

Abstract
Human cytochrome P450 2D6 (CYP2D6) is responsible for the metabolism of approximately 20% of drugs in common clinical use. The CYP2D6 gene locus is highly polymorphic. Many of the polymorphisms have been shown to be clinically relevant and can account for inter-individual differences in the metabolism of specific drugs. In addition to the established sources of variability in CYP2D6-dependent drug metabolism, a recent study in our laboratory identified CYP2D6 in the mitochondria of human liver samples and found that it is metabolically active in this novel location. In the present study we show that mutations are present in the targeting signal region of CYP2D6 that may help to account for the inter-individual variability that was observed previously in the level of the mitochondrial enzyme in human liver samples. These mutations were identified within the ER targeting domain, the proline-rich domain as well as the putative protein kinase A (PKA) and protein kinase C (PKC)-specific phosphorylation sites. In vitro studies demonstrate that the mutations identified in the targeting signals affect the efficiency of mitochondrial targeting of CYP2D6. Since the mitochondrial enzyme has been shown to be active in drug metabolism, this pharmacogenetic variation could play a role in modulating the response of an individual to drug therapy.
Keywords: human mitochondrial CYP2D6, genetic variants, mitochondrial targeting signal, inter-individual variations
Cytochrome P450 2D6 (CYP2D6) has been shown to be involved in the metabolism of approximately 20% of drugs in common clinical use [1]. These drugs belong to a wide spectrum of therapeutic classes including analgesics, antidepressants, antiarrhythmics, antipsychotics, antihypertensives, and β-adrenergic receptor antagonists [2]. The CYP2D6 gene locus is highly polymorphic and more than 112 allelic variants have been described to date (http://www.imm.ki.se/CYPalleles/cyp2d6.htm). The human population can be classified into four general phenotypes of CYP2D6 drug-metabolizing activity according to the number of functional CYP2D6 alleles that are present. Extensive metabolizers (EMs) have two functional CYP2D6 alleles, intermediate metabolizers (IMs) have one functional allele, poor metabolizers (PMs) have no functional alleles, and ultrarapid metabolizers (UMs) have three or more functional alleles that arise by gene duplication [3–5].
Many of the polymorphisms identified in the CYP2D6 gene locus have been shown to be clinically relevant and can account for inter-individual differences in the metabolism of specific drugs [6]. The genetic polymorphism of CYP2D6 has been implicated as a likely reason for adverse drug effects and poor treatment outcome in antidepressant, cardiovascular, and cancer therapy treatment regimes [7–10]. In general these studies found that PMs were at increased risk of adverse drug effects [7] or therapeutic failure due to decreased production of active metabolites [8,9]. UMs, on the other hand, were generally at increased risk of decreased efficacy of drugs due to rapid metabolism and inactivation [7,10]. Although the use of pharmacogenetic approaches have not yet progressed to the point where they can be used to design individualized treatment strategies, there are positive indications that this would be beneficial in the future treatment of various disease conditions [6,11].
In addition to the established sources of variability in CYP2D6-dependent drug metabolism, a recent study in our laboratory identified CYP2D6 in the mitochondria of human liver samples and found that it is metabolically active in this novel location [12]. We found that there was significant variability between individuals as to the level of the mitochondrial enzyme as well as its metabolic activity [12]. Earlier studies in our laboratory demonstrated that several other cytochrome P450 enzymes (CYPs), namely CYP1A1, 2B1, and 2E1, are bimodally targeted to mitochondria and microsomes following induction with xenobiotics [13–17]. These studies helped to establish the existence of “chimeric signals,” composed of a mitochondrial targeting signal localized immediately adjacent to the endoplasmic reticulum (ER) targeting domain at the N-terminus of these cytochrome P450 proteins. The mitochondrial-localized CYPs are capable of efficiently interacting with mitochondrial adrenodoxin (Adx) and adrenodoxin reductase (AdxR) to catalyze drug metabolism [14,18,19]. P450 MT2, the mitochondrial-targeted form of CYP1A1, was shown to exhibit altered substrate specificity as compared to the microsomal enzyme [14,20].
In the present study, we have identified polymorphisms in the targeting signal regions of CYP2D6 that could potentially contribute to the large amount of inter-individual variability that was observed previously in the level of the mitochondrial enzyme [12]. Screening of a human liver bank using a reverse genetics approach enabled us to identify several variant forms with mutations within the ER targeting domain, the Proline-rich domain as well as the protein kinase A (PKA) and protein kinase C (PKC)-specific phosphorylation sites. These domains have been shown to be important in the mitochondrial targeting of CYP2D6 and the other CYPs that have been studied in this laboratory [12–17]. In vitro studies demonstrate that the mutations identified in the targeting signal region of CYP2D6 affect the efficiency of mitochondrial targeting. Since the mitochondrial enzyme has been shown to be active in drug metabolism, this pharmacogenetic variation could play a role in modulating the response of an individual to drug therapy.
Materials and Methods
Isolation of Total RNA and Characterization of Human CYP2D6 Variants
Total RNA was isolated from human livers using TRIzol reagent as per the supplier’s protocol (Invitrogen, Carlsbad, CA). Reverse transcription was performed with 20 µg total RNA and an antisense primer (5’-TCGAATTCCTGAGGAAGCGAGGGTCGTCGTACTCG-3’) corresponding to nucleotides 585–610 of the cDNA sequence. PCR was performed to amplify the 5’-terminal 600 nucleotides of CYP2D6 from each liver sample. The constructs were engineered to contain 5’ HindIII and 3’ EcoRI sites and cloned into a PCR TOPO II vector (Invitrogen). The nucleotide sequences of WT and mutant cDNA constructs were confirmed by sequencing.
PCR amplification and analysis of human liver genomic DNA
Genomic DNA was isolated from human liver samples as described by Strauss [21]. Briefly, 1 g liver tissue was minced, frozen in liquid nitrogen, and ground to a fine powder using a mortar and pestle. The powdered tissue was suspended in 12 ml digestion buffer (10 mM Tris-Cl (pH 8.0) containing 100 mM NaCl, 25 mM EDTA, 0.5% SDS, and 0.1 mg/ml proteinase K) and incubated with shaking for 20 h. The sample was extracted twice with phenol/chloroform/isoamyl alcohol (25:24:1, v/v) and one time with chloroform/isoamyl alcohol (24:1, v/v). The DNA was then precipitated using ½ volume of 7.5 M ammonium acetate and 2 volumes of 100% ethanol. The pellet was rinsed with 70% ethanol and then resuspended in 10 mM Tris-Cl (pH 7.5) buffer containing 0.1 mM EDTA at a concentration of 1 mg/ml. DNase-free RNase was added to the sample at a concentration of 1 µg/ml and incubated at 37°C for 1 h. The DNA was then re-extracted and precipitated as described above. PCR amplification was performed using a forward primer (5’-AGAAGGGCACAAAGCGGGAACTGG-3’) corresponding to nucleotides 1873–1895 of intron 3, and a reverse primer (5’-TGCAGAGACTCCTCGGTCTCTC-3’) corresponding to nucleotides 2118–2139 of intron 4. The amplicon was cloned into PCR TOPO II vector (Invitrogen) and analyzed by nucleotide sequencing.
Real-Time PCR for detection of Full-length and Exon 3 Skipped Splice Variants
Total RNA was isolated from human livers using TRIzol reagent as per the supplier’s protocol (Invitrogen). cDNA was generated from 5 µg RNA using the cDNA Archive kit (Applied Biosystems, Foster City, CA), and 100 ng cDNA was used as a template in each reaction. Quantification of the full length and splice variant forms of CYP2D6 in relation to endogenous control was performed by real-time PCR using both Taqman probes (Applied Biosystems) and SYBR Green dye (Applied Biosystems) in an ABI Prism 7300 sequence detection system (Applied Biosystems). A TaqMan gene expression assay that was designed to detect just the exon 3-skipped splice variant form (Hs02576167_m1, Applied Biosystems) was used. The data were normalized to 18S rRNA content using a Taqman assay (Applied Biosystems).
For SYBR Green amplification, a single primer set was designed that would amplify both the exon 3-intact transcripts and exon 3-skipped splice variant forms of CYP2D6. Using the forward primer, 5’-ATCACCCAGATCCTGGGTTTC-3’, and reverse primer, 5’-ATCACGTTGCTCACGGCTTT-3’, exon 3-intact CYP2D6 was detected as a 242 bp amplicon while the exon 3-skipped splice variant form was detected as an 89 bp amplicon. In this case, human Actin was amplified for use as an endogenous control (Applied Biosystems). CYP2D6 amplification was monitored using the sequence detection system and terminated when all samples were found to be in the exponential phase of amplification (approximately cycle 31). Actin amplification was allowed to proceed until completion, when all samples were in the plateau phase (cycle 40), and then used for normalization of cDNA content between the samples. CYP2D6 amplification products were resolved on an 8% polyacrylamide TBE (Tris/borate/EDTA) gel and the gel was stained with ethidium bromide. Imaging analysis was performed using VersaDoc 3000 imaging system (Bio-Rad, Hercules, CA). Digital image analysis was performed using QUANTITY ONE, version 4.5.
Construction of CYP2D6-DHFR fusion constructs
CYP2D6 variant constructs identified in human liver samples were fused to dihydrofolate reductase (DHFR) by ligating the DNA sequence encoding the N-terminal 178 amino acids of the CYP2D6 variants in-frame with the mouse DHFR coding sequence through an EcoRI linker in the PGEM7zf vector (Promega, Madison, WI). cDNAs for the exon 3 skipped and exon 4 deletion variants were fused to DHFR by ligating the sequence encoding the N-terminal 127 and 175 amino acids, respectively, to the DHFR sequence in PGEM7zf vector. In each case, the forward primer used for amplification was 5’-GATAAGCTTACCGCAGGTATGGGGCTAGAAGCACTG-3’. The reverse primer used to amplify the full-length and exon 3-skipped variants was 5’-TAGAATTCCAAGAGACCGTTGGGGCGAAAG-3’. The reverse primer used to amplify the exon 4 deletion variant was 5’-TAGAATTCGGCGATCACGTTGCTCACGGC-3’.
In vitro Transport of Nascent Protein into Isolated Mitochondria
Sp6 or T7 polymerase-coupled rabbit reticulocyte lysate (RRL) transcription-translation systems (Promega) were used to translate cDNA constructs in pGEM7zF (Promega) and PCR TOPO II (Invitrogen) vectors in the presence of [35S]Met as described before [13]. In some cases, translation products were phosphorylated using a method described previously [22]. Translation products were pre-incubated with the catalytic subunit of protein kinase A (Invitrogen) 0.37 µg/50 µl reaction and 100 µM ATP for 30 min at 30°C.
Mitochondria were freshly isolated from rat liver using a previously described method [13] with some modifications. The rat was euthanized by CO2 asphyxiation and the liver was immediately perfused with ice cold saline. Liver was homogenized in 10 volumes (w/vol) of sucrose-mannitol buffer containing BSA (20 mM Hepes, pH 7.5, containing 70 mM sucrose, 220 mM mannitol, 2 mM EDTA, and 0.5 mg/ml BSA). The homogenate was centrifuged twice at 600 g for 10 min to pellet nuclei and cell debris. The supernatant was then centrifuged at 8,000 g for 15 min to pellet mitochondria. The pellet was washed three times in sucrose-mannitol buffer without BSA (20 mM Hepes, pH 7.5, containing 70 mM sucrose, 220 mM mannitol, 2 mM EDTA) and the final pellet was suspended in the same buffer. Protein estimation was carried out using the method of Lowry et al [23]. Cytochrome c oxidase activity was assessed and the mitochondria were found to contain nearly 80% of total tissue cytochrome c oxidase activity. The mitochondrial preparation was also found to have less than 0.5% of microsome-specific NADPH cytochrome P450 reductase activity.
Import of 35S-labeled translation products into the freshly isolated mitochondria was carried out using the system described by Gasser et al. [24] and modified by Bhat et al. [25] and Addya et al. [13]. Trypsin digestion (150 µg trypsin/mg mitochondrial protein) of mitochondria was performed for 20 min on ice. Control mitochondria were incubated similarly without added trypsin. Soybean trypsin inhibitor (1.5 mg/mg protein) was added to all samples to terminate the reactions. Mitochondria from both trypsin-treated and untreated samples were re-isolated by pelleting through 0.8M sucrose, and the proteins were subjected to SDS-PAGE followed by fluorography.
Results
Screening for Human CYP2D6 Variant Forms
A genetic screen was performed to investigate whether there are any polymorphisms in the targeting signals of CYP2D6 that could account for the large amount of inter-individual variability that was observed previously in the level of the enzyme associated with the mitochondria [12]. Our previous studies with other members of CYP family 2, namely CYP2B1 and CYP2E1, showed that the N-terminal 160 amino acid region of the protein was capable of directing the protein bimodally to both ER and mitochondria [15,17,19]. In addition, our recent study with CYP2D6 showed that the mitochondrial targeting signal is localized between residues 23–33 [12]. Therefore, we focused our screening strategy on the region encoding the N-terminal 200 amino acids of CYP2D6.
Total RNA isolated from human liver tissues was used to amplify the first 200 amino acid coding region by RT-PCR. The amplified DNA was cloned and sequenced in order to identify any mutations present in the putative targeting domains. Of the 20 liver samples screened, five samples showed varied point mutations or splicing defects in the CYP2D6 sequence and these samples were selected for further analysis. The full complement of variant CYP2D6 sequences identified in these samples is listed in Table 1. Schematics of the CYP2D6 constructs with point mutations and representations of the identified splice variants are also shown (Figs. 1A, 1B). A variant of CYP2D6 that has three amino acid substitutions, P34S, L91M, and H94R, was identified in both HL123 and 141 (Fig. 1A, Table 1). In HL123 these mutations were identified in the full-length sequence, while in HL141 they were present alongside a splicing defect in which the first 12 amino acids of exon 4 were deleted (Fig. 1B, Table 1). In the initial genetic screening by cDNA sequence analysis, we did not detect the exon 3-skipped splice variant mRNAs in these liver samples; however, RT-PCR (data not shown) and real-time PCR analysis (Fig. 3A, B) performed later, both confirmed the presence of the exon 3-skipped variant in these liver samples. Analysis of HL127 resulted in the detection of two different CYP2D6 variants: a full-length wild-type sequence as well as an exon 3-skipped splice variant in which there is a substitution of S70G. Likewise, in HL130, two different variants were identified: a full-length wild-type sequence and an exon 3-skipped splice variant with a substitution of I12M in the ER targeting domain. HL136 had a substitution of D97N present alongside the exon 3-skipped splicing defect.
Table 1.
Summary of point mutations and splice variants identified in the 5’ 600 bp region of CYP2D6 cDNAs isolated from human liver samples.
| Human Liver Sample |
Number of Constructs Identified |
Nucleotide Change |
Effect | Allele Nomenclature |
|---|---|---|---|---|
| 123 | 1 | 100C>T; 974C>A; 984A>G |
P34S; L91M; H94R |
CYP2D6*4a |
| 127 | 2 | 1. 911A>G | 1. S70G, exon 3 skipped |
|
| 2. None | 2. Wild-type | |||
| 130 | 2 | 1. 36A>G | 1. I12M, exon 3 skipped |
|
| 2. None | 2. Wild-type | |||
| 136 | 1 | 992G>A | D97N, exon 3 skipped |
|
| 141 | 1 | 100C>T; 974C>A; 984A>G; 1846G>A |
P34S. L91M, H94R, exon 4 deletion (first 12 amino acids) |
CYP2D6*4A, B, F, G, H, Jb |
This construct has point mutations characteristic of the *4 group of alleles, but lacks the splicing defect that is seen in all other members of this group.
The complement of point mutations identified in this construct and the presence of a splicing defect is similar to that found in a variety of *4 group members. Sequencing of the full-length cDNA would be required to determine which subgroup this construct belongs to.
Fig. 1.

Identification of genetic variants of CYP2D6 with mutations in putative targeting signals. (A and B) Total RNA was extracted from human liver samples and the first 600 nucleotides of the coding region were amplified by RT-PCR. The amplified DNA was cloned and sequenced. (A) Schematic representations of point mutations identified in the human liver samples. (B) Schematic representations of splice variants identified. The deleted or skipped exon regions are shown as hatched boxes.
Fig. 3.

Real-time PCR analysis of CYP2D6 full-length and exon 3-skipped splice variant in human liver samples. (A) Relative quantification of the exon 3-skipped splice variant form of CYP2D6 mRNA by realtime PCR using TaqMan gene expression assay kit (Hs02576167_m1, Applied Biosystems). Data represent the mean ± SEM for three separate estimates. (B) Detection of both the full length and exon 3-skipped splice variant forms of CYP2D6 mRNAs using SYBR Green dye chemistry (Applied Biosystems). Exon 3-intact CYP2D6 was detected as a 242 bp amplicon while the exon 3-skipped splice variant form was detected as an 89 bp amplicon. CYP2D6 amplification products were resolved on an 8% polyacrylamide TBE gel and the gel was stained with ethidium bromide. Human Actin was amplified for use as an endogenous control (Applied Biosystems). Densitometric analysis was performed and the level of exon 3-skipped splice variant (89 bp amplicon) was calculated as a percentage of total transcripts detected (242 bp amplicon + 89 bp amplicon).
The Genetic Basis for Splice Variant Identified in HL141
The mutations identified in sample HL141 are very similar to those designated as CYP2D6*4 in the CYP2D6 allele nomenclature database. The G1846A mutation in the intron 3 splice acceptor site (present in this group of alleles) has been implicated as the cause of a splicing defect; however, the precise nature of this splicing defect remains unknown. Therefore, in order to understand the molecular basis of the 12 amino acid exon 4 deletion identified in HL141, we amplified the genomic DNA in the region of the intron 3 splice acceptor site and sequenced it. PCR amplification of the 266 bp fragment and comparison of the nucleotide sequence with the WT sequence (GenBank accession number M33388) (Figs. 2A, 2B), revealed the mutation G1846A in the intron 3 splice acceptor site.
Fig. 2.

Determination of genetic basis for splice variant identified in HL141. Genomic DNA from nucleotide 1873 (intron 3) to nucleotide 2139 (intron 4) of sample HL141 was amplified by PCR, cloned into PCR TOPO II vector and sequenced. (A) Agarose gel picture showing the 266 bp amplified product. Lane 1, 100 bp DNA ladder (New England Biolabs, Ipswich, MA). Lanes 2–4, 266 bp product amplified from genomic DNA of sample HL141. Lane 5, PCR negative control with no template added. (B) Alignment of HL141 amplified DNA sequence with CYP2D6 WT sequence, GenBank accession number M33388.
Identification of Exon 3 Skipped Splice Variant Using Real-Time PCR
Real-time PCR was used to investigate the relative abundance of the exon 3-skipped splice variant in the five human liver samples that were selected for further analysis following the initial genetic screen. A TaqMan gene expression assay was utilized to detect the splice variant form of CYP2D6. This assay was designed to amplify the boundary between exons 2 and 4 of CYP2D6, which is present only in the exon 3-skipped splice variant (RefSeq NM_001025161.1). In this case, HL130 was found to have the highest level of CYP2D6 splice variant mRNA, followed by HL127 and then HL123 (Fig. 3A). HL141 has an intermediate level of splice variant mRNA while HL136 has the lowest level among the human liver samples tested (Fig. 3A).
An alternate method of amplification utilizing SYBR Green dye chemistry was used to confirm these findings and to extend the results to investigate the abundance of exon 3-skipped splice variant relative to exon 3-intact transcripts in each of the liver samples. A set of primers was designed to amplify both of the transcripts. The exon 3-intact form amplifies at 242 bp, while the exon 3-skipped variant amplifies at 89 bp. The amplification of CYP2D6 was monitored and terminated during the exponential phase. Actin was amplified as an endogenous control and used to normalize cDNA content between the samples. The products were resolved on a polyacrylamide TBE gel to visualize the relative levels of exon 3-intact and exon 3-skipped splice variant in each liver sample (Fig. 3B). The relative abundance of splice variant detected between the different liver samples using this method of amplification (Fig 3B) was very similar to the TaqMan amplification results (Fig. 3A).
When comparing the abundance of splice variant relative to exon 3-intact transcripts in the different liver samples, it is clear that HL130 has the highest level of both exon 3-intact and exon 3-skipped CYP2D6 mRNA. In this sample, the splice variant represents approximately 43% of the total transcript pool (Fig. 3B). Interestingly, HL127 has the second highest level of exon 3-skipped splice variant among all the liver samples, but it has lowest level of exon 3-intact transcripts. In this liver, the splice variant transcript was found to represent 61% of the total CYP2D6 transcripts detected (Fig. 3B). In HL123, HL136, and HL141, the exon 3-intact CYP2D6 transcript was significantly more abundant than the splice variant. HL123 and HL141 have similar levels of exon 3-intact transcript, but HL123 has a higher level of splice variant as compared to HL141. In HL123, 34% transcripts are exon 3-skipped splice variant, whereas, in HL141, 30% of the transcripts are splice variants (Fig. 3B). HL136 has the second lowest level of exon 3-intact transcripts and the lowest level of splice variant transcripts among all the liver samples. In this liver, the splice variant represents 39% of the total CYP2D6 transcripts detected (Fig. 3B).
Effect of Mutations on Mitochondrial Targeting of CYP2D6 in vitro
In order to determine whether the mutations in the targeting signals affect the mitochondrial targeting of CYP2D6, the N-terminal region of the wild-type and variant constructs was fused to dihydrofolate reductase (DHFR), a cytosolic protein, and used for in vitro import into isolated rat liver mitochondria. This experiment tests the strength of the targeting signals in the CYP2D6 variant constructs by evaluating their ability to transport a cytosolic reporter protein, DHFR, into the mitochondria. The fusion constructs consist of the N-terminus of the CYP2D6 variants (Table 1) fused to DHFR through an EcoRI linker region. For the constructs that have no splicing defect present, 178 amino acids have been fused to DHFR (Fig. 4A). However, in the case of the exon 3 skipped variants 127 amino acids were fused to DHFR, because 51 amino acids comprising exon 3 are missing in this variant (Fig. 4A). For the exon 4 deletion variant 175 amino acids were fused to DHFR (Fig. 4A). In this case, 12 amino acids have been deleted from exon 4 but a larger fragment was amplified for this variant so the overall number of amino acids is similar to the wild-type full-length constructs. When these fusion proteins were translated in vitro there was a clear difference in the mobility of the exon 3 skipped variant which has an apparent molecular mass of about 33 kDa (Fig. 4B). The full length constructs, which have no splicing defect, have an apparent molecular mass of 40 kDa (Fig. 4B). The construct in which 12 amino acids have been deleted from exon 4 shows no difference in mobility as compared to the full-length constructs (Fig. 4B).
Fig. 4.


Effect of CYP2D6 genetic variation on mitochondrial targeting in vitro. (A) The N-termini of CYP2D6 variants (Table 1) were fused to DHFR through an EcoRI linker region. The full-length constructs were generated by fusing the N-terminal 178 amino acids of CYP2D6 to DHFR; the exon 3 skipped variants have 127 amino acids fused to DHFR; the exon 4 deletion variant is composed of 175 amino acids of CYP2D6 fused to DHFR. (B) Size comparison of each translation product used in the import experiment. 35S-labeled translation products were resolved by SDS-PAGE. (C–G) The 35S-labeled fusion proteins were used for in vitro import into isolated rat liver mitochondria. Trypsin digestion (150 µg/ml) of mitochondria was performed for 20 min on ice. Proteins (200 µg each) were subjected to SDS-PAGE and fluorography. “C” represents control experiments in which total protein bound and imported into mitochondria is present, “T” represents trypsin treated mitochondria in which only the protein imported into mitochondria is present. PKA refers to experiments in which the translation products were pre-incubated with the catalytic subunit of protein kinase A, 0.37 µg/50 µl reaction, prior to import. In the lanes marked “In,” 20% of the counts used as input for the import reactions have been loaded. Densitometric measurements were performed in order to determine the extent of import for each construct following trypsin treatment. This has been labeled as % import. The values were determined by calculating the level of each fusion protein in the mitochondria as a percentage of input. (C) Import of WT full-length CYP2D6-DHFR fusion constructs from HL 127 and HL130 are compared. (D) Import of exon 3-skipped splice variant constructs with varied point mutations (as labeled) from HL127, HL130, and HL136 are compared. (E) Import of constructs with P34S; L91M; H94R point mutations from HL123 and HL141 are compared. In HL123 the point mutations are present in the full-length construct, whereas in HL141 they are present alongside the exon 4 splicing defect in which 12 amino acids of exon 4 have been deleted. (F) Import of WT CYP2D6. (G) Import of positive control, Su9-DHFR, in which the presequence of subunit 9 of N. crassa F0F1 ATPase has been fused to DHFR.
The 35S-labeled fusion proteins were incubated with freshly isolated rat liver mitochondria to allow import to occur; resistance to limited protease digestion was used as a criterion for assessing protein import. Full-length (178 amino acid) wild-type sequences of both HL127 and 130 fused to DHFR exhibited a low level of mitochondrial import. PKA-mediated phosphorylation increased import approximately three-fold for both constructs (Fig. 4B). The exon 3-skipped splice variants (127 amino acids) isolated from the same liver samples with either a point mutation of S70G, corresponding to HL127, or an ER targeting domain mutation of I12M, corresponding to HL130, had a similar level of import as the full-length constructs in the absence of phosphorylation and the level of import increased approximately two-fold following PKA-mediated phosphorylation (Fig. 4C). In contrast, the exon 3-skipped splice variant isolated from HL136, which expressed a point mutation of D97N had a very low level of import both in the presence and absence of phosphorylation (Fig. 4C). The full-length sequence containing three point mutations —P34S, L91M, and H94R— isolated from HL123 had a low level of import under basal conditions, but this import increased four-fold in the presence of PKA-mediated phosphorylation (Fig. 4D). The extent of mitochondrial import for this construct was very similar to that of the full-length wild-type constructs (Fig. 4B). The construct corresponding to HL141 (159 amino acids), which has the same three point mutations as sample HL123 in addition to a splicing defect in which the first twelve amino acids of exon 4 have been deleted, showed a very low level of import in the presence and absence of phosphorylation (Fig. 4D).
WT CYP2D6 was used as a positive control for the in vitro import experiments. As shown previously [12], the WT protein targets to mitochondria at a moderate level in the absence of phosphorylation and this increases by approximately 21% in the presence of PKA-mediated phosphorylation (Fig. 3F). We also used Su9-DHFR as a positive control in these experiments. This construct is a fusion of two sequences: the presequence of subunit 9 of Neurospora crassa F0F1-ATPase, and dihydrofolate reductase. The presequence used here is a classic mitochondrial targeting signal that gets cleaved after entry into mitochondria. In this in vitro system, only the cleaved portion (27 kDa) remains after import into mitochondria and trypsin treatment (Fig. 3G).
Correlation between targeting of mutant constructs in vitro in isolated mitochondria and in vivo in human liver samples
The extent of mitochondrial targeting detected in vitro for the variant constructs correlates well with the mitochondrial CYP2D6 contents of the various liver samples from which the cDNA variants were identified (Figs 3C–E, Table 2). All of the constructs identified in HL127 and HL130 were found to target well to mitochondria in vitro following PKA-mediated phosphorylation (Figs. 3C,D). Immunoblot data also demonstrates that CYP2D6 targets strongly to mitochondria in these liver samples (Table 2). Likewise, the mutant construct identified in HL123 was found to target well to mitochondria following phosphorylation in vitro (Fig. 3E), and this liver sample was also found to have significant mitochondrial CYP2D6 in the western blot analysis (Table 2). On the other hand, the exon 3-skipped splice variant identified in HL136 was found to target poorly to mitochondria in the in vitro import experiments (Fig 3D), and CYP2D6 was found to be present in a relatively low level in the mitochondria in the liver sample (Table 2, [12]). The construct isolated from HL141 was also found to target poorly to mitochondria in vitro (Fig. 3E) and there was almost no CYP2D6 detected in mitochondria isolated from this liver sample (Table 2, [12]).
Table 2.
Distribution of CYP2D6 between mitochondria and microsomes isolated from human liver samplesc.
| Liver Sample | Mitochondria (% total CYP2D6) |
Microsomes (% total CYP2D6) |
|---|---|---|
| HL123 | 46.1 | 53.9 |
| HL127 | 47.3 | 52.7 |
| HL130 | 47.4 | 52.6 |
| HL136d | 20 | 80 |
| HL141d | 1.1 | 98.9 |
Measured by densitometric analysis of immunoblots performed on mitochondria and microsomes isolated from human liver samples.
Densitometric measurements for distribution of CYP2D6 between mitochondria and microsomes in HL136 and HL141 were published earlier in [12].
Discussion
The identification of five different natural CYP2D6 variants with mutations in the putative targeting domains of CYP2D6 provides a foundation for investigating the physiological relevance of mitochondrial CYP2D6 in human liver (Figs. 1A, 1B, Table 1). The variants with P34S, L91M, and H94R mutations (HL123 and 141) are of interest because the Pro-rich domain has been suggested to be important for activation of the cryptic mitochondrial targeting signal [15,17]. This variant has been documented previously and is designated CYP2D6*4 (http://www.imm.ki.se/CYPalleles/cyp2d6.htm). We identified two different variants with this complement of point mutations, one intact cDNA (HL123) and one splice variant (HL141). All of the alleles described under CYP2D6*4 in the CYP2D6 allele nomenclature database have a splicing defect alongside these point mutations; however, the exact nature of the splicing defect has not yet been described in the literature. We report here that the splicing defect in this group of alleles is the deletion of the first twelve amino acids of exon 4, which we identified in HL141. Numerous studies have identified the 1846G>A mutation at the 3’ end of intron 3 as the likely cause of the splicing defect and have predicted that this would cause a shift in the splice acceptor site by one nucleotide [26–29]. However, instead of the frameshift that would be expected from a shift in the splice acceptor site by one nucleotide, our results show that the splice acceptor site is shifted by 36 nucleotides resulting in the loss of 12 amino acids from exon 4. There is an AG dinucleotide sequence at the 3’end of the deleted region of this variant that could potentially serve as a splice acceptor site when the 1846G>A mutation is present and abolishes the consensus site (Fig. 5).
Fig. 5.
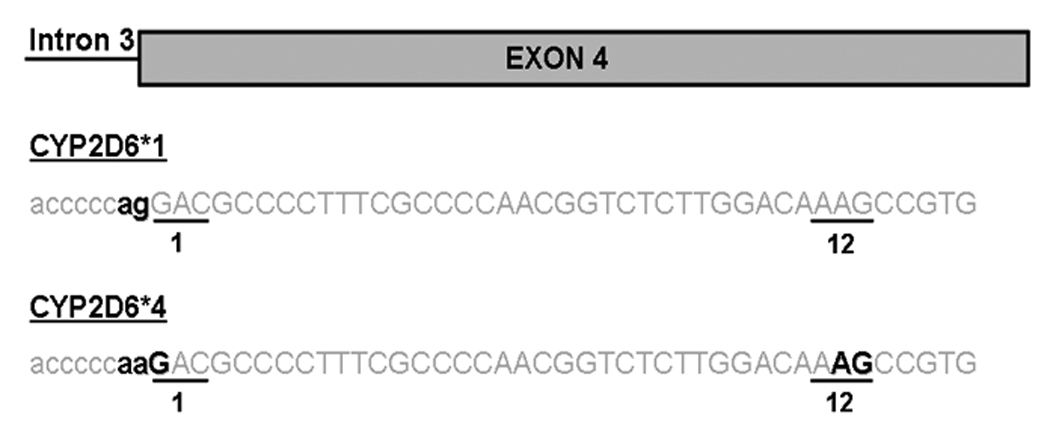
A schematic representation of the potential splicing mechanism for the 12 amino acid deletion in exon 4.
The exon 3-skipped splice variant is particularly interesting with regard to mitochondrial targeting because exon 3 encodes two PKA sites (S135 and S148) as well as three putative PKC sites (S137, T138, S168) as predicted by NETPHOSK 1.0 [30]. We showed earlier that PKA-mediated phosphorylation at S135 contributes to the mitochondrial targeting of CYP2D6 but found that it is probably not the only site involved [12]. The role of PKC-mediated phosphorylation in the mitochondrial targeting of CYP2D6 has not yet been investigated. The exon 3-skipped splice variant has been identified in human liver in several previous studies [31,32]; however, the molecular mechanism for its formation is unknown. An information theory-based analysis [29] predicts that a point mutation within exon 3, 1758G>T identified in CYP2D6*8 and 1758G>A identified in CYP2D6*14, could decrease the strength of the exon 3 donor splice site. According to the crystal structure of CYP2D6 [33], the deleted region in this variant form corresponds to helices C and D of CYP2D6 structure and includes residues important in formation of the active site cavity (Gly118, Val119, Phe120, Leu121, Ala122), catalytic activity (Phe120), as well as heme binding (Trp128, Arg132). Thus, the deletion would likely have a significant effect on the catalytic activity of the enzyme.
The exon 3-skipped splice variant was identified at the mRNA level in each human liver sample tested; however, the level of the splice variant varied significantly between the liver samples (Figs. 3A,B). The relative quantification of exon 3-skipped splice variant transcripts using the TaqMan assay was largely confirmed by the parallel analysis using SYBR green dye chemistry (Figs. 3A,B). Any variability is most likely due to a different efficiency of amplification between the different primer pairs used. The use of SYBR Green dye chemistry for amplification of CYP2D6 was advantageous in that it permitted visualization of the relative levels of full-length and exon 3-skipped variant within each human liver sample analyzed (Fig. 3B). As a result of this analysis, it became clear that in HL127 the exon 3-skipped splice variant is the predominant CYP2D6 transcript, representing 61% of the total transcripts detected. In all other liver samples tested, the transcripts with exon 3 intact are present at higher levels than the exon 3-skipped splice variant, with the splice variant ranging from 30% to 43% of the total CYP2D6 transcripts detected (Fig. 3B). Denson et al screened 96 human liver samples and found that the exon 3-skipped splice variant was present in very variable levels, ranging from 1%–94% of the total CYP2D6 transcripts in 90% of the samples [31]. The precise factors which regulate this variability remain unknown. Additionally, in our experience, the level of exon 3-skipped variants is most apparent in RT-PCR based analysis, as we have been unable to reliably detect this molecular change at the protein level by SDS-PAGE analysis.
In vitro mitochondrial targeting assays using DHFR fusion proteins suggested that known mutations and deletions within the N-terminal 200 amino acid region of CYP2D6 can affect the level of mitochondrial targeting (Fig. 4). The majority of mutant constructs identified in the genetic screen targeted to mitochondria at a similar level as the wild-type CYP2D6 DHFR fusion protein. All of these constructs demonstrated a low level of import into mitochondria in the absence of phosphorylation, and a significant increase in mitochondrial targeting following PKA-mediated phosphorylation (Figs. 3C–E). However, the presence of D97N point mutation in the exon 3-skipped splice variant (HL136) greatly decreased its responsiveness to PKA-mediated phosphorylation (Fig. 3D). Likewise, the construct that contains the three point mutations P34S, L91M, H94R in addition to the deletion of the first twelve amino acids of exon 4 (HL141) was also unresponsive to PKA-mediated phosphorylation. This could be the result of the major conformational change that we predict to be caused by the 12 amino acid deletion in exon 4. We previously reported that PKA-mediated phosphorylation enhances mitochondrial import of CYP2D6 [12]; however, the exact role of this phosphorylation in the mitochondrial targeting of CYP2D6 remains unclear. The correlation between the in vitro import results and the immunoblot analysis of mitochondria and microsomes isolated from the human liver samples suggests that the in vitro targeting may be partially predictive of the mitochondrial targeting in vivo. Overall, these results suggest that further investigation of CYP2D6 variant forms could prove enlightening both with regard to the mitochondrial targeting mechanism and their function.
The exact reason for the high level of inter-individual variability in the level of the mitochondrial enzyme remains unclear; however, given the significant number of polymorphic variants with mutations in the putative targeting domains, it is tempting to speculate that the presence of particular mutations and the possible involvement of other physiological factors, e.g. phosphorylation, may determine the level of mitochondrial CYP2D6. In view of the volume of data suggesting modulation of cAMP levels under various stress conditions, this is a likely cause of inter-individual variations in mitochondrial CYP2D6 levels. Since the mitochondrial enzyme has been shown to be active in drug metabolism, this pharmacogenetic variation could play a role in modulating individual responses to drug therapy.
Acknowledgements
We are thankful to Domenico Galati for help with cDNA cloning. This research was supported by NIH grants R01 GM34883 (NGA) and R37 CA090426 (FPG) and training grant T32 GM007170.
ABBREVIATIONS
- bp
base pair
- CYP
cytochrome P450 (EC 1.14.14.1)
- CPR
NADPH-cytochrome P450 reductase (EC 1.6.2.4)
- DHFR
dihydrofolate reductase (EC 1.5.1.3)
- ER
endoplasmic reticulum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.13)
- HL
human liver sample
- PKA
protein kinase A (EC 2.7.11.11)
- PKC
protein kinase C (EC 2.7.11.13)
- RRL
rabbit reticulocyte lysate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bertz RJ, Granneman GR. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet. 1997;32:210–258. doi: 10.2165/00003088-199732030-00004. [DOI] [PubMed] [Google Scholar]
- 2.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry, Naunyn. Schmiedebergs. Arch. Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 3.Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am. J. Hum. Genet. 1997;60:284–295. [PMC free article] [PubMed] [Google Scholar]
- 4.Zanger UM, Fischer J, Raimundo S, Stüven T, Evert BO, Schwab M, Eichelbaum M. Comprehensive analysis of the genetic factors determining expression and function of hepatic CYP2D6. Pharmacogenetics. 2001;11:573–585. doi: 10.1097/00008571-200110000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Griese EU, Zangar UM, Brudermanns U, Gaedigk A, Mikus G, Morike K, Stüven T, Eichelbaum M. Assessment of the predictive power of genotypes in the in-vivo catalytic function of CYP2D6 in a German population. Pharmacogenetics. 1998;8:15–26. doi: 10.1097/00008571-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 2008;392:1093–1108. doi: 10.1007/s00216-008-2291-6. [DOI] [PubMed] [Google Scholar]
- 7.Rau T, Wohlleben G, Wuttke H, Thuerauf N, Lunkenheimer J, Lanczik M, Eschenhagen T. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants – a pilot study. Clin. Pharmacol. Ther. 2004;75:386–393. doi: 10.1016/j.clpt.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 8.Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, Jin Y, Storniolo AM, Nikoloff DM, Wu L, Hillman G, Hayes DF, Stearns V, Flockhart DA. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther. 2006;80:61–74. doi: 10.1016/j.clpt.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Schroth W, Antoniadou L, Fritz P, Schwab M, Muerdter T, Zanger UM, Simon W, Eichelbaum M, Brauch H. Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J. Clin. Oncol. 2007;25:5187–5193. doi: 10.1200/JCO.2007.12.2705. [DOI] [PubMed] [Google Scholar]
- 10.Kirchheiner J, Heesch C, Bauer S, Meisel C, Seringer A, Goldammer M, Tzvetkov M, Meineke I, Roots I, Brockmöller J. Impact of the ultrarapid metabolizer genotype of cytochrome P40 2D6 on metoprolol pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2004;76:302–312. doi: 10.1016/j.clpt.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Tomalik-Scharte D, Lazar A, Fuhr U, Kirchheiner J. The clinical role of genetic polymorphisms in drug-metabolizing enzymes. Pharmacogenomics J. 2008;8:4–15. doi: 10.1038/sj.tpj.6500462. [DOI] [PubMed] [Google Scholar]
- 12.Cook Sangar M, Anandatheerthavarada HK, Tang W, Prabu SK, Martin MV, Dostalek M, Guengerich FP, Avadhani NG. Human liver mitochondrial CYP2D6: individual variations and implications in drug metabolism. FEBS J. 2009 doi: 10.1111/j.1742-4658.2009.07067.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Addya S, Anandatheerthavarada HK, Biswas G, Bhagwat SV, Mullick J, Avadhani NG. Targeting of NH2-terminal-processed microsomal protein to mitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2. J. Cell Biol. 1997;139:589–599. doi: 10.1083/jcb.139.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anandatheerthavarada HK, Addya S, Dwivedi RS, Biswas G, Mullick J, Avadhani NG. Localization of multiple forms of inducible cytochromes P450 in rat liver mitochondria: immunological characteristics and patterns of xenobiotic substrate metabolism. Arch. Biochem. Biophys. 1997;339:136–150. doi: 10.1006/abbi.1996.9855. [DOI] [PubMed] [Google Scholar]
- 15.Anandatheerthavarada HK, Biswas G, Mullick J, Sepuri NB, Otvos L, Pain D, Avadhani NG. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic AMP-dependent phosphorylation at ser128. EMBO J. 1999;18:5494–5504. doi: 10.1093/emboj/18.20.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhagwat SV, Biswas G, Anandatheerthavarada HK, Addya S, Pandak W, Avadhani NG. Dual targeting property of the N-terminal signal sequence of P450 1A1. Targeting of heterologous proteins to endoplasmic reticulum and mitochondria. J. Biol. Chem. 1999;274:24014–24022. doi: 10.1074/jbc.274.34.24014. [DOI] [PubMed] [Google Scholar]
- 17.Robin MA, Anandatheerthavarada HK, Biswas G, Sepuri NB, Gordon DM, Pain D, Avadhani NG. Bimodal targeting of microsomal CYP2E1 to mitochondria through activation of an N-terminal chimeric signal by cAMP-mediated phosphorylation. J. Biol. Chem. 2002;277:40583–40593. doi: 10.1074/jbc.M203292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anandatheerthavarada HK, Amuthan G, Biswas G, Robin MA, Murali R, Waterman MR, Avadhani NG. Evolutionarily divergent electron donor proteins interact with P450MT2 through the same helical domain but different contact points. EMBO J. 2001;20:2394–2403. doi: 10.1093/emboj/20.10.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robin MA, Anandatheerthavarada HK, Fang JK, Cudic M, Otvos L, Avadhani NG. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N-terminus and requires mitochondrial specific electron transfer proteins for activity. J. Biol. Chem. 2001;276:24680–24689. doi: 10.1074/jbc.M100363200. [DOI] [PubMed] [Google Scholar]
- 20.Boopathi E, Anandatheerthavarada HK, Bhagwat SV, Biswas G, Fang JK, Avadhani NG. Accumulation of mitochondrial P450MT2, NH(2)-terminal truncated cytochrome P4501A1 in rat brain during chronic treatment with β-naphthoflavone. A role in the metabolism of neuroactive drugs. J. Biol. Chem. 2000;275:34415–34423. doi: 10.1074/jbc.M004431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss WM. Preparation of genomic DNA from mammalian tissue, Chapter 2, Unit 2.2. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA JA, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1998. [Google Scholar]
- 22.Koch JA, Waxman DJ. P450 phosphorylation in isolated hepatocytes and in vivo. Methods Enzymol. 1991;206:305–315. doi: 10.1016/0076-6879(91)06100-h. [DOI] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin-Phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 24.Gasser SM, Daum G, Schatz G. Import of proteins into mitochondria. Energy-dependent uptake of precursors by isolated mitochondria. J. Biol. Chem. 1982;257:13034–13041. [PubMed] [Google Scholar]
- 25.Bhat NK, Avadhani NG. The transport and processing of carbamyl phosphate synthetase-I in mouse hepatic mitochondria. Biochem. Biophys. Res. Commun. 1984;118:514–522. doi: 10.1016/0006-291x(84)91333-0. [DOI] [PubMed] [Google Scholar]
- 26.Gough AC, Miles JC, Spurr NK, Moss JE, Gaedigk A, Eichelbaum M, Wolf CR. Identification of the primary gene defect at the cytochrome P450 CYP2D locus. Nature. 1990;347:773–776. doi: 10.1038/347773a0. [DOI] [PubMed] [Google Scholar]
- 27.Hanioka N, Kimura S, Meyer UA, Gonzalez FJ. The human CYP2D locus associated with a common genetic defect in drug oxidation: a G1934 – A base change in intron 3 of a mutant CYP2D6 allele results in an aberrant 3’ splice recognition site. Am. J. Hum. Genet. 1990;47:994–1001. [PMC free article] [PubMed] [Google Scholar]
- 28.Kagimoto M, Heim M, Kagimoto K, Zeugin T, Meyer UA. Multiple mutations of the human cytochrome P450IID6 gene (CYP2D6) in poor metabolizers of debrisoquine. Study of the functional significance of individual mutations by expression of chimeric genes. J. Biol. Chem. 1990;265:17209–17214. [PubMed] [Google Scholar]
- 29.Rogan PK, Svojanovski S, Leeder JS. Information theory-based analysis of CYP2C19, CYP2D6, CYP3A5 splicing mutations. Pharmacogenetics. 2003;13:207–218. doi: 10.1097/00008571-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 31.Denson J, Wu Y, Yang W, Zhang J. Inter-individual variation of several cytochrome P450 2D6 splice variants in human liver. Biochem. Biophys. Res. Commun. 2005;330:498–504. doi: 10.1016/j.bbrc.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Zhuge J, Yu YN. Three new alternative splicing variants of human cytochrome P450 2D6 mRNA in human extratumoral liver tissue. World J. Gastroenterol. 2005;10:3356–3360. doi: 10.3748/wjg.v10.i22.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxbrow AK, Lewis CJ, Tennant MG, Modi S, Eggleston DS, Chenery RJ, Bridges AM. Crystal structure of human cytochrome P450 2D6. J. Biol. Chem. 2006;281:7614–7622. doi: 10.1074/jbc.M511232200. [DOI] [PubMed] [Google Scholar]