

Abstract
Brain ischemia causes more extensive injury in hyperglycemic than normoglycemic subjects, and the increased damage is to astroglia as well as neurons. In the present work, we found that in cortical astrocytes from rat or mouse, reoxygenation after hypoxia in a medium mimicking interstitial fluid during ischemia increases hemichannel activity and decreases cell-cell communication via gap junctions as indicated by dye uptake and dye coupling, respectively. These effects were potentiated by high glucose during the hypoxia in a concentration-dependent manner (and by zero glucose) and were not observed in connexin 43−/− astrocytes. The responses were transient or persistent after short and long periods of hypoxia, respectively. The persistent responses were associated with a progressive reduction in cell viability that was prevented by La3+ or peptides that block connexin 43 (Cx43) hemichannels or by inhibition of p38 MAP kinase prior to hypoxia-reoxygenation but not by treatments that block pannexin hemichannels. Block of Cx43 hemichannels did not affect the reduction in gap junction mediated dye coupling observed during reoxygenation. Cx43 hemichannels may be a novel therapeutic target to reduce cell death following stroke, particularly in hyperglycemic conditions.
Keywords: Stroke, Diabetes, Cell cultures, Connexin channels, Astrocytic death
INTRODUCTION
Stroke is a major cause of death in industrialized countries and results from a transient or permanent reduction in cerebral blood flow produced, in most cases, by arterial occlusion or local thrombosis, i.e., focal ischemia (Dirnagl et al., 1999). Severe and/or prolonged reduction in cerebral blood flow leads to deprivation of oxygen and glucose as well as build-up of potentially toxic substances. Even a brief period of ischemia induces depletion of intracellular ATP paralleled by a progressive reduction of electrochemical gradients across the plasma membrane (Silver et al., 1997) followed by metabolic changes characteristic of necrosis and/or apoptosis (Benjelloun et al., 2003; Walton et al., 1999).
During and after transient ischemia, death is more prominent in neurons than in astrocytes primarily due to the neurotoxic effect of secreted glutamate (Choi et al., 1987). In addition, metabolic differences between neurons and astroglia are likely to contribute. The relative insensitivity of astrocytes to oxygen/glucose deprivation (OGD) has been ascribed to their ability to switch from aerobic to anaerobic metabolism (Peuchen et al., 1996); nevertheless, prolonged ischemia will also impact their functioning and eventually kill them (Sugawara et al., 2002). Many astrocytic functions are impaired by sublethal ischemic insults, and these functions could be critical for neuronal viability (Orellana et al., 2009). Astrocytes provide metabolic and structural support and control the extracellular concentrations of glutamate, K+, and H+ (Fields and Stevens-Graham 2002).
Many of the functions of astrocytes are facilitated by spatial buffering mediated through gap junctions between astrocytes (Giaume et al., 2007). Gap junctions are membrane specializations that provide a cytoplasmic pathway between contacting cells that is permeable to molecules up a size of ~1.4 nm diameter (and may or may not be charge selective). Generally, gap junctions are aggregates or plaques that contain a few tens to thousands of cell-cell channels. Each gap junction channel spans two apposed membranes and is formed by a hemichannel or connexon contributed by each membrane (Sáez et al., 2003). Each hemichannel is composed of six protein subunits termed connexins (Cxs), a highly conserved family encoded by 21 genes in human and 20 in mouse with orthologs in other mammals (Cruciani and Mikalsen 2005). Connexins are widely expressed in mammalian tissues (Saez et al., 2003); astrocytes express primarily Cx43 with lesser amounts of Cx26 and Cx30 (Orellana et al., 2009), and gap junctions between them are permeable to both positive and negative ions and small molecules, including second messengers such as Ca2+, inositol 1,4,5-trisphosphate and cyclic nucleotides, ATP, transmitters, e.g., glutamate, and energy producing metabolites, e.g., glucose and lactate (Tabernero et al., 2006).
Recently, the presence of functional connexin hemichannels in nonjunctional membrane of astrocytes was demonstrated by several experimental approaches (Sáez et al., 2005). The use of exogenous expression systems permitted the study of their electrophysiological and permeability properties and the mechanisms that control their activity. Under appropriate physiological conditions, hemichannels release physiologically relevant quantities of signaling molecules (e.g., ATP, glutamate, NAD+ and PGE2) to the extracellular milieu (Sáez et al., 2005). In vitro ischemia-like conditions enhance hemichannel activity in astrocytes and many other cell types (Orellana et al., 2009). Opposite to its action on hemichannels, in vitro ischemia-like conditions reduce gap junctional communication between astrocytes (Orellana et al., 2009). Thus, during the last decade it has become clear that connexins in astrocytes can play either a protective or a deleterious role in neuronal and glial survival during or after ischemia (Orellana et al., 2009).
It has long been known that hyperglycemia during acute brain ischemia increases the extent of tissue injury in animals (Myers and Yamaguchi 1977) and in humans (Kagansky et al., 2001). Previous studies ruled out increased lactate (Cronberg et al. 2004; Lin et al. 1995) and glutamate concentrations (Cronberg et al., 2004) as crucial determinants of this added injury. In hippocampal slices, glucose itself or in combination with acidosis mediates the detrimental effects (Cronberg et al., 2004), but the mechanisms remain unknown. The protocol of chemical ischemia that we used previously increases activity of Cx43 hemichannels, which accelerates cell death (Contreras et al., 2004). However, the metabolic inhibitors used were irreversible and for a more physiological procedure we subjected astrocyte cultures to oxygen deprivation in the presence of various concentrations of glucose in a saline mimicking interstitial fluid during ischemia (Bondarenko and Chesler 2001). We then assayed hemichannel activity following reperfusion with normoxic and normoglycemic solutions. We found that hypoxia did induce opening of hemichannels that occurred largely after return to normoxic and normoglycemic conditions and that the effect was greater after hypoxia in hyperglycemic conditions.
MATERIALS AND METHODS
Reagents and antibodies
SuperSignal kit for enhanced chemiluminescence (ECL) detection, anti-rabbit IgG antibody-conjugated to horseradish peroxidase, Sulfo-NHS-SS-biotin, and immobilized NeutrAvidin were purchased from Pierce. Previously described Gap26, Gap27, 10panx1, and E1b peptides (Evans and Leybaert 2007; Pelegrin and Surprenant 2006; Wang et al., 2007) were obtained from NeoMPS, SA. (Strasbourg, France). HEPES, H2O (W3500), LaCl3 (La3+), ethidium bromide (EtdBr), Lucifer yellow (LY), 1, 4-dithiothreitol (DTT), SB202190 and oATP were purchased from Sigma-Aldrich (St. Louis, MO, USA). Minimum Eagle’s medium (MEM), Dulbecco’s Minimum Eagle’s medium (DMEM), HCO3− free/F-12 medium, fetal bovine serum (FBS), normal goat serum (NGS), penicillin, streptomycin and trypsin-EDTA were obtained from GibcoBRL (Grand Island, NY, USA). Hoechst 33342 and Rhodamine B dextran 10 kDa (Rdex) were obtained from Molecular Probes, Inc. (Eugene, OR, USA). Where applicable the provider’s protocol was followed.
Animals
Newborn (P1-P2) Sprague-Dawley rats were obtained from the Animal Institute of the Pontificia Universidad Católica de Chile, and mice were maintained in the animal facility of the Institut de Biologie at the Collége de France. Mouse astrocyte cultures were made from newborn (P1–P2) animals. Normal cells were obtained from OF1 mice (Charles River, L’Arbresle, France). Astrocyte cultures lacking Cx43 were also prepared from the cortices of P0 littermates from Cx43+/− breedings. Each brain was treated separately, giving rise to cultures of Cx43−/−, Cx43+/−, or Cx43+/+ astrocytes. Cx43 expression was first characterized by immunocytochemical labeling and confirmed afterwards by genotyping performed as described for Cx43−/− mice (Theis et al., 2001). Genotyping was performed from a tissue sample, using PCR analysis, as described below.
All experimental protocols were approved by the Ethical committee of the Pontificia Universidad Católica de Chile or carried out in accordance with the European Community Council Directives of November 24th 1986 (86/609/EEC) and all efforts were made to minimize the number of animals used and their suffering.
Cell cultures
For experiments in Chile, cortical astrocytes were obtained from neonatal rats or mice. Briefly, meningeal tissue was removed, and the neocortex was minced and incubated at 37°C for 30 min in Ca2+-free PBS containing trypsin (0.5%) and EDTA (5 mM). Trypsin was then removed, and tissue immersed in MEM medium supplemented with 10% horse serum and containing 1 mg/ml bovine pancreas DNase I, and triturated using a Pasteur pipette. The dissociated cells were centrifuged, and the pellet was resuspended in MEM supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin sulfate, and the cells were plated in plastic bottles (25 ml, Nunc Clone, Marienfeld, Germany). Confluent cells were replated on 90-mm plastic culture dishes (Nunc Clone), with 5×105 cells per plate for biotinylation or 2.5 × 103 cells per glass coverslip (12 mm in diameter and 1.3–1.7 thick). Cells were grown at 37°C in a 5% CO2/95% air atmosphere at nearly 100% relative humidity. All experiments were carried out when cells reached ~80% confluence. Cells were always fed the day before an experiment.
For experiments in Paris, astrocyte cultures were prepared from the cortex of Cx43−/− and Cx43+/+ mice. Briefly, cells were seeded (2×105 cells per well) on glass coverslips placed inside 16-mm diameter 4 well plastic plates (NunClon, Nunc) in DMEM, containing glucose (5 mM), penicillin (5 U/ml) and streptomycin (5 μg/ml) (Invitrogen, Carlsbad, CA, USA) and 10% FCS (Hyclone, Logan, UT, USA). The medium was changed twice a week. The mouse genotype was determined by PCR analysis. Briefly, a mouse tissue sample was digested in buffer (KCl 50 mM; MgCl2 1.5 mM; Tris-HCl 10 mM, pH = 8.0; IGEPAL CA-630 0.45%; Tween-20 0.45%) containing Proteinase K (500 μg/ml; Promega, Madison, WI, USA) at 56°C over night. After digestion, 1 μl of the supernatant, containing mouse DNA, was added in 24 μl of primer solution (1:1000 in pure water). Two sets of primers were used: one for the Cx43 wild-type gene, a 22 mer forward oligonucleotide and a 25 mer reverse oligonucleotide (5′-CCCCACTCTCACCTATGTCTCC-3′ and 5′-ACTTTTGCGCCTAGCTAGCTATCCC-3′, respectively); the second for the LacZ insert in the Cx43 coding region, a 22 mer foward oligonucleotide and a 22 mer reverse oligonucleotide (5′-GGCATACAGACCCTTGGACTCC-3′ and 5′-TGCGGGCCTCTTCGCTATTACG-3′, respectively). The PCR reaction was achieved using “PCR ready to go” kit (Amersham Biosciences, Saclay, France) with the solution described above, following the instructions of the kit. DNA was firstly annealed at 94°C and then amplified at 55°C for 40 cycles. The PCR products were analyzed by electrophoresis in a 2% agarose gel and stained with ethidium (Etd). The specific amplified sequences were 550 and 850 bp long for the mutant gene and wild-type gene, respectively.
All primary cultures were highly enriched in astrocytes as determined by the fraction reactive to antiGFAP (~99%) and very small fraction labeled by the microglia marker isolectin B4 (< 1%).
Human cervix carcinoma (HeLa) cells (ATCC, CCL-2, Rockville, MD, USA) transfected with cDNA encoding rat Cx43 with EGFP attached to the C terminus (Cx43-EGFP) were grown in DMEM medium containing 100 μg/ml streptomycin, 100 U/ml penicillin, and 500 μg/ml G-418 and supplemented with 10% FBS. The medium was replaced at 2-day intervals. Subconfluent cells were routinely harvested by trypsinization and plated on glass coverslips or on 60 mm plastic culture dishes (Nunc Clone). Parental and Cx43-EGFP cells were kept at 37°C in a 5% CO2/95% air atmosphere at nearly 100% relative humidity. Cx43-EGFP cells could be recognized by their fluorescence emission at 530 nm and excitation at 489 nm.
Hypoxia-reoxygenation protocol
Astrocyte cultures were subjected to 3 or 6 h of hypoxia in an artificial cerebrospinal fluid medium (previously bubbled with 5% CO2/95% N2 for 15 min) that mimics the interstitial ionic concentrations of ischemic brain [“ischemic saline”; (Bondarenko and Chesler 2001)] (in mM: 51 NaCl, 65 K-gluconate, 0.13 CaCl2, 1.5 MgCl2, 10 HEPES, pH 6.8) containing different glucose concentrations (in mM: 0, 5, 12, 17, 22, 27, 32 or 37). Hypoxia was induced by placing the cultures inside an incubator chamber and removing the air with a CO2/N2 (5%, 95%) flow for 7 min, after which the chamber was closed for 3 or 6 h as previously described (Martínez and Sáez 2000). Then, reoxygenation was allowed in control saline (MEM with 5 mM glucose or different concentrations where appropriate and 10% FBS at 37°C in a 5% CO2/95% air atmosphere at nearly 100% relative humidity).
Dye Coupling
Cells plated on glass coverslips were bathed with recording medium (HCO3−-free F-12 medium buffered with 10 mM HEPES, pH 7.2) and permeability mediated by gap junctions was tested by evaluating the transfer to neighboring cells of LY microinjected into one cell. The cultures were observed on an inverted microscope equipped with xenon arc lamp illumination and a Nikon B filter (excitation wavelength 450–490 nm; emission wavelength above 520 nm). LY (115 mM in 150 mM LiCl) was microinjected through a glass microelectrode by brief overcompensation of the negative capacitance circuit in the amplifier to cause oscillations until the impaled cell was brightly fluorescent. Three minutes after dye injection, cells were observed to determine whether dye transfer occurred. The coupling index was calculated as the mean number of cells to which the dye spread excluding injected cells from which there was no spread and was expressed as percent of control [Dye coupling (%)]. In all experiments, dye coupling was tested by injecting a minimum of 10 cells.
Dye uptake and time-lapse fluorescence imaging
For visualization of Etd dye uptake by “snapshot”, control and treated cells were exposed to 5 μM EtdBr for 10 min at 37°C. Then, cells were washed with Hank’s balanced salt solution (in mM: NaCl: 137; KCl: 5.4; Na2HPO4: 0.34; KH2PO4: 0.44, CaCl2: 1.2 at pH 7.4), and examined by epifluorescence (518 nm excitation and 605 nm emission) on an inverted microscope (Daiphot-Nikon) equipped with a CCD camera (Nikon) associated with image analysis software (Lucia-Nikon). Images of Etd uptake were analyzed with the image J program (NIH software).
For time-lapse fluorescence imaging, cells plated on glass coverslips were washed twice in phosphate-buffered saline (PBS) solution, pH 7.4. All chemicals were dissolved in ultra pure H2O. Cells were exposed to Locke’s solution (containing: 154 mM NaCl, 5.4 mM KCl, 2.3 mM CaCl2, 5 mM HEPES and pH 7.4) with 5 μM of Etd. Phase-contrast and fluorescence microscopy with time-lapse imaging were used to record cell appearance and fluorescence intensity changes in each condition. Changes were monitored using an imaging system equipped with a Retga 1300I fast-cooled monochromatic digital camera (12-bit) (Qimaging, Burnaby, BC, Canada), monochromator for fluorophore excitation, and METAFLUOR software (Universal Imaging, Downingtown, PA) for image acquisition and analysis. The cultures on glass coverslips were placed in an Olympus BX 51W1I upright microscope with water immersion lenses. Regions of interest were placed over random cells and background subtracted. Fluorescence was recorded every 30 sec. To test for changes in slope, regression lines were fitted to points before and after various treatments using the Excel program and mean values of slopes were compared using GraphPad Prism software.
Western blot analysis
Cultures were rinsed twice with a saline solution (Hank’s), pH 7.4, and cells were scraped off the dish with a rubber policeman and harvested in a solution containing inhibitors of proteases (200 mg/ml soybean trypsin inhibitor, 1 mg/ml benzamidine, 1 mg/ml ε-aminocaproic acid, 2 mM PMSF) and phosphatases (20 mM Na4P2O7 and 100 mM NaF). The suspension was centrifuged (14,000 rpm for 2 min at 4°C), the pellet was resuspended in 50 ml of the solution containing protease inhibitors and sonicated (Ultrasonic cell disrupter, Microson). Proteins were measured in aliquots of cell lysates with the Bio-Rad protein assay. Samples were mixed with Laemmli buffer and either stored at −80°C or resolved in 8% SDS/PAGE. After separating the proteins by electrophoresis, they were electrotransferred to nitrocellulose sheets. The sheets were incubated in PBS-BLOTTO (5% nonfat milk in PBS) overnight at 4°C to block nonspecific binding, and then incubated with primary antibody overnight at 4°C, followed by washing in PBS and incubation with secondary goat anti-rabbit IgG antibody conjugated to horseradish peroxidase for 1 h at room temperature. Immunoreactivity was detected by ECL by using the SuperSignal kit. Resulting immunoblot signals were scanned, and densitometric analysis was performed with SCION IMAGE software.
Cell surface biotinylation and quantitation
Cell cultures (90-mm dishes) were washed three times with ice-cold Hank’s saline solution, pH 8.0, and 3 ml of sulfo-NHS-SS-biotin (0.5 mg/ml) was added to each cell culture followed by incubation for 30 min at 4°C. Cells were washed three times with ice-cold saline plus 15 mM glycine, pH 8.0, to quench unreacted biotin. Then, cells were harvested as for Western blots described above. Presence of 1% SDS during this part of the preparation (1) did not affect the yield of Cx43. An excess of immobilized NeutrAvidin was added (1 ml of NeutrAvidin per 3 mg of biotinylated protein, as recommended by the providers), and the mixture was incubated for 1 h at 4°C. Then, 1 ml of washing buffer (saline solution, pH 7.2 plus 0.1% SDS and 1% Nonidet P-40) was added; the mixture was centrifuged for 2 min at 14,000 rpm at 4°C; and the supernatant was removed and discarded. This washing procedure was repeated three times. After the final wash, the supernatant was removed, and 40 ml of saline solution, pH 2.8, plus 0.1 M glycine was added to release the proteins from the biotin; the pellet was resuspended, and the mixture was centrifuged at 14,000 rpm for 2 min at 4°C. The supernatant was placed in an Eppendorf tube (1.5 ml), and the pH was adjusted immediately by adding 10 μl of 1 M Tris, pH 7.5. Relative levels of Cx43 present in each sample were measured by Western analysis as described above.
Immunofluorescence and confocal microscopy
For all immunostaining experiments, cells grown on coverslips were fixed at room temperature with 2% paraformaldehyde for 30 min, washed three times with PBS, incubated in 0.1 M PBS-glycine three times for 5 min each, and then in 0.1% PBS-Triton X-100 containing 10% NGS for 30 min. To identify astrocytes versus microglia, we used anti-GFAP and isolectin B4, respectively). We first incubated cells for 2 h at room temperature with anti-GFAP monoclonal antibody (IgG1, 1:500) diluted in 0.1% PBS-Triton X-100 with 2% NGS. After three rinses in 0.1% PBS-Triton X-100, cells were then incubated for 50 min at room temperature with both goat anti-mouse Alexa Fluor 355 (1:1500) and isolectin GS-IB4 (1:100), diluted in the same solution as the primary antibody. To label Cx43, cells were incubated with the anti-Cx43 monoclonal antibody (1:500) for 1 h at room temperature. After three washes, cells were incubated for 50 min at room temperature with goat anti-mouse Alexa Fluor 488. After several washes, coverslips were mounted in Fluoromount and examined with an upright microscope equipped with epifluorescence (Eclipse E800, Nikon). To visualize double immunostaining, a confocal laser-scanning microscope (TBCS SP2; Leica, Wetzlar, Germany) was used. Stacks of consecutive confocal images taken with a 63 X objective at 500 nm intervals were acquired sequentially with two lasers (argon 488 nm and helium/neon 543 nm), and Z projections were reconstructed using Leica confocal software. The immunofluorescence images were analyzed with ImageJ (NIH). The images were obtained as a stack in which each image had an optical thickness of 1 μm; each image was put into grey scale and digitized. Afterwards, the ImageJ feature for analysis of particles was used, and the Feret’s diameter was measured to quantify the diameter of Cx43 immunoreactive puncta. The Feret’s diameter is the longest measured distance between any two points along the ROI boundary.
Cell death quantification
Astrocyte membrane breakdown was evaluated by incorporation of Rhodamin B dextran (Rdex, 10 kD) after each treatment of interest. Astrocytes cultures were incubated for 3 min with Rdex (100 μM) to label damaged cells and Hoechst 33342 (1 μM) to label all the cells. The cultures were rinsed five times in PBS and then the staining was evaluated using an Olympus BX 51W1 microscope
Data analysis and statistics
Data are reported as means ± SEM. The statistical analyses were performed using GraphPad Prism software. Effects on dye coupling and dye uptake were analyzed by one- or two way ANOVA, followed by a multiple comparisons Tukey’s test or the Bonferroni correction, respectively. In all cases, a P value less than 0.05 was considered statistically significant.
RESULTS
Hypoxia-reoxygenation increases hemichannel activity assessed by dye uptake, an effect potentiated by high glucose during the hypoxia
Previous studies using metabolic inhibition demonstrated an increase in Cx43 hemichannel activity in cortical astrocytes (Orellana et al., 2009). The inhibitors used, antimycin A and iodoacetate, are not readily reversible, and here we have used a more physiologic procedure of hypoxia-reoxygenation that has the added advantage of allowing investigation of an equivalent of reperfusion (Martínez and Sáez 2000). With this method, astrocyte cultures were deprived of oxygen in the presence of varying concentrations of glucose and then returned to normal (control) medium with 5 mM glucose. We used “ischemic saline” during the hypoxic period (see Methods), which better mimics the interstitial fluid composition during in vivo ischemia and results in greater ischemic damage to astrocytes in culture (Bondarenko and Chesler 2001). We first evaluated the effect of hypoxia in normal glucose on hemichannel activity measured as uptake of ethidium ion (5 μM Etd, added to the medium as EtdBr). Astrocytes under control conditions showed a low rate of Etd uptake (Fig. 1A, B) as previously demonstrated (Contreras et al., 2004; Retamal et al., 2006). After 3 h in ischemic saline with low oxygen and normal glucose (5 mM) and 1 h reoxygenation in control saline and 5 mM glucose, uptake relative to control was almost unchanged (Fig. 1A, C, I). With the same protocol but high glucose (27 mM) present only during hypoxia, uptake had increased to ~150% of control at the time of reoxygenation, suggesting that the cell permeabilization response began during the hypoxic period. Uptake then increased to > 300% of control at 1 h before recovering to near control at 2.5 h. Hypoxia in medium containing zero glucose increased uptake to an intermediate degree (Fig. 1I). When the hypoxic period was 6 h in ischemic saline with 0, 5, or 27 mM glucose, the effects were similar but persistent throughout the 3 h reoxygenation period in 5 mM glucose (Fig. 1E–H, J). Dye uptake was elevated (to ~190% of control) at the start of the reoxygenation after hypoxia in 27 mM glucose and increased to ~430% of control after 1 h reoxygenation in control saline with 5 mM glucose (Fig. 1E and J). After 6 h hypoxia in 5 mM glucose and 1 h reoxygenation in 5 mM glucose, the Etd uptake rate was increased to ~220% (p < 0.001) of control (Fig. 1E and J). Uptake after 3 or 6 h hypoxia in different glucose concentrations and 1 h reoxygenation in 5 mM glucose depended on glucose concentration during the hypoxia, and was greater at zero as well as high glucose (Fig. 1K).
Figure 1. Hypoxia-reoxygenation increases rate of Etd uptake by rat astrocytes; an effect potentiated by high glucose.

(A) Time-lapse measurements of Etd uptake in rat astrocytes under control conditions (○) and starting at 1 h reoxygenation after 3 h hypoxia in 5 mM ( ) or 27 mM glucose (●). Gap 26 (200 μM), a Cx43 hemichannel blocker, was applied after 12 min of Etd uptake measurement. (B–D) Fluorescence micrographs of Etd uptake (10 min exposure to dye after 1 h reoxygenation) in astrocytes under control conditions (B) and at 1 h reoxygenation after 3 h hypoxia in 5 (C) or 27 mM glucose (D). (E) Time-lapse measurements of Etd uptake in astrocytes under control conditions (○) and starting at 1 h of reoxygenation after 6 h of hypoxia in 5 () or 27 mM glucose (●). (F–H) Fluorescence micrographs of Etd uptake (10 min exposure to dye) in astrocytes under control conditions (F) and at 1 h reoxygenation after 6 h hypoxia in 5 (G) or 27 mM glucose (H). (I) Averaged data normalized to control (dashed line) of Etd uptake rate by astrocytes measured at the beginning of reoxygenation (time 0) and after increasing periods of reoxygenation following 3 h hypoxia in 0 mM (○),5 mM () or 27 mM glucose (●). Zero time is at the beginning of reoxygenation. *** p < 0.001, (●) vs (); ††† p < 0.001, (●) vs (○); £££ p < 0.001, (○) vs (). (J) Etd uptake rate of astrocytes after increasing reoxygenation periods following 6 h hypoxia in 0 mM (○),5 mM (), or 27 mM glucose (●). Zero time is at the beginning of reoxygenation. *** p<0.001, ** p<0.005, * p<0.05; (●) vs (); ††† p<0.001, †† p<0.005, † p<0.05; (●) vs (○); £ p<0.05; (○) vs (). (K) Etd uptake rate at 1 h reoxygenation following 3 (□) or 6 h (■) of hypoxia in different glucose concentrations. *** p<0.001, * p<0.05; (■) vs (□). Each value corresponds to mean ± S.E. of 20 cells in a representative of five experiments. Bar = 60 μm.
) or 27 mM glucose (●). Gap 26 (200 μM), a Cx43 hemichannel blocker, was applied after 12 min of Etd uptake measurement. (B–D) Fluorescence micrographs of Etd uptake (10 min exposure to dye after 1 h reoxygenation) in astrocytes under control conditions (B) and at 1 h reoxygenation after 3 h hypoxia in 5 (C) or 27 mM glucose (D). (E) Time-lapse measurements of Etd uptake in astrocytes under control conditions (○) and starting at 1 h of reoxygenation after 6 h of hypoxia in 5 () or 27 mM glucose (●). (F–H) Fluorescence micrographs of Etd uptake (10 min exposure to dye) in astrocytes under control conditions (F) and at 1 h reoxygenation after 6 h hypoxia in 5 (G) or 27 mM glucose (H). (I) Averaged data normalized to control (dashed line) of Etd uptake rate by astrocytes measured at the beginning of reoxygenation (time 0) and after increasing periods of reoxygenation following 3 h hypoxia in 0 mM (○),5 mM () or 27 mM glucose (●). Zero time is at the beginning of reoxygenation. *** p < 0.001, (●) vs (); ††† p < 0.001, (●) vs (○); £££ p < 0.001, (○) vs (). (J) Etd uptake rate of astrocytes after increasing reoxygenation periods following 6 h hypoxia in 0 mM (○),5 mM (), or 27 mM glucose (●). Zero time is at the beginning of reoxygenation. *** p<0.001, ** p<0.005, * p<0.05; (●) vs (); ††† p<0.001, †† p<0.005, † p<0.05; (●) vs (○); £ p<0.05; (○) vs (). (K) Etd uptake rate at 1 h reoxygenation following 3 (□) or 6 h (■) of hypoxia in different glucose concentrations. *** p<0.001, * p<0.05; (■) vs (□). Each value corresponds to mean ± S.E. of 20 cells in a representative of five experiments. Bar = 60 μm.
The increases in uptake were not observed, if the cells were maintained in control saline throughout the hypoxia-reoxygenation period independent of the glucose concentration (not shown); this result suggests that the differences in ionic concentration during ischemia play a significant role in the permeability changes consistent with earlier observations on damage to neurons in hippocampal slice cultures (Cronberg et al., 2004; Rytter et al., 2003). The long period of hypoxia required to see significant changes in cultured astrocytes as compared to just 5 min of ischemia followed by reperfusion in hyperglycemic animals (Muranyi et al. 2006) might be explained by changes in astrocyte phenotype under the culturing conditions as well as interactions between neurons and glia in vivo.
Increasing osmotic pressure of the 5 mM glucose saline by adding sucrose equimolar to the added glucose did not significantly enhance the hypoxia-reoxygenation induced increases in dye uptake (not shown). Furthermore, the hypoxia induced decrease in dye coupling to be described below (Fig. 4) was unaffected by the osmotic pressure differences.
Figure 4. Hypoxia-reoxygenation reduces dye coupling in rat astrocytes; an effect potentiated by high glucose.

(A–D) Fluorescence micrographs of LY coupling in astrocytes under control conditions (A), at 1 h of reoxygenation after 3 h hypoxia in 5 (B) or 27 mM glucose (C), and at 1 h of reoxygenation after 6 h hypoxia in 27 mM glucose (D). The (*) in panels A–D denotes the cell microinjected with LY. Bar = 60 μm. (E) Average number of astrocytes to which LY spread from an injected cell after different reoxygenation periods following 3 h of hypoxia in 5 (), 27 (●) or 0 mM (○) glucose (expressed as percentage of the number in control conditions, dashed line). Zero time is at the beginning of reoxygenation. ** p<0.005, *** p<0.001, (●) v/s (). (F) LY coupling by astrocytes after increasing reoxygenation periods following 6 h hypoxia in 5 (), 27 (●) or 0 mM (○) glucose. *** p<0.001, (●) vs (●); ††† p<0.001, †† p<0.005, (●) vs (○); ## p<0.005, # p<0.05; () vs (○). (C) LY coupling at 1 h reoxygenation after 3 (□) or 6 h (■) hypoxia in different glucose concentrations. * p<0.05; (■) vs (□). For each graph values are means ± S.E. of 10 cells in a representative experiment of five; separate cultures were used for each time point.
We found no differences in Etd uptake if cells were bathed in 5 or 27 mM glucose before or after the hypoxic episode (Fig. 2A); thus, the effects of high and zero glucose begin during the hypoxic period. Furthermore, there was no difference between reoxygenation in normal and in ischemic saline (not shown). To determine whether the astrocyte permeabilization induced by hypoxia in high glucose was mediated by Cx43 hemichannels, we applied various blocking agents during dye uptake after 3 h hypoxia in 27 mM glucose and 1 h reoxygenation in 5 mM glucose. We used La3+ or specific “connexin mimetic peptides” (Gap26 or Gap27), which have the same sequence of a region of the first or second extracellular loops of Cx43, respectively (Evans and Leybaert 2007). The application of La3+ (200 μM) or Gap26 (200 μM) during Etd uptake greatly reduced the rate induced by hypoxia-reoxygenation (Fig. 1A and E); also, Gap27 (200 μM), La3+ (200 μM) or Gap26 (200 μM) prior to Etd application reduced the percentage of labeled cells below control values in “snapshot” experiments (Fig. 2B).
Figure 2. High or zero glucose levels during hypoxia increase astroglial Etd uptake after reoxygenation, an effect independent of high glucose before or after the hypoxic period; uptake is mediated exclusively by Cx43 hemichannels.

(A) Averaged data normalized to control (dashed line) of Etd uptake rate by rat astrocytes after 3 h hypoxia in 0 mM (left), 5 mM (middle) or 27 mM glucose (right) and 1 h reoxygenation and maintained 24 h before hypoxia and during the 1 h reoxygenation with 5 mM (white bar), 27 mM glucose (dashed white bar) or with the two possible combinations of one before and the other after (black and gray bars). * p < 0.001; with respect to control. (B) Percentage of Etd-positive cells in snapshot pictures of mouse astrocytes compared to control (dashed line) after 3 h hypoxia in 27 mM glucose and 1 h reoxygenation. Cells were then exposed to 5 μM Etd for 20 min and snapshots were taken. In parallel experiments, cells were simultaneously exposed for 20 min to Etd and a connexin hemichannel blocker, La3+ (200 μM), Gap26 (200 μM) or Gap27 (200 μM), or a Px1 hemichannel blocker, 10panx1 (200 μM) or a P2X channel blocker, oATP (200 μM). The number of Etd-positive cells in astrocyte cultures of Cx43−/− mice at 1 h of reoxygenation following 3 h hypoxia in 27 mM glucose is also shown.
Recently, it was demonstrated that cultured astrocytes express pannexin1 (Px1) in vitro (Huang et al. 2007). However, in primary astrocytes Px1 was not found at the cell surface (Huang et al. 2007). To investigate the possible contribution of Px1 hemichannels in post-hypoxic dye uptake, we used an extracellular loop peptide (10pnx1) that has been reported to block Px1 hemichannels (Pelegrin and Surprenant 2006), but it was without effect on uptake induced in mouse astrocytes by 3 h hypoxia in high glucose and 1 h reoxygenation (Fig. 2B). Since Px1 hemichannels can be activated by extracellular ATP acting on P2X7 receptors (Pelegrin and Surprenant 2006), we also studied the effect of oATP, a P2X7 receptor blocker; again, there was no effect on hypoxia-reoxygenation induced Etd uptake (Fig. 2B). Similar results were obtained in rat astrocytes (not shown). Thus, these findings suggest that Cx43, but not Px1 hemichannels mediate the increase in astrocyte permeability induced by hypoxia in high glucose and reoxygenation. Further evidence was obtained using astrocytes from Cx43−/− mice. Subjecting these cells to 3 h of hypoxia in 27 mM glucose and 1 h of reoxygenation did not cause an increase in Etd uptake like that observed in wild type astrocytes (Fig. 2B), supporting the inference that astroglial permeabilization occurred specifically through Cx43 hemichannels.
We also used HeLa cells transfected with Cx43-EGFP to evaluate if hemichannel permeability induced by hypoxia occurs in another cell type and whether the effect could be reproduced in an exogenous expression system. These cells show basal Etd uptake at the resting potential and exhibit unitary events with conductance and pharmacological sensitivity predicted for Cx43 hemichannels (Contreras et al., 2003). As reported, cells expressing more Cx43-EGFP showed faster Etd uptake (Fig. 3A–C and D). Already at 1 min of reoxygenation following 6 h of hypoxia in 5 and 27 mM glucose Etd uptake was increased to ~250% and 520% of control, respectively (Fig. 3G); these increases almost certainly occurred during the hypoxic period. In both cases complete recovery occurred after 2–3 h reoxygenation (Fig. 3G). The increase in Etd uptake was due to Cx43 hemichannels, since it was sensitive to La3+ (Fig. 3F) and was directly related to Cx43-EGFP fluorescence in both control cells and in cells subjected to hypoxia-reoxygenation (p < 0.001, r2 = 0.93 and p < 0.001, r2 = 0.94, respectively, Fig. 3E). In summary, hypoxia induced increase in hemichannel mediated dye uptake occurs in another cell type expressing an exogenous connexin, Cx43-EGFP. The effects are as large as in astrocytes, but in contrast are reversible. The greater resistance of the HeLa cells may be related to their tumor origin and selection for survival with poor circulation (Anderson et al., 2006).
Figure 3. Hypoxia-reoxygenation increases Cx43-hemichannel activity in HeLa cells expressing Cx43-EGFP; an effect potentiated by high glucose.

Etd uptake was measured in mixed cultures of Cx43-EGFP and parental cells (5 μM Etd). (A) Fluorescence micrograph showing different degrees of EGFP expression. (B) Etd uptake (10 min incubation in 5 μM) after 6 h hypoxia in 27 mM glucose (6 h hypox/27 mM) and 1 h reoxygenation. (C) Merge of EGFP and Etd fluorescence. (D) Time course of Etd uptake in Cx43-EGFP cells (1–4 in A) and parental HeLa cells (5–7 in A) after 6 h hypoxia in 27 mM glucose and 1 h reoxygenation. (E) Rate of Etd uptake was proportional to Cx43-EGFP fluorescence for seven cells after 6 h hypoxia in 27 mM glucose and 1 h reoxygenation (●) and for six cells in control conditions (○). (F) Time-lapse measurements of Etd uptake in Cx43-EGFP cells under control conditions (○) and after 6 h hypoxia in 5 () or 27 mM (●) glucose and 1 h reoxygenation. (G) Averaged Etd uptake normalized to control (dashed line) by Cx43-EGFP cells for different reoxygenation periods after 6 h hypoxia in 5 (○) or 27 mM (●) glucose. Each point corresponds to the mean ± S.E. of 20 cells in a representative experiment of four. Bar = 10 μm.
Hypoxia-reoxygenation reduces dye coupling between astrocytes, an effect potentiated by high glucose during the hypoxia
In the central nervous system of vertebrates, gap junctional coupling of astrocytes is prominent and plays important roles in ionic homeostasis, thus helping to ensure the normal functioning of neurons (Simard and Nedergaard 2004). In view of data presented thus far and of previous studies showing that a hypoxic episode can cause delayed and transient reduction in dye coupling of cortical astrocytes in culture (Martínez and Sáez 2000), we investigated whether astroglial coupling is more sensitive to hypoxia in high glucose.
Since leakage of the microinjected dye (LY) to evaluate intercellular communication via gap junctions might occur through hemichannels, particularly in view of their increased opening after hypoxia-reoxygenation, we measured dye coupling in both the absence and the presence of 200 μM La3+ in the extracellular solution. La3+ did not significantly modify the intercellular diffusion of microinjected LY in astrocytes under control conditions or after hypoxia in normal or high glucose followed by reoxygenation in normal glucose (not shown). Three hours of hypoxia in 27 mM glucose and ischemic saline followed by reoxygenation reduced dye coupling compared to control (Fig. 4A, C, E). The maximal effect induced by 27 mM glucose occurred at 1 to 1.5 h of reoxygenation with ~60% reduction in dye coupling compared to control (Fig. 4E). In addition, hypoxia in 5 mM glucose or in zero glucose reduced dye coupling by ~20% and ~40% of control, respectively, at 1 h of reoxygenation, less than the effect in 27 mM glucose (Fig. 4A–C, E). The inhibitory effects on dye coupling induced by 6 h hypoxia in 27 mM glucose, normal glucose or zero glucose were more pronounced than those induced by 3 h of hypoxia with the same glucose concentrations (Fig. 4D, F). Unlike the transient reduction induced by 3 h of hypoxia, 6 h of hypoxia in 27 mM glucose induced a persistent reduction in dye coupling reaching about ~90% inhibition at 1 h of reoxygenation and recovering very little by 3.5 h reoxygenation (Fig. 4F). Similar to the concentration dependence of the effect of glucose during hypoxia on subsequent hemichannel mediated dye uptake (Fig. 1K), glucose during 3 or 6 h of hypoxia reduced dye coupling after 1 h reoxygenation in a concentration-dependent manner with a moderate effect at zero glucose, a smaller effect at 5–12 mM glucose, and then an effect increasing to a plateau at 27–37 mM glucose (Fig. 4G).
Hypoxia-reoxygenation increases the level of Cx43 on the cell surface, an effect potentiated by high glucose
During metabolic inhibition, an in vitro model of ischemia, Etd uptake is ascribed to an increase in Cx43 hemichannel activity, largely accounted for by elevated levels of surface hemichannels with little change in open probability (Retamal et al., 2006). To test if reoxygenation after hypoxia in high glucose affects the surface levels of Cx43, cell surface proteins were biotinylated, isolated with NeutrAvidin beads, and resolved by Western blotting. After 3 h hypoxia and 1 h reoxygenation the levels of surface Cx43 depended on the concentration of glucose present during the hypoxia period; they were unaffected by 5 mM glucose but increased by zero and 22–37 mM glucose (Fig. 5A, left panel, B). The increases in surface Cx43 levels were delayed and transient; they were significantly higher at 1 h than at 40 min reoxygenation, and had returned to control levels by 3 h of reoxygenation (Fig. 5C, left panel, D). Levels of surface Cx43 were also elevated in astrocytes exposed to 1 h reoxygenation after 6 h of hypoxia in zero glucose or in 22 mM glucose or higher but not in 5 mM glucose (Fig. 5E, left panel, F). After 6 h hypoxia in 27 mM glucose, surface Cx43 was increased after a shorter period of reoxygenation (20 min) compared to the increase induced by reoxygenation after 3 h hypoxia (40 min) (Fig. 5G, left panel, H). Moreover, after 6 h hypoxia the surface Cx43 levels remained high for at least 3 h reoxygenation, whereas after 3 h hypoxia and 3 h reoxygenation, recovery was complete.
Figure 5. Hypoxia-reoxygenation increases the level of surface Cx43 in astrocytes, an effect potentiated by high glucose.

Cultured astrocytes were subjected to 3 (A–D) or 6 h (E–H) hypoxia (hypox) in presence of different glucose concentrations and then to reoxygenation (reox) for different periods of time. Representative western blots of levels of surface (left panels in A, C, E, and G) and total Cx43 (right panels) are shown. (A, E) The left panels show levels of surface Cx43 present in astrocytes under control conditions, i.e., not subjected to hypoxia (lane 1) or subjected to 1 h of reoxygenation after 3 (A) or 6 h (E) hypoxia in 0 (lane 2), 5 (lane 3), 22 (lane 4), 27 (lane 5) and 37 mM (lane 6) glucose. The right panels show the levels of total Cx43 under the same conditions along with α-tubulin as a loading control. The Cx43 phosphorylated (P1–P2) and nonphosphorylated (NP) forms are indicated on the left of each gel. (B, F) Quantitation of data exemplified by A and E; surface and total Cx43 after 3 (B) or 6 h (F) hypoxia in the indicated glucose concentrations followed by 1 h reoxygenation in 5 mM glucose. Total levels were normalized according to the levels of α-tubulin detected in each lane. (C, G) Surface and total Cx43 after 3 (C) or 6 h (G) hypoxia in 27 mM glucose and the indicated times of reoxygenation. (D, H) Quantitation of data exemplified by C and G; surface and total Cx43 after 3 (D) or 6 h (H) hypoxia in 27 mM glucose and the indicated times of reoxygenation. Total levels were normalized according to the levels of α-tubulin detected in each lane.
Under all conditions described above, the total levels of Cx43 were not affected (Fig. 5A, C, E, G, right panels; B, D, F, H). Furthermore, in all Western blot analyses of surface or total Cx43 levels the characteristic phosphorylated P1-P2 bands were predominant, and the unphosphorylated NP band was less intense (Fig. 5A, C, E, G), suggesting that the treatments did not reduce the expression or increase the degradation of Cx43 or induce dephosphorylation of Cx43 detectable by this technique.
Hypoxia-reoxygenation causes disappearance of large Cx43 aggregates and increases GFAP expression
To examine further the altered Cx43 distribution in the cells, we applied indirect immunofluorescence to astrocytes under conditions that induced the greatest changes in hemichannel activity and gap junctional coupling. In control astrocytes Cx43 labeling was heterogeneous including areas of different shape and size; the larger ones are likely correspond to gap junction plaques or internalized gap junctions, and the smaller ones are likely trans Golgi vesicles for Cx43 delivery to the plasma membrane and small junctional regions internalized and targeted for degradation (Fig. 6A and Fig. 1S). In astrocytes subjected to 6 h of hypoxia in 5 mM glucose and 1 h of reoxygenation, Cx43 immunoreactivity was not located in big continuous areas, and labeling was of smaller intracellular puncta, possibly vesicles (Fig. 6B and Fig. 1S). The larger areas had likely been internalized as small vesicles (Gaietta et al. 2002). No obvious difference in Cx43 immunoreactivity was observed between astrocytes subjected to 6 h hypoxia in the presence of 27 mM glucose and those subjected to 6 h hypoxia in 5 mM glucose (1 h reoxygenation, Fig. 6C and Fig. 1S). Cells subjected to 6 h hypoxia with 5 or 27 mM glucose and 1 h reoxygenation showed an increase in GFAP immunoreactivity and in the number of astroglial processes (Fig. 6C), two properties that are characteristic of astroglial activation (Pekny and Nilsson 2005).
Figure 6. Hypoxia-reoxygenation causes disappearance of the larger Cx43 immunoreactive areas found in control cells and increases astroglial activation during reoxygenation.
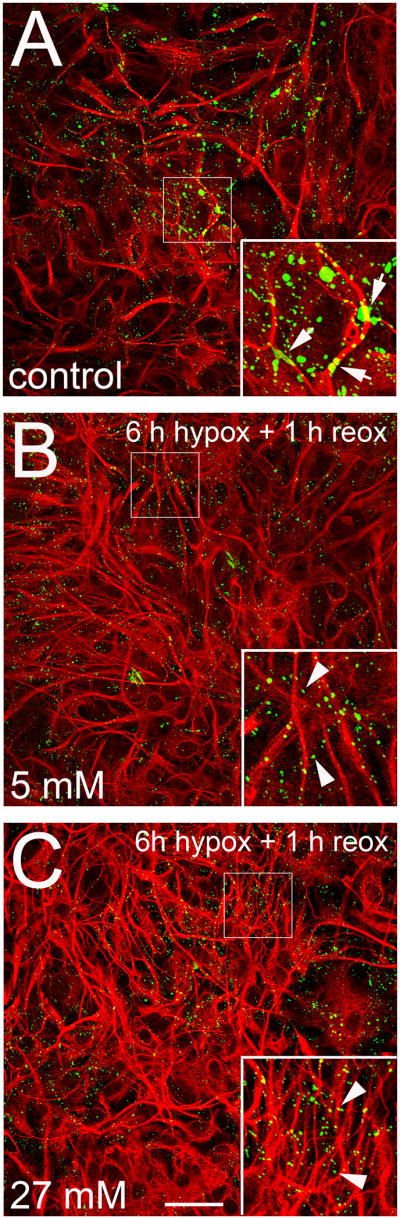
Representative confocal images depicting Cx43 (green) and GFAP (red) immunolabeling of astrocytes under control conditions (A) or at 1 h reoxygenation after 6 h hypoxia in 5 (B) or 27 mM (C) glucose. The larger Cx43 labeled areas in A may represent gap junction plaques (arrows); Cx43 labeling in B and C is only of smaller areas (arrow heads). Increased GFAP labeling in B and C indicates increased activation, which appears more pronounced for hypoxia in 27 mM glucose. Insets: 2X magnification of the indicated area of each panel. Bar = 50 μm.
Increase in dye uptake induced by hypoxia-reoxygenation is reversed by DTT, but decrease in dye coupling is not
Redox potential modulates the activity of Cx43 hemichannels in astrocytes (Retamal et al., 2006; Retamal et al., 2007b). Reducing agents reverse the increase in membrane permeability through hemichannels induced by metabolic inhibition or by application of nitric oxide donors or proinflammatory cytokines (Retamal et al., 2007b). However, reducing agents increase membrane permeability under normoxic conditions (Retamal et al., 2006; Retamal et al., 2007b). To evaluate the redox state of Cx43 hemichannels in astrocytes subjected to hypoxia with normal or high glucose, we measured dye uptake after 3 or 6 h of hypoxia and 1 h reoxygenation and then applied DTT (10 mM) during time lapse measurement of Etd uptake. In control conditions, the application of 10 mM DTT increased Etd uptake to ~400% of control (Fig. 7A, B). However, in cells that were exposed to 3 h of hypoxia in 27 mM glucose and 1 h reoxygenation, DTT applied during Etd uptake did not affect the uptake rate, whereas following 6 h of hypoxia in 27 mM glucose and 1 h reoxygenation, DTT reduced the rate, suggesting that the hemichannels had been oxidized (Fig. 7A, B). La3+ largely blocked the increases in uptake rate, whether they were induced by DTT or by hypoxia-reoxygenation. The reduction in dye coupling induced by 6 h hypoxia in 27 mM glucose and 1 h reoxygenation (Fig. 4D, F) was not reversed by DTT (Fig. 7C). These observations suggest that 6 h hypoxia in high glucose and 1 h reoxygenation oxidizes at least some of the hemichannels or an associated molecule and that the processes involved in the regulation of Cx43 hemichannels and gap junction channels during hypoxia-reoxygenation are different.
Figure 7. Changes in hemichannel and gap junction channel activity induced after hypoxia in high glucose and reoxygenation are differentially affected by dithiothreitol (DTT), but both are blocked by a p38 MAPK inhibitor.

(A) Time-lapse measurements of Etd uptake in astrocytes under control conditions (○), and starting at 1 h reoxygenation after 3 () or 6 h (●) hypoxia in 27 mM glucose. After uptake rate was established (7 min), cells were exposed to DTT (10 mM) and then DTT + La3+ (200 μM). Increase in uptake rate caused by DTT in control conditions was largely blocked by La3+. Increase after 3 h hypoxia and reoxygenation was not affected by DTT but was blocked by La3+. Increase after 6 h hypoxia was reduced by DTT and blocked by La3+. (B, C) Averaged data normalized to control (dashed line) of Etd uptake rate (B) or LY coupling between cells (C) at 1 h reoxygenation following 6 h hypoxia in 27 mM glucose. DTT was applied 7 min after initiating the Etd uptake measurement at 1 h of reoxygenation. SB202190, a p38 MAP kinase inhibitor (10 μM), was applied 1 h before the hypoxia. Significance is with respect to dye uptake after 6 h hypoxia and 1 h reoxygenation or control dye coupling. ***: p<0.001. (C) LY coupling as percent of control coupling (dotted line, 100%) after 6 h hypoxia in 27 mM glucose and 1 h reoxygenation.
Changes in hemichannel and gap junction channel activity induced by hypoxia-reoxygenation are p38 MAPK dependent
Because Cx43 hemichannels and gap junction channels in astrocytes treated with proinflammatory cytokines are modulated through a p38 MAPK-dependent pathway (Retamal et al., 2007a), and hypoxia-reoxygenation is a proinflammatory condition, we examined whether the changes in hemichannels and gap junctions induced by hypoxia-reoxygenation in high glucose are mediated by a p38 MAPK pathway. The addition of SB202190, an inhibitor of p38 MAPK, 1 h before the hypoxia largely prevented the Etd uptake increase induced by 6 h hypoxia in 27 mM glucose and 1 h of reoxygenation (Fig. 7B). Moreover, the same treatment with SB202190 prevented the decrease in dye coupling induced by the same hypoxia-reoxygenation protocol (Fig. 7C).
The increased hemichannel activity induced by hypoxia-reoxygenation, which is potentiated by high glucose, promotes astroglial death
To examine if the rise in hemichannel activity induced by hypoxia-reoxygenation is deleterious, we evaluated astroglial viability measured by exclusion of Rdex at different reoxygenation periods after 3 or 6 h hypoxia in normal or high glucose. Due to its large size, Rdex is taken up only by cells with disrupted membranes (Kondo et al., 2000). Under control conditions and after 3 h hypoxia in 5–27 mM glucose and 6 h reoxygenation, few astrocytes took up Rdex (Fig. 8A, G). However, after 6 h of hypoxia in 5 mM glucose and 6 h reoxygenation, ~20 % of the astrocytes took up Rdex and were, by this measure, dead (Fig. 8D, G), and after 6 h hypoxia in 27 mM glucose and 3–6 h reoxygenation, ~40 – 45% of astrocytes were dead (Fig. 8E, G). Cell death induced by 6 h of hypoxia in high glucose was greatly reduced by the hemichannel blockers, Gap26, Gap27 or La3+, applied at the start of the reoxygenation period (Fig. 8H). The p38 MAPK inhibitor, SB202190, which reduces hypoxia-reoxygenation opening of Cx43 hemichannels, was also protective. A peptide reported to act on pannexin hemichannels, 10panx1, was without effect, as was a somewhat longer peptide, E1b (Pelegrin and Surprenant 2006; Wang et al., 2007). In addition, probenecid, which blocks Px1 hemichannels (Silverman et al. 2008), was not protective.
Figure 8. Connexin hemichannel activity induced by hypoxia-reoxygenation leads to astroglial death.

(A–F) Fluorescence micrographs of Rdex (red) uptake and Hoechst 33342 nuclear staining (blue) in rat astrocytes under control conditions (A), at 6 h of reoxygenation after 3 h hypoxia in 5 (B, 3 h hypox/5 mM) or 27 mM glucose (C, 3 h hypox/27 mM) or at 6 h reoxygenation after 6 h hypoxia in 5 mM glucose (D, 6 h hypox/5 mM) or 27 mM glucose (E, 6 h hypox/27 mM). (F) Gap26 (300 μM) applied at the start of reoxygenation after 6 h of hypoxia in 27 mM glucose prevented uptake of Rdex at 1 h reoxygenation. (G) Quantitation of cell death measured by Rdex staining as percentage of total cells identified with Hoechst 33342 at 0, 1, 3, and 6 h reoxygenation following 3 or 6 h hypoxia in 5 or 27 mM glucose. * p<0.001, compared to control. (H) Effect of Gap26 (200 μM), Gap27 (200 μM), La3+ (200 μM), 10panx1 (200 μM), E1b (200 μM), probenecid (2 mM Prob), and SB202190 (10 μM SB) on cell death induced by 6 h reoxygenation following 6 h hypoxia in 27 mM glucose. *** p<0.001, compared to the effect induced by 6 h hypoxia/27mM + 6 h reox. (I) Gap26 had no effect on the reduction in dye coupling induced by 6 h reoxygenation following 6 h hypoxia in 27 mM glucose. Each bar corresponds to the mean ± S.E. of four experiments. Bar in F: 50 μm.
To determine whether the reduction in astroglial death induced by Gap26 was associated with block of hemichannels or an effect on gap junctional coupling, we applied Gap26 during the 6 h of reoxygenation after 6 h hypoxia in high glucose and then evaluated dye coupling. Although Gap26 reduced astrocyte death (Fig. 8F, H), it did not affect the reduction in dye coupling induced by hypoxia-reoxygenation (Fig. 8I). Taken together these observations indicate that astroglial death resulting from hypoxia-reoxygenation is linked to the increase in Cx43 hemichannel permeability rather than to pannexin hemichannels or the reduction in gap junctional communication.
DISCUSSION
The present work shows that hypoxia-reoxygenation can enhance activity of Cx43 hemichannels and reduce gap junctional coupling of cortical astrocytes in culture. The effects are potentiated by high as well as zero glucose during the hypoxic period. The effects of high glucose also occur in HeLa cells expressing exogenous Cx43-EGFP. High or zero glucose during the hypoxia enhances the increase in Cx43 hemichannel activity, but high glucose before or after the hypoxia has no effect. Much of the increase in uptake can be accounted for by increased levels of surface Cx43 hemichannels without an increase in their open probability. Increased hemichannel activity decreases astrocyte survival after hypoxia-reoxygenation, and the reduced gap junction communication does not by itself decrease astrocyte viability when hemichannels are blocked.
Several lines of evidence indicate that the increase in dye uptake induced by hypoxia-reoxygenation in astrocytes is due to Cx43 hemichannels. First, hemichannel activity induced by hypoxia in high glucose is detected in wild type but not in Cx43−/− astrocytes. Second, pharmacological treatments known to inhibit Cx43, but not pannexin hemichannels (e.g., La3+ and Gap26), block dye uptake and prevent astroglial death induced by hypoxia in high glucose. Third, the increase in dye uptake induced by hypoxia is not observed in Cx43−/− astrocytes. Fourth, two pannexin mimetic peptides, 10panx1 and E1b, as well probenecid, which are presumptive pannexin hemichannel blockers, do not affect dye uptake.
To examine the generality of the Cx43 response, we tested whether Cx43-EGFP hemichannels expressed in HeLa cells were similarly sensitive to hypoxia-reoxygenation in high glucose. Reoxygenation after hypoxia increase the activity of Cx43-EGFP hemichannels, and the response is potentiated by high glucose concentrations present during the hypoxic episode. Therefore, this aspect of the response to hypoxia and high glucose conditions may be cell type independent.
Differences in intensity, duration, and/or “quality” (absence of or high glucose) of an ischemic episode determine whether there is no lasting response, induction of “ischemic tolerance”, also termed ischemic preconditioning, or damage, which may appear with some delay (Gidday, 2006). In our experiments, 6 h hypoxia induced greater effects on hemichannels and gap junctional communication than 3 h hypoxia, and the effects were more pronounced at high and zero glucose during the hypoxia period (difference in quality) and could be irreversible in the case of hemichannel activity after 6 h hypoxia and 6 h of reoxygenation. Previous studies showed that metabolic inhibition with iodoacetate and antimycin A, an in vitro ischemia model, increases Cx43 hemichannel opening and reduces gap junctional coupling of cortical astrocytes (Contreras et al., 2004), but in this model there is no analog of reperfusion, and all changes occur in normoxia. The present study demonstrates that hypoxia in a saline that mimics the interstitial ionic concentrations of ischemic brain (Bondarenko and Chesler 2001) followed by reoxygenation in control saline causes changes in Cx43 based channels similar to those induced by metabolic inhibition, but largely delayed until the period of reoxygenation. The hypoxia-reoxygenation procedure is more physiologic and allows the temporal dissection of changes occurring upon reoxygenation from those during the absence of oxygen. In our experiments, most of the reduction in gap junctional communication, like the increase in hemichannel activity, occurred during reoxygenation, which is in agreement with previous reports on gap junctions using similar conditions (Orellana et al., 2009).
The opposite changes in hemichannel opening and gap junction communication are unlikely to be directly related. The current view is that gap junctions are degraded by internalization of small regions of the entire junction thickness and then transported to lysosomes (Gaietta et al., 2002). Internalization of large plaques into one or the other of the coupled cells is also reported (Piehl et al., 2007). There is no indication that docked hemichannels can be separated from each other and left in the membrane where they might open in the absence of a docked hemichannel in an apposed membrane. However, if junction formation is reduced, there may be more hemichannels in the membrane (VanSlyke and Musil 2005), and a indirect relation between increased hemichannel opening and reduction in coupling is possible.
Astrocytes bathed with saline containing normal glucose (5 mM) did not show enhanced dye uptake or reduced coupling at the onset of reoxygenation after 3 h hypoxia, and by these measures hypoxia in normal glucose is less deleterious than hypoxia at zero or a high glucose concentration in agreement with work of others performed on neurons (Cronberg et al., 2004; Goldberg and Choi 1993; Pringle et al., 1997). Delayed changes were also greater when the hypoxia was in zero or high glucose. Hypoxia in high glucose, during but not before or after the hypoxia, induced similar changes in Cx43 based channels. The effect of zero glucose might be explained by loss of the normal glucose effect mentioned above. The effect of high glucose concentration showed a concentration-dependent relationship, presumably caused by glucose itself and not hypertonicity, since equimolar sucrose concentrations failed to enhance the hemichannel activity.
Several mechanisms could contribute to increased hemichannel activity measured as dye uptake: increased opening of hemichannels already present in the plasma membrane, increase in number of hemichannels in the membrane by increased insertion or decreased internalization, and increase in the permeability of open hemichannels. Glucose during the hypoxia increased the surface levels of Cx43 after 1 h reoxygenation in a concentration-dependent manner, perhaps enough to account for the increase in permeability considering the differences in the techniques of measurement.
The reduction in dye coupling induced by hypoxia-reoxygenation cannot be attributed to a reduction in Cx43 levels or altered phosphorylation state detectable by changes in electrophoretic mobility, because at different periods of reoxygenation, the Cx43 total levels and pattern of immunoreactive bands were similar to those of control cells. However, immunofluorescence labeling showed a marked decrease in structures compatible with gap junction plaques after 6 h hypoxia in normal or high glucose and 1 h reoxygenation, suggesting that internalization and degradation of gap junctions might explain the reduction in dye coupling.
Hypoxia-reoxygenation is a proinflammatory condition, and high glucose enhances the effects on hemichannels and gap junction channels. Furthermore, these findings parallel recent reports showing that gap junction channels and hemichannels are oppositely regulated in cell lines transfected with Cx43 (Orellana et al., 2009). In addition, we investigated the signaling pathways involved in the opposite modulation of Cx43 based channels by hypoxia-reoxygenation and found that p38 MAPK inhibition reduced the effect of hypoxia-reoxygenation on both Cx43 hemichannels and gap junctions. This observation is in agreement with previous reports showing that the opposite regulation of the two types of connexin channel in astrocytes treated with TNF-α plus IL-1β, another proinflammatory condition, depends on the p38 MAP kinase pathway (Retamal et al., 2007a). p38-MAP kinase activity is needed to induce increased activity of Cx43 HCs (Schalper et al., 2008), possibly using a Ca2+-dependent pathway, (Mu et al., 2008) or as consequence of oxidant stress (Guyton et al., 1996).
Furthermore, hemichannel-mediated dye uptake induced by metabolic inhibition or treatment with NO donors in astrocytes is blocked by DTT without changing Cx43 hemichannel phosphorylation as indicated by the electrophoretic mobility (Retamal et al., 2006). In contrast, under normoxic conditions DTT increases the open probability of hemichannels in astrocytes and in Cx43-transfected HeLa cells, and a gradual transition between the two responses was observed during metabolic inhibition (Retamal et al., 2007b). Here, we found that hemichannel-mediated Etd uptake induced by 3 h of hypoxia in high glucose and 1 h of reoxygenation was insensitive to DTT, but after 6 h of hypoxia and 1 h reoxygenation, DTT reduced Etd uptake. A possible explanation for these different DDT sensitivities of hemichannel-mediated Etd uptake is that 3 h of hypoxia followed by reoxygenation do not induce a drastic unbalance between levels of free radical scavengers (i.e., reduced glutathione) and generation of NO, but after 6 h hypoxia and reoxygenation there is depletion of free radical scavengers and persistent generation of NO.
The irreversible increase in hemichannel activity induced by 6 h hypoxia in high glucose might be expected to increase astroglial death, and hemichannel blockers delay membrane breakdown following irreversible metabolic inhibition (Contreras et al., 2004). We examined the incorporation of Rdex, indicative of loss of membrane integrity, at different periods of reoxygenation following 3 or 6 h of hypoxia in high glucose. By this measure we observed cell death that was greater after longer periods of hypoxia at higher glucose concentrations.
Although these results were obtained from cultures of cortical glial cells, they provide a new insight into the role of astroglial connexins in neuronal survival. Loss of neuronal protection by astrocytes could result from dysregulation of their gap junctions and hemichannels. A reduction in gap junctional communication impairs the spatial buffering capacity of astroglial networks, and an increase in hemichannel activity could in turn affect the viability of other cells through a paracrine mechanism. In agreement, ischemic saline in high glucose enhances the release of ATP in retinal cell cultures (Costa et al., 2008). Thus, excessive release or deficient uptake of signaling molecules such as ATP and other hemichannel permeant molecules, e.g., glutamate, arachidonic acid byproducts, could enhance neuronal excitotoxicity. Cx43 gap junction channels and hemichannels are regulated oppositely by hypoxia in high glucose and reperfusion, and both kinds of modulation may be relevant in related pathologies, such as diabetes and stroke.
Supplementary Material
Figure 1S. Quantification of Cx43 immunoreactivity reduction induced by hypoxia-reoxygenation. Quantification of the frequency of Cx43 puncta with small (A, 0 to 4 μm diameter) and large (B, 3 to 6 μm) diameter in astrocytes under control conditions (left panels) or at 1 h reoxygenation after 6 h hypoxia in 5 (middle panels) or 27 mM glucose (right panels).
Acknowledgments
This work was partially supported by CONICYT (24080055 to JAO), FONDECYT (1070591 to JCS) and NIH (NS37402 and NS45287 to MVLB) and INSERM/CONICYT (CG and JCS) grants.
References
- Anderson KM, Tsui P, Guinan P, Rubenstein M. The proliferative response of hela cells to 2-deoxy-D-glucose under hypoxic or anoxic conditions: an analogue for studying some properties of in vivo solid cancers. Anticancer Res. 2006;26:4155–4162. [PubMed] [Google Scholar]
- Benjelloun N, Joly LM, Palmier B, Plotkine M, Charriaut-Marlangue C. Apoptotic mitochondrial pathway in neurones and astrocytes after neonatal hypoxia-ischaemia in the rat brain. Neuropathol Appl Neurobiol. 2003;29:350–360. doi: 10.1046/j.1365-2990.2003.00467.x. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001;34:134–142. doi: 10.1002/glia.1048. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Sáez JC, Bukauskas FF, Bennett MV. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad Sci U S A. 2003;100:11388–11393. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa G, Pereira T, Neto AM, Cristovao AJ, Ambrosio AF, Santos PF. High glucose changes extracellular adenosine triphosphate levels in rat retinal cultures. J Neurosci Res. 2008;87:1375–1380. doi: 10.1002/jnr.21956. [DOI] [PubMed] [Google Scholar]
- Cronberg T, Rytter A, Asztely F, Soder A, Wieloch T. Glucose but not lactate in combination with acidosis aggravates ischemic neuronal death in vitro. Stroke. 2004;35:753–757. doi: 10.1161/01.STR.0000117576.09512.32. [DOI] [PubMed] [Google Scholar]
- Cruciani V, Mikalsen SO. The connexin gene family in mammals. Biol Chem. 2005;386:325–332. doi: 10.1515/BC.2005.039. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Evans WH, Leybaert L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun Adhes. 2007;14:265–273. doi: 10.1080/15419060801891034. [DOI] [PubMed] [Google Scholar]
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A. Glia: the fulcrum of brain diseases. Cell Death Differ. 2007;14:1324–1335. doi: 10.1038/sj.cdd.4402144. [DOI] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Huang Y, Grinspan JB, Abrams CK, Scherer SS. Pannexin1 is expressed by neurons and glia but does not form functional gap junctions. Glia. 2007;55:46–56. doi: 10.1002/glia.20435. [DOI] [PubMed] [Google Scholar]
- Kagansky N, Levy S, Knobler H. The role of hyperglycemia in acute stroke. Arch Neurol. 2001;58:1209–1212. doi: 10.1001/archneur.58.8.1209. [DOI] [PubMed] [Google Scholar]
- Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol. 2000;32:1859–1872. doi: 10.1006/jmcc.2000.1220. [DOI] [PubMed] [Google Scholar]
- Lin B, Busto R, Globus MY, Martinez E, Ginsberg MD. Brain temperature modulations during global ischemia fail to influence extracellular lactate levels in rats. Stroke. 1995;26:1634–1638. doi: 10.1161/01.str.26.9.1634. [DOI] [PubMed] [Google Scholar]
- Martínez AD, Sáez JC. Regulation of astrocyte gap junctions by hypoxia-reoxygenation. Brain Res Brain Res Rev. 2000;32:250–258. doi: 10.1016/s0165-0173(99)00086-7. [DOI] [PubMed] [Google Scholar]
- Mu D, Zhang W, Chu D, Liu T, Xie Y, Fu E, Jin F. The role of calcium, P38 MAPK in dihydroartemisinin-induced apoptosis of lung cancer PC-14 cells. Cancer Chemother Pharmacol. 2008;61:639–645. doi: 10.1007/s00280-007-0517-5. [DOI] [PubMed] [Google Scholar]
- Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes. 2006;55:349–355. doi: 10.2337/diabetes.55.02.06.db05-0654. [DOI] [PubMed] [Google Scholar]
- Myers RE, Yamaguchi S. Nervous system effects of cardiac arrest in monkeys. Preservation of vision. Arch Neurol. 1977;34:65–74. doi: 10.1001/archneur.1977.00500140019003. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Sáez PJ, Shoji KF, Schalper KA, Palacios-Prado N, Velarde V, Giaume C, Bennett MV, Sáez JC. Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid Redox Signal. 2009;11:369–399. doi: 10.1089/ars.2008.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peuchen S, Duchen MR, Clark JB. Energy metabolism of adult astrocytes in vitro. Neuroscience. 1996;71:855–870. doi: 10.1016/0306-4522(95)00480-7. [DOI] [PubMed] [Google Scholar]
- Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle AK, Iannotti F, Wilde GJ, Chad JE, Seeley PJ, Sundstrom LE. Neuroprotection by both NMDA and non-NMDA receptor antagonists in in vitro ischemia. Brain Res. 1997;755:36–46. doi: 10.1016/s0006-8993(97)00089-9. [DOI] [PubMed] [Google Scholar]
- Retamal MA, Cortés CJ, Reuss L, Bennett MV, Sáez JC. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc Natl Acad Sci U S A. 2006;103:4475–4480. doi: 10.1073/pnas.0511118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamal MA, Froger N, Palacios-Prado N, Ezan P, Sáez PJ, Sáez JC, Giaume C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007a;27:13781–1392. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamal MA, Schalper KA, Shoji KF, Bennett MV, Sáez JC. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc Natl Acad Sci U S A. 2007b;104:8322–8327. doi: 10.1073/pnas.0702456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytter A, Cronberg T, Asztely F, Nemali S, Wieloch T. Mouse hippocampal organotypic tissue cultures exposed to in vitro “ischemia” show selective and delayed CA1 damage that is aggravated by glucose. J Cereb Blood Flow Metab. 2003;23:23–33. doi: 10.1097/01.WCB.0000034361.37277.1B. [DOI] [PubMed] [Google Scholar]
- Sáez JC, Berthoud VM, Brañes MC, Martínez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Sáez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711:215–224. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalper KA, Palacios-Prado N, Retamal MA, Shoji KF, Martínez AD, Sáez JC. Connexin hemichannel composition determines the FGF-1-induced membrane permeability and free [Ca2+]i responses. Mol Biol Cell. 2008;19:3501–3513. doi: 10.1091/mbc.E07-12-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Deas J, Erecinska M. Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience. 1997;78:589–601. doi: 10.1016/s0306-4522(96)00600-8. [DOI] [PubMed] [Google Scholar]
- Silverman W, Locovei S, Dahl GP. Probenecid, a Gout Remedy, Inhibits Pannexin 1 Channels. Am J Physiol Cell Physiol. 2008;295:C761–C767. doi: 10.1152/ajpcell.00227.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877–896. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Lewen A, Noshita N, Gasche Y, Chan PH. Effects of global ischemia duration on neuronal, astroglial, oligodendroglial, and microglial reactions in the vulnerable hippocampal CA1 subregion in rats. J Neurotrauma. 2002;19:85–98. doi: 10.1089/089771502753460268. [DOI] [PubMed] [Google Scholar]
- Tabernero A, Medina JM, Giaume C. Glucose metabolism and proliferation in glia: role of astrocytic gap junctions. J Neurochem. 2006;99:1049–1061. doi: 10.1111/j.1471-4159.2006.04088.x. [DOI] [PubMed] [Google Scholar]
- Theis M, Mas C, Doring B, Kruger O, Herrera P, Meda P, Willecke K. General and conditional replacement of connexin43-coding DNA by a lacZ reporter gene for cell-autonomous analysis of expression. Cell Commun Adhes. 2001;8:383–386. doi: 10.3109/15419060109080758. [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Musil LS. Cytosolic stress reduces degradation of connexin43 internalized from the cell surface and enhances gap junction formation and function. Mol Biol Cell. 2005;16:5247–5257. doi: 10.1091/mbc.E05-05-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton M, Connor B, Lawlor P, Young D, Sirimanne E, Gluckman P, Cole G, Dragunow M. Neuronal death and survival in two models of hypoxic-ischemic brain damage. Brain Res Brain Res Rev. 1999;29:137–168. doi: 10.1016/s0165-0173(98)00053-8. [DOI] [PubMed] [Google Scholar]
- Wang J, Ma M, Locovei S, Keane RW, Dahl G. Modulation of membrane channel currents by gap junction protein mimetic peptides: size matters. Am J Physiol Cell Physiol. 2007;293:C1112–1119. doi: 10.1152/ajpcell.00097.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S. Quantification of Cx43 immunoreactivity reduction induced by hypoxia-reoxygenation. Quantification of the frequency of Cx43 puncta with small (A, 0 to 4 μm diameter) and large (B, 3 to 6 μm) diameter in astrocytes under control conditions (left panels) or at 1 h reoxygenation after 6 h hypoxia in 5 (middle panels) or 27 mM glucose (right panels).