

Abstract
Objective
To determine longevity in Rett syndrome (RTT) from a large cohort.
Study design
The North American Database allows the examination of longevity in a large cohort of individuals with RTT from the US and Canada. This database contains information on 1928 individuals (85.5% typical, 13.4% atypical, and 1.1% with MECP2 mutations but not RTT. Kaplan-Meier analyses were performed to assess longevity.
Results
Earlier decennial cohorts had better survival than recent cohorts, most participants surviving into middle age. Comparing overall survival between typical and atypical RTT, those with typical RTT had greater mortality than those with atypical RTT across the observed lifespan (p<.0001). Comparing survival for individuals with RTT and identified mutations with those with MECP2 not known, those with unknown MECP2 status had greater mortality than those with mutations (Log-Rank test, p<.0001).
Discussion
This analysis provides strong evidence for significant longevity in Rett syndrome and indicates the need for careful planning for long-term care of these women. The disproportionately greater survival seen in earlier time periods and in atypical RTT may be attributed to more severely affected individuals dying before diagnosis in the former and greater numbers with milder variants (preserved speech and delayed onset) in the latter.
Keywords: MECP2, mutations, genetics, survival, Kaplan-Meier
Rett syndrome (RTT) is a neurodevelopmental disorder characterized by cognitive impairment, communication dysfunction, stereotypic movement disorder, and growth failure, as first described in 1966 by Andreas Rett (1). The diagnosis of RTT is based on consensus clinical criteria updated in 2001 (2). More than 200 mutations have been identified in the methyl-CpG-binding protein 2 (MECP2) gene; eight common mutations representing 60% or more of individuals fulfilling consensus criteria (3, 4). Individuals with RTT are not capable of independent living.
RTT has a prevalence ranging from 1:10-20,000 females. Incidence values are limited, varying from 0.43-0.71/10,000 females in France (5) to 1.09/10,000 females in Australia (6). Information on longevity is limited. Unpublished observations indicated approximately 70% survival at age 35 versus 98% in the general female population in the US (Glaze and Percy, Baylor College of Medicine). Population-based data from Australia yielded nearly 78% survival at 25 years versus 99.96% in Australian females generally (6).
In order to assess longevity for RTT in the United States and Canada, members of the International Rett Syndrome Association (IRSA) were surveyed (7), providing a robust database with respect to diagnosis, mutation status, date of birth, and date of death, if applicable. This report demonstrates the potential for prolonged survival in individuals with RTT and suggests the need for careful planning for long-term care as well as continued observation of the effects of improved clinical management on longevity.
Methods
IRSA mailed a structured survey to 2,994 members in the US (2,836) and Canada (158) requesting specific information including date of birth; date of death, if applicable; diagnosis (typical RTT, variant or atypical RTT, not RTT, or unknown), discipline of diagnosing physician; mutation testing results, if performed, and testing laboratory; reason why diagnostic testing was not performed; and contact information (Appendix; available at www.jpeds.com). No response was received from 1,439, a significant number of whom did not receive surveys due to faulty addresses, yielding 1,555 (52%) completed surveys. The mail outs occurred by bulk rate routing, preventing an accurate count of surveys not reaching their recipients. In addition to the completed surveys, similar data were gathered from the patient databases at Baylor (310) and Greenwood Genetic Center (28) and from the Canadian RTT database (61), each of which is part of IRB-approved protocols at the respective institutions. After careful editing of males or participants who failed to meet consensus criteria for RTT and did not have a MECP2 mutation (26), 1,928 female participants were included in the survival analyses. For longevity analyses, 21 individuals identified as have a MECP2 mutation but not RTT were excluded from the analysis.
All statistical analyses were conducted using SAS 9.1 (SAS Institute, Cary, NC). Univariate analyses were done using Chi-square and t-test for difference of proportions, and SAS PROC LIFETEST was used to generate Kaplan-Meier survival curves with statistical inference done with the Log-Rank test to compare survival curves across strata and the Log-Rank test and Wilcoxon trend test to examine trends in survival over time. Figure 1 was generated using Stata/SE 9.2 (StataCorp, College Station, TX). The research protocol was reviewed and approved by the University of Alabama at Birmingham Institutional Review Board.
Figure 1. Survival Patterns for Rett Syndrome by Decade of Birth.

Product limit survival function is estimated for six separate epochs: 1935-1959; 1960-1969; 1970-1979; 1980-1989; 1990-1999; and 2000-2009. The resulting curves demonstrate the potential for survival into middle age.
Results
The North American database is composed of individuals who fulfill criteria for typical or variant RTT or who do not meet RTT criteria but have a MECP2 mutation. The diagnoses were made by a pediatric neurologist, a developmental pediatrician, a geneticist, or a general pediatrician. The number of participants and their distribution by diagnosis are shown in Table I. Distribution by diagnosis was 85.5% typical, 13.4% atypical, and 1.1% with MECP2 mutations but not fulfilling criteria for RTT. MECP2 testing was performed on 1165 (60%) individuals; however, mutation results were known by the parents only in 1053 of 1165 (91%). As the parents did not know the testing laboratory, it was not possible to ascertain the mutation status in 112 individuals. Of 1053 individuals for whom mutation results were available, 915/1053 (87%) had a mutation. These 915 included 800 of 870 (92%) individuals with typical RTT, 94 of 162 (58%) individuals with atypical RTT, and 21 of 21 individuals with a mutation but without features of RTT.
Table 1. Participants (1928) and MECP2 Mutations in North American.
| Database by Diagnosis | ||
|---|---|---|
| Diagnostic Group | Diagnostic Distribution (%) | Mutation Distribution (%) |
| Typical | 1648 (85.5 %) | 800/870 (92%) |
| Atypical | 259 (13.4 %) | 94/162 (58%) |
| Not RTT (MECP2 positive) | 21 (1.1 %) | 21/21 (100%) |
Among all participants studied, a total of 305 deaths (15.8%) had occurred as of January 1, 2006. Survival differed significantly between typical (295 out of 1649 died) − 17.9% mortality and atypical diagnosis (10 out of 258 died) − 3.9% mortality (p<.0001 by t-test for difference of proportions). The distribution of participants with typical RTT diagnosis by decade of birth, and age at death or survival as of January 1, 2006 is shown in Table II. In examining the distribution of typical and atypical diagnoses by decade of birth (data not shown), no statistical difference was observed (p=0.14, Chi-square, 5 df = 8.32). However, the proportion of atypical individuals appears to increase over time, rising from approximately 10% among those born prior to 1970 to approximately 20% among those born after 1999.
Table 2. Age at Death or Survival to January 1, 2006 by Decade of Birth among Cases with Typical Rett Syndrome Diagnosis.
| Decade of Birth | Total Cases | Age at Death | Alive on January 1, 2006 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1.0 - 4.9 | 5.0 - 9.9 | 10.0 - 19.9 | 20.0 - 29.9 | 30.0 - 39.9 | 40.0 - 49.9 | 50.0+ | No. | Pct. | ||
| 1935-1959 | 30 | 0 | 0 | 2 | 2 | 5 | 5 | 3 | 13 | 43.3 |
| 1960-1969 | 99 | 0 | 1 | 5 | 10 | 20 | 3 | 0 | 60 | 60.6 |
| 1970-1979 | 286 | 0 | 3 | 28 | 32 | 9 | 0 | 0 | 214 | 74.8 |
| 1980-1989 | 511 | 8 | 21 | 63 | 18 | 0 | 0 | 0 | 401 | 78.5 |
| 1990-1999 | 525 | 13 | 20 | 21 | 0 | 0 | 0 | 0 | 471 | 89.7 |
| 2000-2005 | 198 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 195 | 98.5 |
| Total | 1649 | 24 | 45 | 119 | 62 | 34 | 8 | 3 | 1354 | 82.1 |
Survival trends among all RTT participants studied are shown in Figure 1. Survival patterns were significantly different between the respective groups (p-value for Log-Rank test <.0003), and both the Log-Rank test for trend and the Wilcoxon trend test were statistically significant (p<.0001). The number of individuals available for analysis did not support decade-by-decade analysis of survival patterns by type of RTT diagnosis or MECP2 status. When overall survival patterns were compared between typical and atypical RTT participants, we found that those with typical RTT had significantly greater mortality than did those with atypical RTT, at all ages across the observed lifespan (p<.0001). These patterns are shown in Figure 2.
Figure 2. Overall Survival of Atypical (2A) and Typical (2B) Rett Syndrome.
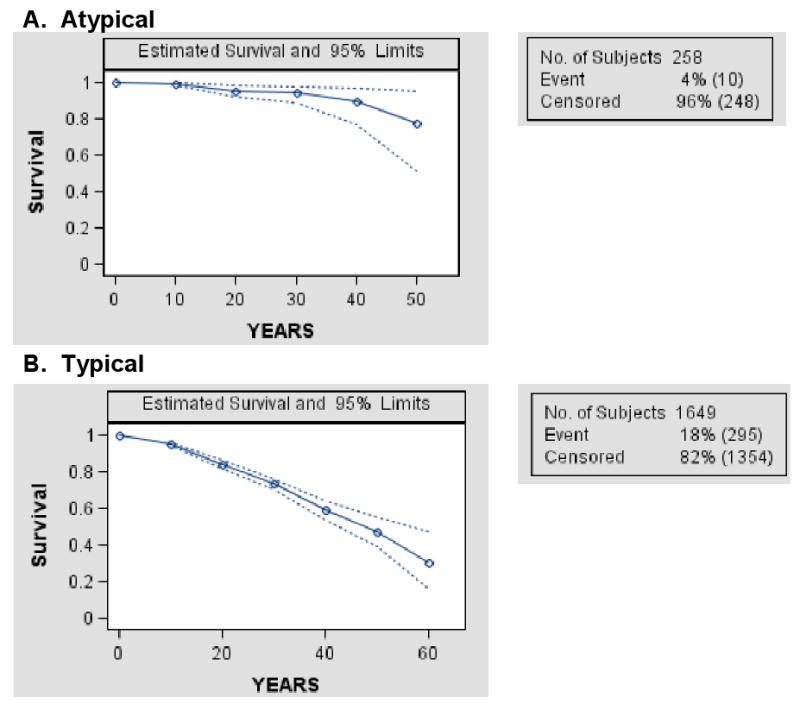
Kaplan-Meier curves indicate significantly better survival for individuals with atypical versus typical Rett syndrome (Log-rank test p< 0.0003). Nevertheless, the potential for survival into middle age is demonstrated for both groups.
We also found that individuals with atypical RTT had significantly better survival than did those with typical RTT, at all ages across the lifespan, and that those with identified mutations also had improved survival compared to those with MECP2 status not known (data not shown). We also examined survival patterns for atypical RTT participants regarding the presence of a mutation. Although survival did not differ significantly across the mutation, MECP2 not known, and no mutation participants, those with MECP2 not known tended to have increased mortality by age 30 and beyond (data not shown). Available data were insufficient to characterize survival patterns by race/ethnicity.
Discussion
In this analysis, we examined patterns of RTT survival among individuals in North America born prior to 1960 to the present. From the most recent to the earliest period, a general pattern of better survival exists the earlier the decade of birth. Given that clinical management has improved considerably from the period when RTT was first recognized, one might expect improved survival among each successive cohort.
We speculate that the observed pattern may have occurred because as molecular genetic tests have become widely available, substantially all individuals with RTT are now being identified in the first years to decade of life. More severely affected individuals with this disorder were more likely to escape diagnosis in the earlier time periods, leading to study populations with greater survival due to milder clinical involvement. Similarly, the disproportionately greater survival seen in atypical RTT may be attributed to the greater number of individuals with milder variants (preserved speech and delayed onset) and lower number of individuals with the early onset seizure or congenital variants. Again, the more severely affected individuals were more likely to escape diagnosis in early time periods. We speculate that the poorer survival among participants lacking MECP2 data may be explained by a greater number of older individuals on whom mutation testing was less likely to be conducted and that those with known mutations included the milder atypical preserved speech and late onset variants.
The North American Database represents the first comprehensive compilation of information on individuals with RTT or with another diagnosis in association with MECP2 mutations in the US and Canada. As such, it provides a unique resource for expanding our understanding of RTT and for comparison with other national databases. The North American Database is derived principally from members of the IRSA in which membership is voluntary. Therefore, this database is not population-based. Nonetheless, the large number of participants should be generally representative of RTT within the US and Canada. We were unable to examine mortality patterns by cause of death, due to incomplete reporting and the expense involved in using the National Death Index for deaths occurring in the United States.
Although data for direct assessment of the generalizability of our results are unavailable, our findings are concordant with those of Laurvik et al (6), who found overall survival of approximately 78% at age 25 among female RTT participants in the Australian registry. Even though earlier birth cohorts in our study may be incomplete, as more severe cases may have died prior to diagnosis, it is likely that the study participants born in recent years approximate the North American population with RTT. We recommend the establishment of a population-based North American or United States registry, similar to the Australian model, to support future investigations concerning the descriptive epidemiology, genetic/genomic research, health services and outcomes, and clinical interventions for RTT patients.
This study of longevity in RTT, derived from the North American RTT Database, provides strong support for the notion of significant longevity in individuals with RTT. In future, as early diagnosis of Rett syndrome improves along with more effective clinical management, it is possible that longevity will be extended even further. As such, the current results indicate the need for careful planning for the future care of these women as they advance in years and could raise the level of concern among their parents in preparing for this future care. How well success is achieved in this planning must be assessed in future studies.
Acknowledgments
The authors acknowledge the gracious participation and provision of information by families of the reported participants and the critical staff support of the International Rett Syndrome Association. Dr. Mary Lou Oster-Granite, Health Scientist Administrator at NICHD, provided invaluable guidance, support, and encouragement for this rare disease initiative. We also thank Jason Salemi, MPH, for his assistance in preparing Figure 1 for publication.
Supported by NIH grants (RR019478), MRRC grant (HD38985), and funds from the International Rett Syndrome Association and Civitan International Research Center.
List of abbreviations
- RTT
Rett syndrome
- MECP2
methyl-CpG-binding protein 2
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wiener Medizinische Wochenschrift. 1966;116:723–726. [PubMed] [Google Scholar]
- 2.Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6(5):293–297. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- 3.Amir R, Van den Veyver I, Wan M, Tran C, Francke U, Zoghbi H. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 4.Percy AK, Lane JB. Rett syndrome: model of neurodevelopmental disorders. J Child Neurol. 2005;20(9):718–721. doi: 10.1177/08830738050200090301. [DOI] [PubMed] [Google Scholar]
- 5.Bienvenu T, Philippe C, De Roux N, Raynaud M, Bonnefond JP, Pasquier L, et al. The incidence of Rett syndrome in France. Pediatr Neurol. 2006;34(5):372–375. doi: 10.1016/j.pediatrneurol.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148(3):347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 7.Percy AK, Lane JB, Childers J, Skinner S, Annese F, Barrish J, et al. Rett syndrome: North American database. J Child Neurol. 2007;22(12):1338–1341. doi: 10.1177/0883073807308715. [DOI] [PubMed] [Google Scholar]