again (second red horizontal bar). Note the biphasic current upon application of amantadine during washout of a saturating concentration of BL-1743 (during yellow bar in D).
As expected from a reversible second-order binding event, the rate and extent of inhibition observed during the BL-1743 incubation period increased in a concentration-dependent manner (Figure 5). The ensuing traces observed during the amantadine-chase period reflect several processes, including (a) the binding of amantadine to the fraction of channels that were not inhibited in the initial BL-1743 pulse, (b) dissociation of BL-1743 to generate uninhibited channel, and (c) binding of amantadine to newly uninhibited A/M2 channels following dissociation of BL-1743. With low concentrations of BL-1743, little inhibition was achieved; thus, upon addition of 100 μM amantadine the kinetics were dominated by this process, and the trace approached a simple exponential decay (Figure 5A and B). On the other hand, with a 1000 μM BL743 pulse, inhibition was complete and the resulting traces reflected a competition between dissociation of BL-1743 and the on-rate for amantadine-binding, both of which occur with relaxation times in the sec to min time scale under these conditions (Figure 5D). Thus, biphasic kinetics were observed, with the rising phase reflecting the fact that as the BL-1743 dissociates from the channel, amantadine does not bind instantly, leading to a partial recovery followed by inhibition by amantadine at longer times. As expected, intermediate behavior was observed with an initial pulse of 100 μM BL-1743 (Figure 5C).
In the complementary experiments A/M2 channels were first inhibited by various concentrations of amantadine (1–100 μM) for 60 sec; then, BL-1743 (100 μM) was substituted for amantadine in the activating solution for 90 sec (Figure 6). The lower were the amantadine concentrations the higher was the effect of BL-1743 inhibition as evidenced by more current recovered after the 5 min long washout (Figure 6A and B). When amantadine was applied at a saturating concentration (100 μM), substitution with BL-1743 did not add to the existing inhibition by amantadine (Figure 6D).
Figure 6. Inhibition of A/M2 channel activity by consecutive application of amantadine and BL-1743.

A–D, Representative traces of the A/M2 channel activity modulated by consecutive application of amantadine and BL-1743. A/M2 channel activity was induced by the application of the activating solution (pH 5.5, red horizontal bars above the traces). The currents were first inhibited by the application of the activating solution (pH 5.5) containing various concentrations of amantadine (1–100 μM, yellow horizontal bar) for 60 sec. After the maximal inhibition of the channel activity was achieved, BL-1743 (100 μM) was substituted for amantadine (1–100 μM) in the activating for 90 sec (green horizontal bar). The inhibitors were washed out for 5min by the non-activating solution (pH 8.5, second blue horizontal bar) and the pH-induced activity of A/M2 channels was measured again (second red horizontal bar). Note that in no instance was a biphasic current observed during application of BL- 1743 as amantadine was washed-out (yellow bars).
Taken together, these results suggest competition between BL-1743 and amantadine for inhibition of the A/M2 channel and support the conclusion that amantadine inhibits A/M2 channel activity by binding inside the channel pore. The results of this competition study also are in a good agreement with the amantadine inhibition kinetic rates reported by Wang et. al. (25)
The reaction rate constants for the spiran amine compounds were not precisely determined; however, as shown in Figure 4C, the washout rate for the spiran amine 8 was not significantly different from that of amantadine, suggesting it should behave similarly to amantadine.
Voltage dependence of inhibition of A/M2 channel activity by amantadine, BL 1743 and spiran amine
Since spiran amine 8 is a positively charged molecule in aqueous solution over the pH range used in this study, its inhibitory effect on A/M2 channels may depend on membrane voltage. To address this possibility, we tested the voltage dependence of A/M2 channel inhibition by spiran amine 8 and compared it to that of BL-1743, spiro piperidine 20 and amantadine. Previous studies have shown that the inhibition of A/M2 channel activity by BL-1743 does not depend on voltage (9), while voltage dependence of amantadine inhibition has not been addressed directly. We observed that inward and outward A/M2 channel currents measured in the presence of each of the tested compounds were equally inhibited in the voltage range from −40 to +40 mV (Figure 7). These findings indicate that inhibition by none of the tested compounds is voltage dependent.
Figure 7. Voltage dependence of A/M2 inhibition by amantadine, BL-1743, spiran amine.
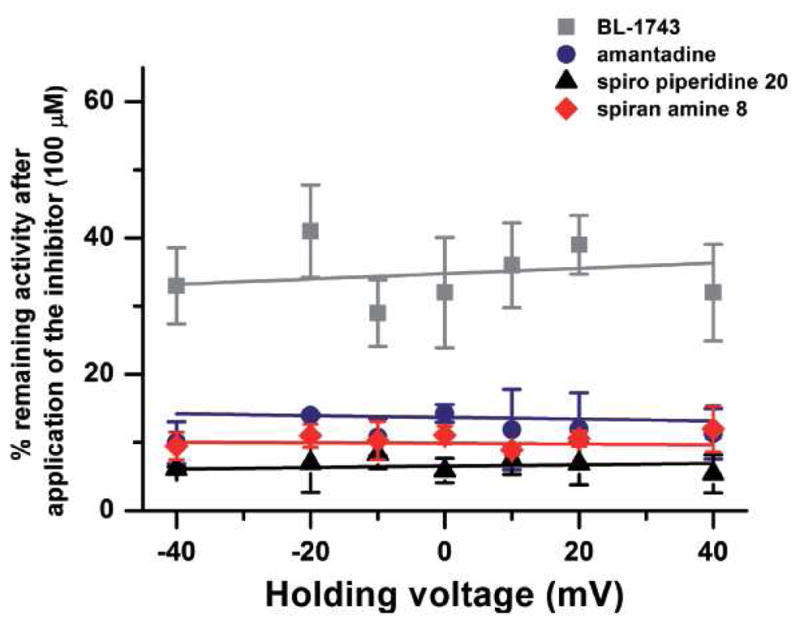
8 and spiro piperidine 20. Inhibition of A/M2 channel activity by amantadine, BL-1743, spiran amine 8 and spiro piperidine 20 was assayed at various holding voltages (from −40mV to +40mV). The mean (±SD) channel activity remaining after the maximal inhibition by each of the inhibitors (100 μM) was achieved was plotted as a function of the holding voltage. The experimental data are the average of three independent experiments. Each point is a mean (±SD) of 6–10 oocytes.
DISCUSSION
As amantadine resistant influenza A strains become prevalent, the need for novel small molecule inhibitors grows. Our previous SAR of the BL-1743 series of compounds revealed one tight binding inhibitor - spiro piperidine 20 with IC50 of 0.9 μM, which has been shown to perturb a larger region in the pore-lining area of wt A/M2 than amantadine (10). In the current study an additional set of compounds was made by converting the spiro-piperidine scaffold to spiran amine, which still retains the conformation of spirocycles for tight binding, but has a slightly more extended structure. Compound 8 from this series shows promising inhibitory activity against two of the naturally occurring A/M2 point mutants: A/M2-L26F and A/M2-V27A (Figure 2). L26 and V27 are the residues closest to the channel exterior (Figure 8) to have been associated with amantadine-resistance in naturally occuring mutants of A/M2 (12, 16, 34). It is interesting to note that, although these L26F and V27A were inhibited by 8, none of the amantadine insensitive mutants located deeper in the pore was sensitive to spirane amine 8 (Table 2). However, of these, only S31N poses a significant clinical threat.
Figure 8. Structure of amantadine complexes with the transmembrane domain of A/M2 protein.

Panel A and B illustrates the crystal structure of the channel (pdb code 3K9J); viewed from the outside of the virus (A) and in a side-on view with on helix removed for clarity (B). Panel C shows a lower resolution model (2KAD) obtained from an analysis solid-state NMR angular and distance restraints. The identities of critical residues are shown below, showing the C-alpha and sidechain atoms.
Figure 8A and 8B illustrates the position of L26 and V27 in the crystal structure of the transmembrane domain of A/M2 complexed with amantadine at low pH, while Figure 8C shows a model of the complex at high pH in phospholipid bilayers obtained by solid state NMR (12). Although there are significant differences between the details of these and other experimental models (35, 36), V27 projects directly towards the pore, while L26 is critical for packing at the helix-helix interface. Thus, changes to residues at these positions are likely to cause changes to the N-terminal region of the channel lumen, precisely at a location where amantadine has been found to bind. We are currently conducting crystallographic and molecular dynamics analyses of these mutants to determine their affect on the channel structure, and also to define the mechanism by which compound 8 is able to inhibit these otherwise drug-resistant variants. However, it is inviting to speculate that the decrease in hydrophobicity and steric bulk associated with the V27A mutation might increase the polarity and pore radius near the N-terminal region of the binding site. In this scenario, the extended length of compound 8 would provide increased steric and physicochemical complementarity to the variants, particularly its amine group projected outward toward the exterior of the virus.
It was previously shown that BL-1743 inhibits A/M2 channel activity by binding inside the channel pore with the κon kinetic similar to that of amantadine, but with nearly 10-fold faster koff kinetic (9, 25). By employing the differences in the kinetic properties of amantadine and BL-1743 inhibition, we tested whether these two compounds compete for the same binding site. Figure 5D shows that when A/M2 channels are occupied with BL-1743, after amantadine substitution for BL-1743 in the recording solution, no additional inhibition by amantadine occurs, and slow re-initiation of amantadine binding starts only as the unbinding of rapidly reversible BL-1743 occurs. These findings are consistent with bound BL-1743 molecules preventing the channel from binding amantadine. Since BL-1743 binds in the pore, this result indicates that amantadine also inhibits A/M2 channel activity by binding in the channel pore.
As amantadine, BL-1743 and spirane amine 8 are all easily protonated in the pH range used in this study, and it is often observed that the efficacy of a positively charged channel inhibitor increases as membrane voltage is made more negative, we tested the voltage dependence of these compounds and of spiro piperidine 20 in the range of −40 to +40 mV. Earlier electrophysiological characterizations of AM2 inhibition by BL-1743 revealed that the inhibition was not voltage dependent (9). The present study confirms the voltage-independence of BL-1743, amantadine, spiro piperidine 20, and spirane amine 8 (Figure 7). This finding is consistent with the location of the binding site occurring on the N-terminal half of the pore in a location exterior (N-terminal in peptide sequence) to the Trp/His gate, over which the transmembrane voltage drop is expected to occur. This binding mode is also consistent with the site inferred from solid state NMR measurements of the perturbation to channel resonances associated with binding of spiro piperidine 20 (10). Furthermore, the existence of mutants conferring BL-1743 resistance (9, 24) suggest that the drug binding site is located on the extracellular-facing portion of the pore, N-terminal of H37. The lack of voltage dependence of amantadine inhibition and failure for intracellularly injected amantadine to inhibit (37) are also consistent with this binding location, and argue against a proposed mechanism of amantadine binding to the outer intracellular-facing side of pore of the protein (21, 22).
Together, these data argue for the previously proposed mode of binding to the pore of the channel (14–17). However, assuming the voltage drop occurs at the His/Trp gate, these data do not differentiate models in which the positively charged ammonium (or guanidinium group of BL-1743) of the drugs are directed toward the viral exterior or downward toward the H37 residues. It also should be born in mind that the lack of a voltage-dependence might occur if the binding of the positively charged inhibitor occurs in an exchange in which one proton is released to the outside of the cell when one inhibitor molecule enters the pore without changing the total charge inside the pore. While this is a formal possibility that is consistent with the fact that the binding of drug occurs with concomitant loss of one or more protons from the H37 residues (38), it seems unlikely that the proton release would obligatorily occur to the exterior, rather than the interior, which is the natural direction of proton flow.
In summary, these results demonstrate the feasibility of designing anti-viral drugs effective against amantadine-resistant escape mutants of influenza A virus. Relatively modest modifications to BL-1743 have now resulted in a potent inhibitor of two naturally occurring A/M2 amantadine insensitive mutants: A/M2-L26F and A/M2-V27A. Moreover, the electrophysiological experiments lend support for the existence of the binding site in the outer portion of the channel pore (12, 15, 16, 23, 34). Thus, three-dimensional models in which the drug is bound in the N-terminal regions of the pore should provide useful starting points for the design of even more tight-binding and broad-spectrum agents.
Supplementary Material
Scheme 4.

Synthesis of spiran 18, 19.
Footnotes
The research was supported by GM56416 grant (W.F.D. and J.W) and NIH research grants R01 AI- 57363 (L.H.P.), R01 AI-20201 (R.A.L), R01 AI-74517 (W.F.D. and J.W). R.A.L. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
Supporting information which describes the chemical synthesis of the compounds and the CD titration assay is available free of charge online at http://pubs.acs.org.
Contributor Information
Victoria Balannik, Email: v-balannik@northwestern.edu.
Jun Wang, Email: juwang@sas.upenn.edu.
Yuki Ohigashi, Email: y-ohigashi@northwestern.edu.
Xianghong Jing, Email: x-jing@northwestern.edu.
Emma Magavern, Email: magavern@mail.med.upenn.edu.
Robert A. Lamb, Email: ralamb@northwestern.edu.
William F. DeGrado, Email: wdegrado@mail.med.upenn.edu.
Lawrence H. Pinto, Email: Larry-Pinto@northwestern.edu.
References
- 1.Cox NJ, Subbarao K. Influenza. Lancet. 1999;354:1277–1282. doi: 10.1016/S0140-6736(99)01241-6. [DOI] [PubMed] [Google Scholar]
- 2.Lagoja IM, De Clercq E. Anti-influenza virus agents: synthesis and mode of action. Med Res Rev. 2008;28:1–38. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 4.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, Lopez-Gatell H, Olivera H, Lopez I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD, Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler- Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov AI, Cox NJ. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science. 2009 doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto LH, Lamb RA. Controlling influenza virus replication by inhibiting its proton channel. Mol Biosyst. 2007;3:18–23. doi: 10.1039/b611613m. [DOI] [PubMed] [Google Scholar]
- 7.Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J Biol Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 8.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci U S A. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu Q, Pinto LH, Luo G, Shaughnessy MA, Mullaney D, Kurtz S, Krystal M, Lamb RA. Characterization of inhibition of M2 ion channel activity by BL-1743, an inhibitor of influenza A virus. J Virol. 1996;70:4246–4252. doi: 10.1128/jvi.70.7.4246-4252.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Cady SD, Balannik V, Pinto LH, DeGrado WF, Hong M. Discovery of spiro-piperidine inhibitors and their modulation of the dynamics of the M2 proton channel from influenza A virus. J Am Chem Soc. 2009;131:8066–8076. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 12.Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J Mol Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furuse Y, Suzuki A, Oshitani H. Large-Scale Sequence Analysis of M Gene of Influenza A Viruses from Different Species: Mechanisms for Emergence and Spread of Amantadine Resistance. Antimicrob Agents Chemother. 2009 doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duff KC, Gilchrist PJ, Saxena AM, Bradshaw JP. Neutron diffraction reveals the site of amantadine blockade in the influenza A M2 ion channel. Virology. 1994;202:287–293. doi: 10.1006/viro.1994.1345. [DOI] [PubMed] [Google Scholar]
- 15.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi M, Cross TA, Zhou HX. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J Phys Chem B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: insights from molecular simulation. Biophys J. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khurana E, Dal Peraro M, DeVane R, Vemparala S, DeGrado WF, Klein ML. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 20.Saito R, Sakai T, Sato I, Sano Y, Oshitani H, Sato M, Suzuki H. Frequency of amantadine-resistant influenza A viruses during two seasons featuring cocirculation of H1N1 and H3N2. J Clin Microbiol. 2003;41:2164–2165. doi: 10.1128/JCM.41.5.2164-2165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci U S A. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jing X, Ma C, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandhi CS, Shuck K, Lear JD, Dieckmann GR, DeGrado WF, Lamb RA, Pinto LH. Cu(II) inhibition of the proton translocation machinery of the influenza A virus M2 protein. J Biol Chem. 1999;274:5474–5482. doi: 10.1074/jbc.274.9.5474. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flaugh ME, Crowell TA, Farlow DS. Acid-catalyzed annelation of .alpha.- alkylaldehydes and .alpha.,.beta.-unsaturated ketones. A one-pot synthesis of 4,4-dimethyl-2- cyclohexen-1-one. The Journal of Organic Chemistry. 1980;45:5399–5400. [Google Scholar]
- 27.Chan Y, Epstein WW. 4,4-DIMETHYL-2-CYCLOHEXEN-1-ONE. Organic Syntheses. 1988;50:496–498. [Google Scholar]
- 28.Turner RM, Lindell SD, Lay SV. A Facile Route to Imidazol-4-Yl Anions and Their Reaction with Carbonyl-Compounds. Journal of Organic Chemistry. 1991;56:5739–5740. [Google Scholar]
- 29.Balannik V, Lamb RA, Pinto LH. The oligomeric state of the active BM2 ion channel protein of influenza B virus. J Biol Chem. 2008;283:4895–4904. doi: 10.1074/jbc.M709433200. [DOI] [PubMed] [Google Scholar]
- 30.Shimbo K, Brassard DL, Lamb RA, Pinto LH. Ion selectivity and activation of the M2 ion channel of influenza virus. Biophys J. 1996;70:1335–1346. doi: 10.1016/S0006-3495(96)79690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeda M, Pekosz A, Shuck K, Pinto LH, Lamb RA. Influenza a virus M2 ion channel activity is essential for efficient replication in tissue culture. J Virol. 2002;76:1391–1399. doi: 10.1128/JVI.76.3.1391-1399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li D, Saito R, Suzuki Y, Sato I, Zaraket H, Dapat C, Caperig-Dapat IM, Suzuki H. In vivo and in vitro alterations in influenza A/H3N2 virus M2 and hemagglutinin genes: effect of passage in MDCK-SIAT1 cells and conventional MDCK cells. J Clin Microbiol. 2009;47:466–468. doi: 10.1128/JCM.00892-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stouffer AL, Nanda V, Lear JD, DeGrado WF. Sequence determinants of a transmembrane proton channel: an inverse relationship between stability and function. J Mol Biol. 2005;347:169–179. doi: 10.1016/j.jmb.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 34.Stouffer AL, Ma C, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure. 2008;16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci U S A. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Venkataraman P, Knopman J, Lamb RA, Pinto LH. The M2 proteins of Influenza A and B viruses are single-pass proton channels. In: Fischer WB, editor. Viral Membrane Proteins: Structure, Function and Drug Design. Kluwer Academic/Plenum Publisher; New York: 2005. pp. 101–111. [Google Scholar]
- 38.Li C, Yi M, Hu J, Zhou HX, Cross TA. Solid-state NMR and MD simulations of the antiviral drug amantadine solubilized in DMPC bilayers. Biophys J. 2008;94:1295–1302. doi: 10.1529/biophysj.107.112482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.