

Abstract
Mammographically dense breast tissue is one of the greatest risk factors for developing breast carcinoma, yet the associated molecular mechanisms remain largely unknown. Importantly, regions of high breast density are associated with increased stromal collagen and epithelial cell content. We set out to determine if increased collagen matrix density, in the absence of stromal cells, was sufficient to promote proliferation and invasion characteristic of a malignant phenotype in non-transformed mammary epithelial cells. We demonstrate that increased collagen matrix density increases matrix stiffness to promote an invasive phenotype. High matrix stiffness resulted in the increased formation of activated 3D-matrix adhesions and a chronically elevated outside-in/inside-out FAK-Rho signaling loop, which was necessary to generate and maintain the invasive phenotype. Moreover, this signaling network resulted in hyperactivation of the Ras-MAPK pathway, which promoted growth of mammary epithelial cells in vitro and in vivo and activated a clinically relevant proliferation signature that predicts patient outcome. Hence, the current data provides compelling evidence for the importance of the mechanical features of the microenvironment and suggest that mechanotransduction in these cells occurs through a FAK-Rho-ERK signaling network with ERK as a bottleneck through which much of the response to mechanical stimuli is regulated. As such, we propose that increased matrix stiffness explains part of the mechanism behind increased epithelial proliferation and cancer risk in human patients with high breast tissue density.
Keywords: Epithelial Morphogenesis, Breast Cancer, Breast Tissue Density, Tumor-Stroma Interaction, Cell-Extracellular Matrix Interaction, Mechanotransduction, 3D-Matrix/Focal Adhesion
INTRODUCTION
Mammographically dense breast tissue is correlated with a greater than four-fold increased risk for developing breast carcinoma (Boyd et al., 1998; Boyd et al., 2001; McCormack & dos Santos Silva, 2006), making it one of the greatest independent risk factors for breast cancer (Boyd et al., 1998; McCormack & dos Santos Silva, 2006). Ductal carcinoma in situ arises overwhelmingly in dense regions of the breast (Ursin et al., 2005), and high tissue density is associated with a greater risk for invasive breast carcinoma (Gill et al., 2006; Habel et al., 2004). Importantly, areas of increased breast density are associated with significantly increased fibrillar collagen deposition (Alowami et al., 2003; Guo et al., 2001; Li et al., 2005). Yet, while there is considerable correlative data identifying breast density as a risk factor for developing carcinoma, and increased stromal collagen has been shown to promote mammary tumor formation in mice (Provenzano et al., 2008b), the molecular mechanisms driving breast density-related tumor formation and progression remain largely unknown.
Breast cells exist in a varying multi-axial load environment that differs during cycles of ductal growth and involution, and depends on body position and activity. During normal daily activities breast tissue can undergo substantial deformation, the magnitude of which depends on breast anatomy and composition, and activity level. Moreover, large deformations can be inflicted during mammographic examination and while undergoing certain medical procedures (see (Plewes et al., 2000; Samani et al., 2001)), yet the effects of these mechanical loads on the ductal epithelium are unknown. Further complicating this issue is the fact that breast adenomas and carcinomas are stiffer than normal tissue (Sarvazyan et al., 1995; Sumi et al., 2000); increasing the mechanical load environment for adjacent cells (Krouskop et al., 1998; Plewes et al., 2000). Hence, an understudied aspect of the epithelial-stromal interaction is the fact that epithelial cells exist in a dynamic mechanical microenvironment, where dense collagenous stroma may increase force transmission to breast cells during tissue remodeling or deformation and increase resistance to cellular contractility. Consistently, such mechanical signals arising from the rigidity of the microenvironment have been shown to play a role in the transformed phenotype of breast epithelial cells in vitro (Paszek et al., 2005; Wozniak et al., 2003).
Focal adhesions (FAs) are sites of integrin-clustering that link the actin cytoskeleton to the extracellular matrix (ECM; (Burridge et al., 1988)). The primary functions of these complexes are to offer physical attachment to the ECM, transduce force between the cell and its microenvironment, and operate as a scaffolding node from which multiple signaling cascades emanate to regulate cell proliferation, survival, and migration (Burridge & Chrzanowska-Wodnicka, 1996; Geiger et al., 2001; Mitra et al., 2005; Playford & Schaller, 2004). It has been demonstrated that application of external force to adhesions or exposure to a “stiff” two-dimensional substrate promotes FA size and strength (Choquet et al., 1997; Galbraith et al., 2002; Pelham & Wang, 1997; Sniadecki et al., 2007); and that Rho-dependent contractile force through the actin cytoskeleton promotes FA assembly (Chrzanowska-Wodnicka & Burridge, 1996; Ridley & Hall, 1992). Furthermore, in fibroblasts, focal adhesion kinase (FAK), a key FA signaling molecule, is necessary for mechanosensing (Geiger et al., 2001; Mitra et al., 2005; Wang et al., 2001a) and becomes phosphorylated during cell deformation (Wang et al., 2001b). Fibroblast deformation promotes FAK activation through phosphorylation on Y397 and Y925, followed by FAK-dependent extracellular signal-regulated kinase (ERK) phosphorylation (Wang et al., 2001b) and proliferation (Wang et al., 2005). FAK(Y397) phosphorylation creates a high-affinity site that is recognized by several Src-homology-2 (SH2) domain-containing proteins including Src and Shc (Schaller et al., 1994; Schlaepfer et al., 1998; Xing et al., 1994). Moreover, FAK Y925 phosphorylation by Src promotes Grb2-FAK interactions which, along with Shc, link FAK to the Ras pathway (Schlaepfer & Hunter, 1996; Schlaepfer et al., 1998). Combined, these studies suggest that the force balance at the cell-matrix junction influences matrix adhesion structure and signaling, and that a functional linkage between FAK, Rho, and ERK exists.
The purpose of this study was to investigate the molecular mechanisms by which dense collagen matrices influence breast cellular phenotype. This has great clinical relevance due to the increased carcinoma risk correlated with high breast tissue density. Importantly, increased breast density is associated with increased epithelial cellularity (Guo et al., 2001; Li et al., 2005) and one prevailing hypotheses for breast density-related carcinoma risk centers on increased epithelial growth that is susceptible to increased mutagenic damage (Martin & Boyd, 2008). As such, we set out to determine if increased collagen matrix density per se, in the absence of stromal cells, was sufficient to alter nontransformed breast cell growth and promote cellular proliferation and invasion. While previous work by Paszek and co-workers (Paszek et al., 2005) elegantly showed that changing substrate stiffness can regulate epithelial phenotype, the molecular mechanisms associated with collagen matrix density-induced mechanoregulation remain largely unknown. In the current work we demonstrate how mechanical signals play a fundamental role in promoting growth in a model of breast tissue density. We find that higher matrix density is associated with increased matrix stiffness that resulted in increased adhesion signaling and chronically elevated activation of a FA-RhoGTPase-MAPK network. Moreover, mechanical stimuli promoted expression of a clinically relevant proliferation gene signature (that is predictive of human patient outcome) and epithelial proliferation through FAK-dependent (Ras pathway-mediated) ERK activation.
RESULTS
Matrix density determines ECM rigidity and regulates epithelial cell phenotype
Although it has been shown that increased collagen matrix density in three-dimensional (3D) culture disrupts tubulogenesis of T47D breast carcinoma cells (Wozniak et al., 2003) and normal acini formation of MCF10A cells (Paszek et al., 2005), the effects of increased collagen density on tubule structures from nontransformed mammary epithelial cells (MECs) has not been examined. To investigate the effects of matrix density on breast cell phenotype, normal murine mammary gland (NMuMG), nontransformed MCF10A, and well-differentiated T47D mammary epithelial cells (MECs) were cultured in three-dimensional (3D) collagen matrices of increasing density. Cells cultured under “optimal” low density (LD) conditions faithfully recapitulated mammary tubule duct-like structure, whereas increasing collagen density disrupted epithelial morphogenesis (Fig 1A, Supp Fig S1). Compared to well-differentiated tubules formed in LD matrices, colonies in high density (HD) matrices were larger and more cell-dense (cell-filled luminal space), expressed markers of destabilized cell-cell adhesion, and possessed an invasive phenotype with cells near the epithelial-ECM interface extending membrane protrusions into the matrix (Supp Figs S1 and S2).
Figure 1. Matrix density-induced stiffness regulates epithelial cell phenotype.
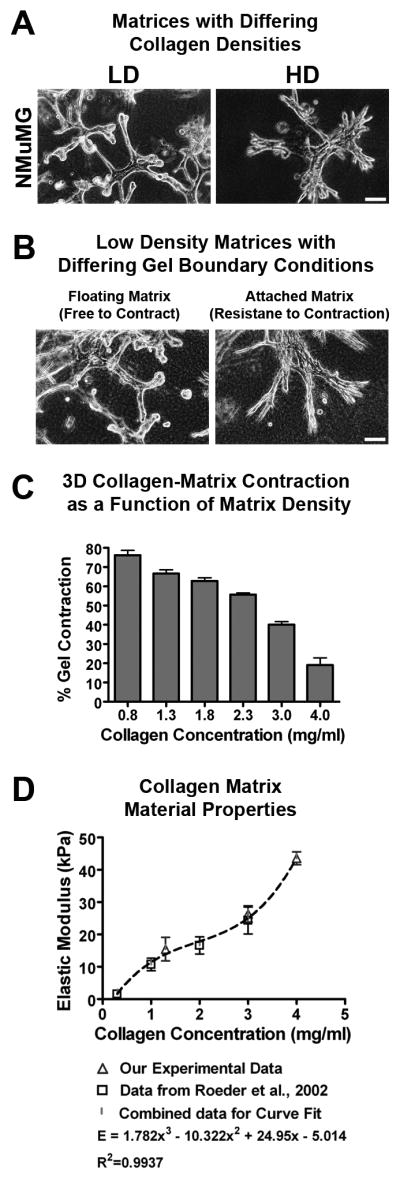
A. Representative micrographs of NMuMG mammary epithelial cells in low-density (LD) and high-density (HD) collagen matrices (density optimized for each cell type as described in the Materials and Methods section). Bar: = 100 μm
B. Representative micrographs of NMuMG cells seeded into LD collagen matrices allowed to freely contract and undergo tubulogenesis (left) or constrained to resist cell-mediated matrix contraction and disrupt differentiation (right). Bar = 100 μm
C. NMuMG-mediated matrix contraction as a function of collagen density after 7 days (n = 6; mean ± SEM).
D. Elastic modulus of collagen matrices at densities used in this study (n = 4; mean ± s.d.), compared to the data of Roeder et al. (2002) for matched collagen matrix conditions.
Interestingly, changing boundary conditions, by leaving LD matrices attached to the culture dish, to increase resistance to cell-mediated collagen gel contraction, re-capitulated the HD phenotype in NMuMG (Fig 1B, Supp Fig 1) and human MCF10A and T47D cells (data not shown), suggesting that the force-balance at the cell-matrix interface influences epithelial behavior. Consistent with this observation, MECs were significantly less able to contract higher density matrices (Fig 1C) due to increased matrix stiffness as the collagen density was increased (~10 to 44 kPa; density 1 to 4 mg/ml; Fig 1D). Importantly, the effect of a higher concentration of collagen gels was not due to ligand concentration; (Supp Fig 3). These results suggest that increased resistance to matrix contraction regulates epithelial behavior, and further support the conclusion that the physical properties of the microenvironment are important regulators of epithelial phenotype.
Matrix stiffness regulates the formation and activation of 3D-matrix adhesions
Using 3D culture models, it has been shown that integrin-containing adhesions play a significant role in regulating epithelial phenotype (Keely et al., 1995; Weaver et al., 1997). Moreover, FAK is overexpressed in human breast cancer (Cance et al., 2000) and is necessary for mammary tumor growth and progression (Lahlou et al., 2007; Provenzano et al., 2008a). Since FAK is also required for mechanosensing and influences cell proliferation, survival, and migration (Burridge & Chrzanowska-Wodnicka, 1996; Geiger et al., 2001; Mitra et al., 2005; Playford & Schaller, 2004), we examined the state of FAK as a function of tissue stiffness.
3D-matrix adhesions were examined in live cells expressing GFP-Vinculin using multiphoton microscopy. Increased matrix stiffness (25 to 44 kPa) resulted in increased adhesion clustering at the cell-matrix interface (Fig. 2A). Consistent with these findings, immunofluorescent microscopy of 3D cultures confirmed that increased matrix stiffness induced vinculin and paxillin-positive 3D-matrix adhesions (Fig 2B). Moreover, quantitative analysis of membrane protrusions confirmed the significantly more invasive phenotype in HD matrices (Figs 2C).
Figure 2. Matrix stiffness promotes 3D-matrix adhesions and FAK signaling.

A. Representative micrographs of live cell multiphoton excitation (MPE) and second harmonic generation (SHG) imaging of NMuMG cells expressing GFP-Vinculin (MPE = pseudo-colored red) cultured in low density (LD) and high density (HD) collagen matrices (SHG = pseudo-colored green) for 7 days. Note the differential vinculin localization as a function of matrix stiffness, with increased 3D-matrix adhesion clustering at the cell-ECM interface in stiffer HD matrices (open arrowheads). Bottom right (LD) and farthest right (HD) open arrowheads indicate the regions magnified 4X. Bar = 25 μm
B. Immunofluorescence analysis of NMuMG cells in LD and HD collagen matrices showing increased localization of two hallmark focal adhesion proteins, vinculin (top) and paxillin (bottom), to 3D-matrix adhesions in HD matrices; with FAK phosphorylated at Y397 (red) co-localized with paxillin in 3D-matrix adhesions under HD conditions (representative of n ≥ 5). Bar = 10 μm
C. Morphometric analysis of NMuMG cells after differentiating in LD or HD matrices, showing significantly increased cell protrusion into HD matrices (nlength ≥ 70, nperimeter ≥ 22 from n ≥ 8 matrices/conditions; mean ± SEM).
D. Western blot analysis and densitometry of pFAK(Y397) levels in lysates from NMuMG cells cultured in LD or HD matrices for 7 days (n ≥ 6; mean ± SEM).
E. Western blot analysis of Src protein that co-precipitated with immunoprecipitated FAK, showing increased FAK-Src association in cells within HD matrices (n = 3; mean ± SEM).
F. Western blot analysis and densitometry of phosphorylated Src levels following immunoprecipitation of Src from NMuMG cell lysates after being cultured in LD or HD matrices for 7 days (n = 3; mean ± SEM).
Notably, FAK phosphorylated at Y397 co-localized with activated paxillin at 3D-matrix adhesions (Fig 2B, Supp Fig S4), and total pFAK(Y397) levels were significantly increased (~2-fold) under HD conditions (Fig 2D). Consistent with the signaling function of this phosphorylation event, complex formation between FAK and its signaling partner, Src, was significantly increased (Fig 2E), with levels of activated Src (Y418-phosphorylated in 3D-matrix adhesions, Supp Fig S4) upregulated ~2-fold (Fig 2F), providing previously unreported evidence that, in addition to FAK, Src is activated and increased FAK-Src are promoted by culture in 3D matrices of increased stiffness. Further, these results strongly suggest a role for FAK in regulating the response to physical signals to and from MECs.
FAK is activated by exogenous force
We speculate that high force at the cell-matrix interface promotes a feedback loop of outside-in and inside-out signaling between FAs and the RhoGTPase pathway, with FAK being a mechano-responsive central regulator of the focal adhesion. To date, reported experiments have focused on the response of MECs to changes in stiffness of the 3D environment or 2D substrate. The mechano-responsive signal transduction following application of exogenous force has not been reported for MECs cultured in 3D matrices. Therefore, in order to test our conclusions that FAK activity is force-responsive in MECs, we applied external mechanical stimuli to MECs cultured on collagen-coated 2D substrates and within 3D collagen matrices. Following the application of substrate deformation, MECs exhibited increased FAK activation (Fig 3A). In both non-transformed (NMuMG and MCF10A) and transformed (MDA-MB-231) cells, the level of pFAK(Y397)-positive FA area was significantly increased >2-fold (Fig 3A). Consistent with this observation, pFAK(Y397) was increased in loaded 3D collagen matrices (Fig 3B). Thus, both normal and transformed MECs are mechano-responsive, responding directly to external force applied to the matrix adhesion sites, supporting the conclusion that the force balance at the cell-matrix interface is a critical regulator of cell behavior.
Figure 3. Application of exogenous force and increased substrate stiffness promotes FAK phosphorylation and a FAK-dependent protrusive phenotype.

A. Substrate strain-induced cell deformation of mammary epithelial cells adhered to type I collagen-coated (soft) silicone elastomer substrates. Static equibiaxial substrate deformation (10%) was applied for 20 minutes. Subsequently, immunofluorescent analysis was performed to detect and quantify changes in pFAK(Y397) positive focal adhesions (nFA ≥ 440 for NMuMG and MDA-MB-231 cells; nFA ≥ 146 for MCF10A cells; * p=0.006 for MCF10A; p=0.0001 for NMuMG and MDA-MB-231; mean ± SEM). Bar = 10μm
B. NMuMG cells cultured within a 3D collagen matrix (3mg/mL) were loaded by deforming (10% axial strain) collagen matrices for 20 minutes. Subsequently, Western blot and densitometry analysis was used to examine significantly (*p=0.0006) increased FAK(Y397) phosphorylation levels following matrix deformation (n ≥ 4).
C. Immunofluorescent analysis for FAK(Y397) phosphorylation (red), actin cytoskeleton architecture (green), and the nucleus (blue) in NMuMG cells cultured on collagen matrices of increasing substrate stiffness (shear modulus was increased by increasing collagen matrix density, range = 1 to 4 mg/mL). Bar = 10 μm
D. pFAK(Y397)-positive focal adhesion area increasing as a function of increasing substrate stiffness (G′; nFA ≥ 125; mean ± SEM)
E. Immunofluorescent analysis of paxillin (top) or FAK(Y397) phosphorylation and actin cytoskeleton architecture (bottom) in NMuMG cells cultured on type I collagen-coated (30 μg/mL) polyacrylamide gels of increasing stiffness (polyacrylamide gels were crosslinked with varying concentrations of bisacrylamide to control stiffness). The nucleus is shown in blue. Bar = 10 μm
F. Fluorescent localization of the actin stress fibers (green) in NMuMG cells cultured on type I collagen-coated (30 μg/mL) polyacrylamide gels of equal stiffness transfected with either non-silencing control siRNA (left) or siRNA targeting FAK (right). siRNA was labeled with Cy3 (red) to ensure examination of siRNA transfected cells. The nucleus is shown in blue. A representative Western blot demonstrating FAK siRNA knockdown (consistently greater than 85%) is shown in the right panel. Arrowheads indicate the regions magnified in panels C, E, and F. Bar = 10 μm.
FAK is necessary for the epithelial response to substrate stiffness
To confirm that MECs are not only mechano-responsive, but also integrate signals from the mechanical environment and respond, we cultured cells on top of 3D collagen gels that have a progressively escalating shear modulus to increase resistance to cell traction force. As substrate stiffness increased (G′: 4.4–16.8 kPa) cells displayed a more protrusive phenotype with increased FAs (Fig 3C), consistent with findings for cells in 3D matrices of increasing elastic modulus (Fig 2). Likewise, pFAK(Y397)-positive FA area increased progressively as a function of substrate stiffness (Fig 3D). Furthermore, while cells on softer surfaces (G′=4.4 kPa) displayed cortical actin localization, cells on stiffer substrates (G′=9.6–16.8 kPa) had actin stress fiber formation (Fig 3C), suggesting a role for Rho in the cellular response to physical stimuli.
To confirm that results obtained by increasing collagen matrix density were not due to presentation of more integrin-binding ligand, we prepared polyacrylamide (PA) gels coated with equal concentrations of collagen. Consistent with results obtained from collagen matrices, MECs cultured on PA gels of increasing stiffness (G′=6.4–18.3 kPa) displayed a more protrusive phenotype, had increased pFAK(Y397)-positive FAs, and actin stress fiber formation (Fig 3E). Using siRNA to specifically achieve >85% knockdown of FAK (Fig 3F-inset), we studied the role of FAK in the cellular response to matrix stiffness. Following FAK knockdown, MECs on stiff (G′=18.3) substrates no longer formed pronounced protrusions and lacked actin stress fiber formation, demonstrating that MECs sense and respond to the mechanical environment in a FAK-dependent manner. Furthermore, inhibition of FAK activity with FRNK, a dominant-negative C-terminal region of FAK, reverted the invasive phenotype associated with stiff HD conditions (Supp. Fig S5), further implicating FAK as a key regulator of the cellular response to mechanical signals.
Inhibition of the Rho/ROCK pathway reverts the HD matrix-induced invasive phenotype
FA signaling can lead to Rho activation that promotes myosin light chain (MLC)-mediated intracellular contractile force, which further promotes focal adhesion clustering and maturation (Choquet et al., 1997; Chrzanowska-Wodnicka & Burridge, 1996; Galbraith et al., 2002; Riveline et al., 2001). Herein, the presence of actin stress fibers in Figure 2, suggests increased Rho activity in response to mechanical signals. We therefore examined the RhoGTPase pathway as a function of matrix density. To confirm that matrix contraction was Rho mediated, we infected cells with Rho(N19) (data not shown) or inhibited Rho with Exoenzyme C3 Transferase, ROCK with H1152, or myosin-based contractility with blebbistatin; all of which significantly reduced matrix contraction (Fig 4A). In addition, since inhibition of the Rho/ROCK pathway can impair proliferation in MECs in stiff collagen matrices (authors’ unpublished results; also see Figure 9) we controlled for cell number by allowed equal cultures of MECs to contract the collagen matrix and then added the inhibitors for 2 hours. Consistent with results from Fig. 4A inhibition of the Rho/ROCK pathway decreased matrix contraction (Fig. 4B), supporting the conclusion that generation of myosin-mediated cellular contractile force is Rho/ROCK-dependent in nontransformed MECs.
Figure 4. Increased matrix stiffness promotes Rho-mediated contractility.

A. Inhibition of Rho with cell-permeable C3 exoenzyme transferase (10 μg/mL), ROCK (which promotes cellular contractility by directly phosphorylating MLC and/or inhibiting MLC phosphatase) with H1152 (2.5 μM), or myosin-based contractility with blebbistatin (10 μM) significantly suppressed NMuMG-mediated matrix contraction (n = 3; mean ± SEM).
B. Inhibition of Rho, ROCK, myosin-based contractility, or the actin cytoskeleton as described in A for 2 hours in contracted matrices resulted in a significant relaxation of the collagen matrix (n = 4; mean ± SEM; *p<0.01).
C. Quantitative analysis of RhoA activation in NMuMG cells in low density (LD) and high density (HD) collagen matrices after 1 hour (normalized by cell number: left) or 7 days (normalized by total Rho protein: right; n ≥ 3, mean ± SEM).. A representative Western blot for total Rho after 7 days is shown (bottom right)
D. Fluorescence staining analysis of NMuMG cells in LD and HD collagen matrices showing increased actin stress fiber formation (green) under HD conditions. Bar = 10 μm
E. Fluorescence staining analysis of substrate strain-induced cell deformation of MCF10A cells (as described in Figure 3) was performed to detect changes in the actin cytoskeleton (green). Immunofluorescent analysis of FAK(Y397) phosphorylation is shown in red and the nucleus in blue. Arrows indicate the regions magnified in the subpanels. Bar = 10 μm
F. Inhibition of Rho with C3 (a, 10 μg/mL), ROCK with H1152 (b, 2.5 μM), myosin-based contractility with blebbistatin (c, 10 μM) or disruption of the actin cytoskeleton with cytochalasin D (d, 1 μM) for 2 hours in cells that has already developed the HD-induced invasive phenotype caused the invasive phenotype to be significantly reverted (f) when compared to controls (a; *p<0.01; mean ± SEM). Bar = 10 μm
Figure 9. Ras pathway activation of ERK is dependent on FAK phosphorylation.

A and B. Western blot analysis and densitometry of ERK phosphorylation levels in lysates from NMuMG cells cultured in HD matrices for 7 days. A: 24 hours before lysis, cells were infected with either Adeno-GFP or Adeno-FRNK-GFP (n ≥ 4; *p<0.02; mean ± SEM). B: Two hours before lysis, Src or PI3K were inhibited with PP2 (10 μM) or LY294002 (25 μM; n = 3; *p<0.005; mean ± SEM).
C and D. Western blot analysis and densitometry of (C) pFAK(Y397) and (D) pFAK(Y925) phosphorylation levels following immunoprecipitation of FAK from NMuMG cell lysates after being cultured in LD or HD matrices for 7 days (n = 3; *p<0.001; mean ± SEM).
E and F. Western blot analysis of (E) SHC and (F) Grb2 proteins that co-precipitated with immunoprecipitated FAK, showing increased FAK-SHC and FAK-Grb2 association in cells within HD matrices (n = 3; *p<0.004; mean ± SEM).
G. Model for the regulation of epithelial growth by matrix density-induced increases in ECM stiffness. As mammary epithelial cells encounter exogenous mechanical force or increased resistance to cellular contractility from stiff high density matrices they respond in a FAK-dependent manner by developing mature focal or 3D-matrix adhesions. Sustained mechanical signaling results in chronic upregulation of a FAK-Rho signaling loop that produces hyperactivation of related pathways, such as the Ras-MAPK pathway. Elevated ERK1/2 phosphorylation feeds back to regulate epithelial phenotype, controls the majority of the mechanically-induced transcriptome shift, and induces transcription of clinically-relevant proliferation associated genes, which results in increased epithelial proliferation.
Quantitative analysis of Rho-GTP levels showed significantly higher Rho activity in cells within HD matrices, when compared to LD conditions (Fig 4C). Furthermore, actin stress fibers were present when cells were cultured in stiff 3D-matrices (Fig. 4D) or experienced cell deformation (Fig. 4E), supporting our conclusions that mechanical signals through 3D-matrix adhesions promotes Rho activation. These results are consistent with a model in which cells response to aberrantly stiffer microenvironments by increasing the magnitude of intracellular contraction until the force balance at the cell-matrix interface reaches an abnormally high tensional homeostasis.
To elucidate the role of Rho in regulating MEC morphology in 3D matrices, previous studies have utilized cells either stably expressing Rho/ROCK mutants or under continuous pharmacological inhibition of the Rho-ROCK pathway to study morphogenesis of epithelial tubules (Wozniak et al., 2003) or acini (Paszek et al., 2005). Herein, we set out to test whether Rho-mediated contractility was necessary to maintain the invasive phenotype associated with elevated isometric tension, by allowing the invasive phenotype to form and then selectively inhibiting the Rho-ROCK-myosin-actin network. Treatment of cells with pharmacological inhibitors directed against Rho, ROCK, myosin-based contractility, or actin structure for 2 hours significantly reverted the invasive phenotype associated with HD microenvironments (Fig 4F). These data suggest that a signaling loop between FAs and the RhoGTPase pathway is maintained by the isometric force balance at the cell-matrix interface. Moreover, in addition to FAK, data suggest that Rho is necessary for mechanotransduction and part of the mechanism by which MECs respond to physical stimuli.
Density-induced matrix stiffness promotes epithelial proliferation
High breast tissue density in human patients is associated with increased epithelial content (Guo et al., 2001; Li et al., 2005) suggesting that high density promotes epithelial growth and/or survival. Herein, we observed that epithelial colonies were larger and more cell-dense in HD matrices, suggesting a more proliferative phenotype (Supp. Fig S1). Consistent with these observations, microarray analysis of cells cultured in LD vs. HD matrices demonstrated a significant shift in the transcriptome (Fig 5A and Supp. Fig. S6). Specifically, there was a significant statistical enrichment for transcripts associated with cell proliferation under HD conditions (Fig 5B). Hierarchical clustering of differentially expressed transcripts, combined with statistical gene ontology enrichment analysis, produced two clusters associated with proliferation (Fig 5C and D), with each cluster containing genes known to be important for breast carcinoma proliferation (Whitfield et al., 2006). In fact, the full set of genes previously identified as the human breast carcinoma-associated “proliferation signature” (Whitfield et al., 2006), which predicts survival and metastasis-free outcome in human patients (Fig. 5E, Supp Fig S7), was upregulated by increased matrix stiffness (Table 1), suggesting that increased collagen density promotes a hyper-proliferative state for nontransformed epithelial cells.
Figure 5. Matrix density-induced stiffness promotes expression of clinically relevant proliferation-signature genes.

A. Principal Component Analysis (PCA) of density regulated genes to reduce the dimensionality of the data sets and examine the extent to which loss matrix density influences the pattern of expression. Data were mean centered then PCA performed from all experimental samples (LD, n=5; HD, n=5), with the 1st Principal Component a measure of average expression.
B. Gene Ontology analysis of the entire LD vs. HD data set showing enriched transcripts associated with proliferation.
C and D. Proliferation node gene clusters generated by hierarchically clustering (significantly) differentially expressed genes from NMuMG cells within LD and HD matrices showing increased expression of genes associated with proliferation. Also shown is Gene Ontology analysis of the primary (C) and secondary (D) clusters demonstrating enrichment for proliferation associated transcripts.
E. Prognostic value of the Proliferation Signature (PS) transcripts shown in Table 1. Using publicly available data from van de Vijver et al., (van de Vijver et al., 2002) we found that the 41 genes divide human breast cancer patients into two main clusters (cluster analysis provided in Supplementary Figure S5,). Kaplan-Meier survival analysis of the two groups shows that the patients differ significantly in both survival and metastasis-free outcome. Average expression in patients with poorer outcome (blue line) was higher for the PS genes while patients with better outcome (orange line) presented lower expression of these genes.
Table 1.
Differentially expressed transcripts implicated in the conserved human breast carcinoma-associated “proliferation signature” (Whitfield et al., 2006) are upregulated due to increased density-induced matrix stiffness (HD/LD). Greater than 95% of these transcripts are significantly reverted to near LD levels following inhibition of ERK1/2 phosphorylation (HD+U0126/HD+DMSO).
| Gene | Fold Increase Due to Density-Induced Matrix Stiffness | Fold Decrease in HD Matrices after ERK Inhibition | |
|---|---|---|---|
| BircN5 | survivin | 2.9 | −3.1 |
| Aurkb | aurora kinase B | 3.4 | −4.0 |
| Cdc6 | cell division cycle 6 | 3.9 | −4.9 |
| Traip | TRAF-interacting protein | 2.5 | −2.3 |
| Chek1 | checkpoint kinase 1 | 3.3 | −3.4 |
| Pttg1 | pituitary tumor-transforming 1 | 2.1 | −1.7 |
| Dnmt1 | DNA methyltransferase 1 | 2.0 | −2.8 |
| Ung | uracil DNA glycosylase | 3.3 | −4.3 |
| Cdc7 | cell division cycle 7 | 3.4 | −5.1 |
| Fen1 | flap structure specific endonuclease 1 | 3.1 | −3.2 |
| Mcm2 | minichromosome maintenance deficient 2 | 2.9 | −3.4 |
| Mcm3 | minichromosome maintenance deficient 3 | 3.9 | −4.8 |
| Mcm4 | minichromosome maintenance deficient 4 | 2.6 | −3.3 |
| Mcm5 | minichromosome maintenance deficient 5 | 4.3 | −4.4 |
| Mcm6 | minichromosome maintenance deficient 6 | 2.3 | −2.2 |
| Orc1l | origin recognition complex, subunit 1-like | 3.0 | −3.8 |
| Pcna | proliferating cell nuclear antigen | 2.0 | −2.2 |
| Prim1 | DNA primase, p49 subunit | 2.4 | −2.8 |
| Rrm1 | ribonucleotide reductase M1 | 2.5 | −2.5 |
| Rrm2 | ribonucleotide reductase M2 | 3.9 | −3.1 |
| Top2a | topoisomerase (DMA) II alpha | 3.0 | −3.5 |
| Mad2l1 | mitotic arrest deficient-like 1 | 2.4 | −2.4 |
| Cenpe | centromere protein E | 3.5 | −4.0 |
| Bub1 | budding uninhibited by benzimidazoles 1 | 2.8 | −3.1 |
| Ctps | cytidine 5′-triphosphate synthase | 1.8 | −1.3 |
| Dhfr | dihydrofolate reductase | 2.4 | −3.0 |
| Tyms | thymidylate synthase | 2.4 | −2.8 |
| Ccna2 | cyclin A2 | 2.9 | −3.4 |
| Ccnb1 | cyclin B1 | 3.0 | −3.0 |
| Ccnd1 | cyclin D1 | 2.0 | −2.2 |
| Ccne1 | cyclin E1 | 2.0 | −2.2 |
| Ccnf | cyclin F | 3.5 | −2.8 |
| Cdc20 | cell division cycle 20 | 2.6 | −2.7 |
| E2f1 | E2F transcription factor 1 | 2.0 | −1.6 |
| E2f2 | E2F transcription factor 2 | 2.4 | −2.3 |
| E2f3 | E2F transcription factor 3 | 1.3 | 0 |
| E2f7 | E2F transcription factor 7 | 3.1 | −4.5 |
| Mki67 | Ki67 monoclonal antibody antigen | 2.5 | −3.0 |
| Plk1 | polo-like kinase 1 | 3.0 | −2.8 |
| Timp1 | tissue inhibitor of metalloproteinase 1 | 2.0 | 0 |
| Mybl2 | myeloblastosis oncogene-like 2 | 4.0 | −4.4 |
| Aurka | aurora kinase A | 3.1 | −3.6 |
| Foxm1 | forkhead box M1 | 2.7 | −2.8 |
To confirm our observations, we performed Ki-67 staining on MECs cultured in LD and HD matrices. Consistent with microarray data, MECs cultured in stiff matrices were significantly more proliferative (Fig 6A). Furthermore, HD matrices promoted tumor growth in vivo. Xenograft experiments with MDA-MB-231 cell-seeded collagen matrices implanted into nude mice showed significantly increased tumor volume in HD matrix-derived tumors over 40 days (Fig 6B). This finding is consistent with previous data showing increased tumor formation in transgenic mice possessing increased stromal collagen (Provenzano et al., 2008b), and confirms that increased collagen density promotes proliferation in both nontransformed and transformed MECs.
Figure 6. Matrix density-induced stiffness promotes proliferation of mammary epithelial cells.
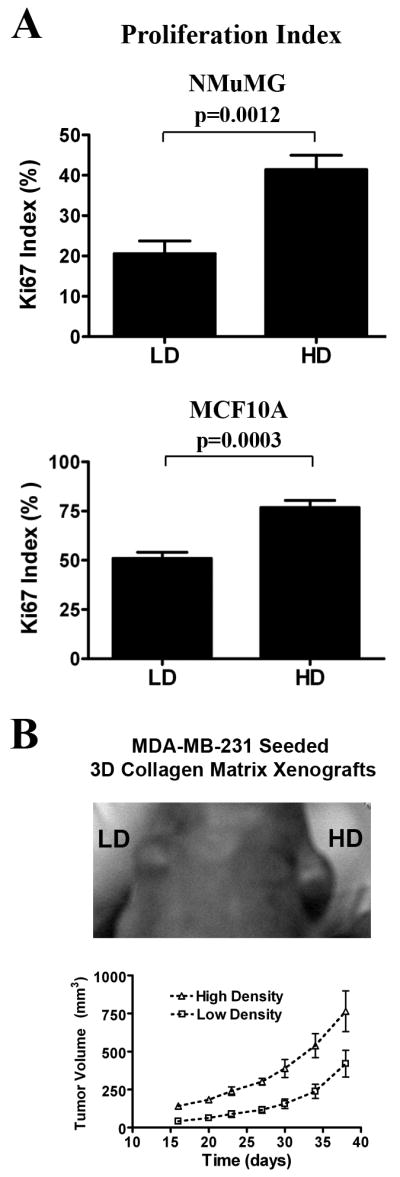
A. Ki-67 index of proliferation potential in NMuMG (top) and MCF10A (supplemented with 50ng/mL HGF to induce tubulogenesis; bottom) cells cultured in LD and HD matrices. Data are mean ± SEM obtained from >300 cells per condition in n ≥ 5 samples per group.
B. Tumor growth in a xenograft model is increased by high matrix density. Human MDA-MB-231 cells were seeded into LD and HD matrices prior to inoculation into nude mice. Each mouse received a LD and HD cell-seeded matrix transplanted into contralateral sides above the 4th inguinal mammary glands. Increased tumor growth resulted from HD xenografts when compared to the LD control (n = 6; mean ± SEM, p<0.01 for each time point after day 15).
Inhibition of ERK phosphorylation suppresses proliferation
The ERK/mitogen-activated protein kinase (MAPK) pathway has been implicated in multiple cellular processes such as proliferation, migration and apoptosis (Dhillon et al., 2007; Turjanski et al., 2007; Yoon & Seger, 2006). Activation of ERK1/2 is sufficient to transform NIH 3T3 cells (Mansour et al., 1994) and pro-growth signals in many human cancers result from hyper-activation of the ERK pathway due to mutation/overexpression of ERK pathway-regulating molecules, such as Ras, receptor tyrosine kinases, or integrins (Dhillon et al., 2007; Giancotti & Ruoslahti, 1999; Turjanski et al., 2007). Furthermore, while work by Paszek et al., (Paszek et al., 2005) showed that in MECs on stiffer basement membrane-coated polyacryamide gels have higher ERK-2 activity in response to EGF stimulation, it is currently not known if changes in the mechanical 3D-microenvironment influence proliferation by altering cytokinesis or promoting activation of more classical signaling pathways, such as MAPK. Therefore, to study the molecular mechanism by which increased proliferation was regulated in MECs cultured in stiff matrices, we examined the role of ERK. Western blot analysis demonstrated a significant increase in ERK1/2 phosphorylation in cells cultured under HD versus LD conditions (Figs. 7A and B). No preference was observed toward either ERK1 or ERK2 activation, as both showed a >2-fold increase in phosphorylation under HD conditions (Fig 7B). Moreover, consistent with our hypothesis that the proliferative phenotype is mechanically regulated, loading 3D matrices promoted ERK1/2 phosphorylation in MECs.
Figure 7. Matrix stiffness induces ERK phosphorylation and results in ERK-dependent membrane protrusion and cell proliferation.

A and B. Western blot analysis (A) and densitometry (B) of phosphorylated ERK1/2 levels (using a phospho-specific antibody that recognizes phosphorylation of the threonine (Thr183) and tyrosine (Tyr185) residues in the activation loops of ERK1 and ERK2) in lysates from NMuMG cells cultured in LD or HD matrices for 7 days (n ≥ 3; mean ± SEM).
C. NMuMG (and MCF10A = data not shown) cells loaded by deforming the collagen matrices as described in Figure 3 show increased ERK1/2 phosphorylation following application of matrix strain (representative of n ≥ 4 samples per condition).
D. Representative Western blot analysis of phosphorylated ERK levels in lysates from NMuMG cells cultured in HD matrices following treatment with 10 μm U0126 or DMSO (control).
E and F. Inhibition of ERK phosphorylation with the MEK inhibitor U0126 (10 μM) for 24 hours in cells that developed the HD-induced invasive phenotype (middle) results in cells that were significantly (F) reverted (right) to the LD phenotype (left). A color version of panel E is available online as Supp. Fig. S9.
G. Ki-67 index of proliferation potential in NMuMG cells cultured in LD and HD matrices. Cells in HD matrices were either treated with DMSO (control) or 10 μm U0126 for 24 hours in cells that had developed the HD-induced invasive phenotype. Data are mean ± SEM obtained from >250 cells per condition in n ≥ 4 samples per group.
To study ERK’s role in mechanotransduction, we treated cells with the MEK inhibitor U0126 to selectively inhibit ERK phosphorylation (Fig. 7D). ERK activation was required to maintain the invasive phenotype resulting from stiff matrices, as the phenotype was reverted after treatment with U0126 (Fig. 7E and F). This effect of MEK/ERK inhibition was possibly through ERK’s regulation of MLC phosphorylation by MLC kinase (Klemke et al., 1997), as this result is consistent with the phenotypic reversion observed following treatment with blebbistatin (Fig 4F). Furthermore, inhibition of ERK phosphorylation significantly decreased stiffness-induced proliferation (Fig. 7G). Hence, ERK is implicated as regulator of both proliferation and invasive phenotype in MECs responding to increased matrix stiffness.
ERK phosphorylation regulates the transcriptional response to increased matrix stiffness
Activated ERK translocates to the nucleus where ERK can regulate the activity of transcription factors to alter gene expression (Dhillon et al., 2007; Turjanski et al., 2007; Yoon & Seger, 2006). Of particular relevance to the malignant phenotype, sustained ERK activation promotes entry into, and progression through, the cell cycle (Chambard et al., 2007; Dhillon et al., 2007). Since ERK inhibition repressed both proliferation and the invasive phenotype of MECs in stiff matrices, we speculated that loss of ERK activity would have a profound effect on the transcriptome, particularly genes associated with proliferation.
Principal component analysis (PCA), to reduce the dimensionality of the data and facilitate a more global analysis of expression trends, demonstrated that the average expression in cells cultured in HD matrices was ‘brought back’ to levels under LD conditions (Fig 8A), indicating a more ‘normal’ state. In fact, inhibition of ERK phosphorylation reverted ~70% of the genes that were differentially expressed by increasing matrix stiffness back toward the expression profile under LD conditions. Hierarchical clustering of differentially expressed transcripts between LD and HD conditions and data from cells in HD matrices treated with either vehicle (DMSO) or U0126, showed ~68% and ~75% of the transcripts that were decreased and increased, respectively, due to HD conditions were near LD levels after ERK inhibition (Fig 8B). When combined, with phenotypic data shown in Figure 6, these results implicate ERK as a central-regulating “bottleneck” in an hour-glass shaped signaling network activated by increased matrix stiffness.
Figure 8. ERK mediated regulation of the HD-induced transcriptome shift.

A. Principal Component Analysis (PCA) of ERK1/2 regulated genes in order to reduce the dimensionality of the data sets as well as determine the extent to which loss of ERK1/2 activity influences the pattern of expression. Data were mean centered then PCA performed from all experimental samples: LD, n=5; HD, n=5; HD+DMSO, n=5; HD+U0126 (10 μM for 24 hours, as described in Figure 7), n=5 arrays. The 1st Principal Component, which may be a measure of average expression, indicates that inhibition of ERK phosphorylation shifts the transcriptome back to near LD levels.
B. Hierarchical cluster of transcripts differentially expressed due to HD matrix conditions over the entire data set (described in panel A) showing that ~70% of the transcripts that are regulated by increased matrix density/stiffness are reverted toward LD levels following ERK inhibition. The majority of the transcripts that were repressed (blue box) due to high matrix density were reverted by inhibition of ERK phosphorylation (turquoise boxes), while the majority of the transcripts that were induced (orange box) due to high matrix density were also reverted by inhibition of ERK phosphorylation (yellow box).
C. Proliferation node gene cluster generated by hierarchically clustering (B) showing reversion of proliferation-associated transcripts following ERK inhibition. The accompanying Gene Ontology analysis of the cluster demonstrates enrichment for proliferation associated transcripts.
D. Computationally predicted transcription factor binding sites (TFBS) that are enriched in the proliferation cluster (C).
Of particular interest to understanding density-regulated epithelial proliferation; transcripts associated with proliferation were significantly decreased following inhibition of ERK phosphorylation Suppression of ERK phosphorylation repressed clinically relevant breast cancer proliferation signature genes (Table 1) that predict human patient outcome (Fig. 5E). Inhibiting ERK activation resulted in significantly reduced expression of >97% of the proliferation signature genes induced by mechanical stimuli from dense ECM; reverting their expression back toward levels associated with normal ductal differentiation (Table 1). Furthermore, differentially expressed transcripts associated with proliferation clustered together following ERK inhibition (Fig. 8C), suggesting that they may share common regulatory mechanisms. Therefore, we computationally inspected the proliferation cluster for enriched transcription factor binding sites (TFBS) to gain insight into ERK’s mechanisms for regulating stiffness-induced proliferation (Fig 8E). Interestingly, many ERK regulated transcription factors (Sp1, HNF4, p53, Spz1, Zic1-3, Pax4, Stat6, and AP4) were previously identified as part of a conserved set of TFs in FAK-regulated mammary carcinoma proliferation and metastasis (Provenzano et al., 2008a). In particular, we previously found that expression of genes associated with the G2-G2/M phases of the cell cycle were regulated by FAK through predicted TFBS for Sp1, p53, p300/CBP, AP4, HNF4, Lmo2-complex, Spz1, Zic1-3, AP2α, Stat6, and Pax4, which are herein also predicted TFBS for ERK-regulated proliferation genes (Fig 8E). In addition, ERK is predicted to regulate TFs associated with earlier phases of the cell cycle (such as E2F, c-Myb, Elk-1; (Chambard et al., 2007; Oh & Reddy, 1999)), consistent with our finding that ERK inhibition resulted in reduced expression of genes associated with each phase of the cell cycle (Table 1). Hence, analysis of TFBS within co-clustered proliferation transcripts suggests that these TFs may be part of the ERK-associated network that is activated in response to mechanical signals, and that FAK is a part of this network.
ERK phosphorylation is regulated by FAK
FAK phosphorylation at Y397 creates a high-affinity site recognized by SH2 domain-containing proteins Src and Shc (Schaller et al., 1994; Schlaepfer et al., 1998; Xing et al., 1994). Subsequent phosphorylation at Y925 by Src promotes Grb2-FAK interactions which, along with Shc, link FAK to the Ras pathway in fibroblasts (Schlaepfer & Hunter, 1996; Schlaepfer et al., 1998). To determine if ERK is regulated by FAK in MECs, we manipulated FAK activity by expressing FRNK, a dominant-negative C-terminal region of FAK. FRNK expression (confirmed with antibodies specific for the C-terminal region of FAK) resulted in decreased FAK(Y397) phosphorylation (Supp. Fig. S8), and significantly inhibited stiffness-induced ERK phosphorylation (Fig. 9A). Furthermore, pharmacological inhibition of Src dramatically repressed ERK phosphorylation, while inhibition of PI3K had no effect (Fig 9B), supporting the conclusion that ERK is regulated by the FAK-Src complex downstream of integrin-mediated signaling.
Notably, in addition to FAK(Y397) phosphorylation (Figs 2D and 9C), FAK(Y925) phosphorylation was increased due to high matrix stiffness (Figure 9D). In conjunction with these increases, the co-precipitation of Shc (Fig 9E) and Grb2 (Fig 9F) with FAK was increased in cells in HD matrices. Both Shc and Grb2 are upstreatm activators of Ras, suggesting that FAK regulates ERK phosphorylation via the Ras pathway (Fig 9G). Hence, we conclude that MECs respond to exogenous mechanical force and increased resistance to cellular contractility with a chronically elevated FAK-Rho signaling loop that results in hyperactivation of the Ras-MAPK pathway, which promotes a hyper-proliferative state (Fig 9G).
DISCUSSION
Breast tissue density and the mechanical microenvironment
High breast tissue density may account for up to 30% of breast cancers (Boyd et al., 1998; Boyd et al., 2001; McCormack & dos Santos Silva, 2006), yet the molecular mechanisms driving this increased risk are not known. Importantly, increased breast density is not only associated with increased epithelial and stromal cellularity but also significantly increased collagen deposition (Alowami et al., 2003; Guo et al., 2001; Li et al., 2005). Therefore, although stromal cell populations (fibroblasts, macrophages, etc.) likely contribute to carcinoma formation and progression in dense breast tissue, the focus of the current study was to examine the influence of stromal collagen on MEC behavior and determine if increased collagen density is sufficient to promote characteristics of a malignant phenotype in nontransformed cells.
Herein, we demonstrate that greater collagen density increased matrix stiffness and that high matrix stiffness promoted proliferation and invasion associated with a malignant phenotype. High stiffness promoted formation of clustered 3D-matrix adhesions containing activated FAK, which was necessary for cells to integrate mechanical signals on stiff substrates (see Supp. Text). In addition to FAK, the cellular response to mechanical signals was also Rho and ERK dependent, and inhibition of the Rho/ROCK pathway or ERK reverted the invasive phenotype associated with HD matrices. Moreover, increased matrix density promoted growth of MECs in an ERK-dependent manner; and ERK activation was FAK/Src dependent. Consequently, the current data emphasize the importance of the mechanical microenvironment and suggest that mechanotransduction in MECs occurs through a FAK Rho ERK signaling network with ERK as a bottleneck through which much of the response to mechanical stimuli is regulated.
Of direct relevance to our findings are recent reports showing that increased resistance to cell contraction promotes a malignant phenotype (Paszek et al., 2005; Wozniak et al., 2003). Supportive of results shown herein, Paszek and co-workers (Paszek et al., 2005) showed that increasing substrate stiffness promotes integrin clustering and FA signaling, and that inhibition of ERK reverts aberrant phenotype due to constitutive-activation of Rho in soft basement membrane gels. Additionally, using unconfined compression testing to determine ECM material properties, the authors (Paszek et al., 2005) showed that increasing collagen concentration (in mixed collagen/basement membrane gels) increased matrix stiffness, which is consistent with results presented herein and data from Roeder and co-workers (Roeder et al., 2002) using tensile testing to determine the elastic modulus. We measured material properties in the tensile direction since it is well established that collagen is predominantly a tensile load bearing protein, with groups of organized fibrils mounting minimal resistance to compression or free bending ((Provenzano & Vanderby, 2006) and references therein). Moreover, when cells contract inside the matrix they face resistance from collagen deformed in the tensile direction (Provenzano et al., 2008c). Hence, it is clear that the data shown here, and in previous studies (Paszek et al., 2005; Wozniak et al., 2003), provide compelling evidence that mechanical signals have a profound influence on epithelial behavior and can promote a malignant phenotype. Importantly, we find that collagen density per se regulates the behavior of pure populations of MECs, even in the absence of stromal cells. Data suggest that MECs respond to stiff matrices with a phenotype and gene expression program consistent with malignant transformation and the in vivo phenotype associated with human breast tissue density (Boyd et al., 1998; Boyd et al., 2001; Gill et al., 2006; Habel et al., 2004; McCormack & dos Santos Silva, 2006). While the in vivo condition is likely more complex, with stromal fibroblasts also stimulated to proliferate by dense matrices and contributing in a feed-forward manner to matrix density and aberrant epithelial behavior, we propose that increased matrix stiffness explains part of the mechanism behind increased epithelial proliferation and cancer risk in human patients with high breast tissue density.
ERK as a central regulating ‘bottleneck’ in the mechanotransduction network
The ERK network transduces signals from extracellular stimuli, such as growth factors or matrix ligands, to regulate cellular processes, such as cell proliferation. In this capacity, ERK serves as a linkage between cell surface receptors and distinctly regulated intracellular targets (Dhillon et al., 2007; Yoon & Seger, 2006). Therefore, in order to carry out the task of regulating a complex variety of intracellular functions, ERK directly interacts with more than 160 known substrates (Yoon & Seger, 2006). Moreover, the MAPK cascade, in total, influences an even greater number of molecules and pathways. In the current work, we demonstrate that ERK functions as a central regulator of transcriptional response to mechanical signals. By inhibiting ERK phosphorylation, the majority of transcripts differentially expressed due to increased matrix stiffness returned to ‘normal’ levels. In addition, HD-induced proliferation was abolished and cellular protrusions into the matrix regressed, essentially reverting epithelial colonies in HD matrices to the LD phenotype. Hence, the cellular response to increased matrix stiffness appears to be largely regulated through ERK, suggesting that ERK acts as a ‘bottleneck’, or point of convergence, in a mechanically activated signaling network possessing an ‘hourglass’ architecture. As such, inhibitors that target ERK, or the ‘upstream’ regulator FAK, may prove to be viable candidates for repressing the increased risk of breast carcinoma due to high breast tissue density.
MATERIALS AND METHODS
A full list of reagents used in this study, as well as additional information for each section of the Materials and Methods can be found in the Supplementary Methods available online.
3D Cell Culture
MCF10A T47D, MDA-MB-231, and NMuMG mammary epithelial cells (MECs) were maintained as described previously (Provenzano et al., 2008c; Wozniak et al., 2003). Each cell type had an optimal collagen density for promoting tubulogenesis that was determined through preliminary experiments. The optimal density for normal growth and differentiation is designated LD (low-density), and compared to HD (high density) conditions that disrupted differentiation. Conditions for each cell type were as follows: MCF10A and T47D (LD=1.3mg/mL; HD=3.0 mg/mL), NMuMG (LD=3.0 mg/mL; HD=4.0 mg/mL).
Xenograft experiments
Mice were maintained at the University of Wisconsin under the approval and guidelines of the Institutional Animal Care and Use Committee. MDA-MB-231 cells were seeded into LD and HD collagen plugs (5*105 cells/plug). Collagen matrices were implanted into female athymic nude mice (BALB/c nu/nu mice, Charles River Laboratories). Each mouse received a LD and HD matrix above the 4th inguinal mammary glands.
Preparation of polyacrylamide substrates
Polyacrylamide substrates were prepared as described by Pelham and Wang (Pelham & Wang, 1997) and Yeung et al., (Yeung et al., 2005).
Multiphoton Laser-Scanning Microscopy
Multiphoton excitation and SHG imaging microscopy were performed as previously described (Provenzano et al., 2008b; Provenzano et al., 2008c).
Material property measurements
Mechanical testing to generate stress-strain data for collagen matrices was performed as described previously (Provenzano et al., 2003) using sample design similar to (Roeder et al., 2002).
Cell/Matrix deformation
For cell deformation on 2D surfaces, a tensile equibiaxial strain (10%) was applied to the collagen-coated substrate (105 MECs/substrate) with a FlexCellR apparatus in an incubator at 37° C with 5% CO2. For tensile loading of 3D matrices, MECs were seeded into matrices formed within Flexcell TissueTrainR culture plates. Uniaxial tensile strain (10%) was applied to the cell-seeded matrices in an incubator at 37°C with 5% CO2.
Immunofluorescence
Immunofluorescence microscopy of cells was performed as described previously (Provenzano et al., 2008b; Wozniak et al., 2003).
Morphometric Analysis
Quantitative analysis of morphology was performed in ImageJ (http://rsb.info.nih.gov/ij/). To quantify membrane protrusion, two morphological indices were generated. Membrane perimeter was quantified in the ECM-facing half of the cell. For membrane protrusion length, the three longest membrane distances from the nucleus centroid that were at least 15 degrees apart were measured. To determine FA area, images were normalized and the threshold limited area of each adhesion measured.
Immunoprecipitation and Western blotting
Immunoprecipitation was performed on cleared lysates at 4°C using 4 μg of antibody (FAK C-20 pAb (Santa Cruz) or 4.47 mAb (Upstate), Src mAb (Upstate), or species-specific control IgG antibodies in co-immunoprecipitation experiments) and 30 μl of Gammabind beads (Amersham Biosciences) per matrix sample. The samples were prepared for SDS-PAGE with the addition of Laemli buffer. Western blotting was performed as described previously (Provenzano et al., 2008a).
Rho GTPase Activation Assay
Rho-A activity was determined using G-LISA™ Luminometric Rho-A assay (Cytoskeleton) according to the manufacture’s direction, with the following exception: lysate volume was increased ≥3-fold to overcome signal loss due to a less concentrated 3D sample.
RNA Isolation and Microarray
Total RNA was isolated from mammary epithelial cells in 3D collagen matrices (LD, n=5; HD, n=5; HD+DMSO, n=5; HD+U0126, n=5) in a manner similar to a previous report (Provenzano et al., 2008a).
Data Analysis and Bioinformatics
Analysis was performed as previously described (Provenzano et al., 2008a).
Statistical Analysis
Two-group data were analyzed with t-tests. Analysis of Variance (ANOVA) followed by the Tukey-Kramer multiple comparison test was used for multi-group data.
Supplementary Material
Acknowledgments
The authors thank Dr. Caroline Alexander and Andreas Friedl for helpful discussions regarding mice and xenograft experiments, Dr Ray Vanderby, Ashish Oza, and Wilmot Valhmu for helpful discussions and equipment access for mechanical testing, and Dr. John G. White for helpful discussions regarding microscopy.
FUNDING
This work was supported by a NIH postdoctoral training grant (T32CA009681) to PPP, and grants from the DOD: W81XWH-04-1-042 (PPP), Am. Cancer Soc.: RSG-00-339CSM (PJK), and NIH: CA076537 (PJK), and EB000184 (KWE).
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare no conflict of interest.
References
- Alowami S, Troup S, Al-Haddad S, Kirkpatrick I, Watson PH. Breast Cancer Res. 2003;5:R129–35. doi: 10.1186/bcr622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd NF, Lockwood GA, Byng JW, Tritchler DL, Yaffe MJ. Cancer Epidemiol Biomarkers Prev. 1998;7:1133–44. [PubMed] [Google Scholar]
- Boyd NF, Martin LJ, Stone J, Greenberg C, Minkin S, Yaffe MJ. Curr Oncol Rep. 2001;3:314–21. doi: 10.1007/s11912-001-0083-7. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Annu Rev Cell Dev Biol. 1996;12:463–519. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Burridge K, Fath K, Kelly T, Nuckolls G, Turner C. Annu Rev Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, Simkins S, Xu L. Clin Cancer Res. 2000;6:2417–23. [PubMed] [Google Scholar]
- Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. Biochim Biophys Acta. 2007;1773:1299–310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Choquet D, Felsenfeld DP, Sheetz MP. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. J Cell Biol. 1996;133:1403–15. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Galbraith CG, Yamada KM, Sheetz MP. J Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Science. 1999;285:1028–32. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Gill JK, Maskarinec G, Pagano I, Kolonel LN. Breast Cancer Res. 2006;8:R30. doi: 10.1186/bcr1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, Khokha R, Boyd NF. Cancer Epidemiol Biomarkers Prev. 2001;10:243–8. [PubMed] [Google Scholar]
- Habel LA, Dignam JJ, Land SR, Salane M, Capra AM, Julian TB. J Natl Cancer Inst. 2004;96:1467–72. doi: 10.1093/jnci/djh260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely P, Fong A, Zutter M, Santoro S. J Cell Science. 1995;108:595–607. doi: 10.1242/jcs.108.2.595. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. J Cell Biol. 1997;137:481–92. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krouskop TA, Wheeler TM, Kallel F, Garra BS, Hall T. Ultrason Imaging. 1998;20:260–74. doi: 10.1177/016173469802000403. [DOI] [PubMed] [Google Scholar]
- Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, McLean GW, Frame MC, Muller WJ. Proc Natl Acad Sci U S A. 2007;104:20302–7. doi: 10.1073/pnas.0710091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Sun L, Miller N, Nicklee T, Woo J, Hulse-Smith L, Tsao MS, Khokha R, Martin L, Boyd N. Cancer Epidemiol Biomarkers Prev. 2005;14:343–9. doi: 10.1158/1055-9965.EPI-04-0490. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Woude GFV, Ahn NG. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Boyd NF. Breast Cancer Res. 2008;10:201. doi: 10.1186/bcr1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack VA, dos Santos Silva I. Cancer Epidemiol Biomarkers Prev. 2006;15:1159–69. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Oh IH, Reddy EP. Oncogene. 1999;18:3017–33. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pelham RJ, Jr, Wang Y. Proc Natl Acad Sci U S A. 1997;94:13661–5. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford MP, Schaller MD. Oncogene. 2004;23:7928–46. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- Plewes DB, Bishop J, Samani A, Sciarretta J. Phys Med Biol. 2000;45:1591–610. doi: 10.1088/0031-9155/45/6/314. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Beggs HE, Keely PJ. American Journal of Pathology. 2008a;173:1551–65. doi: 10.2353/ajpath.2008.080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. BMC Med. 2008b;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Biophys J. 2008c doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Martinez DA, Grindeland RE, Dwyer KW, Turner J, Vailas AC, Vanderby R., Jr J Appl Physiol. 2003;94:314–24. doi: 10.1152/japplphysiol.00340.2002. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Vanderby R., Jr Matrix Biol. 2006;25:71–84. doi: 10.1016/j.matbio.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. Cell. 1992;70:389–99. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD. J Cell Biol. 2001;153:1175–86. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. J Biomech Eng. 2002;124:214–22. doi: 10.1115/1.1449904. [DOI] [PubMed] [Google Scholar]
- Samani A, Bishop J, Yaffe MJ, Plewes DB. IEEE Trans Med Imaging. 2001;20:271–9. doi: 10.1109/42.921476. [DOI] [PubMed] [Google Scholar]
- Sarvazyan AP, Skovaroda AR, Emelianov SY, Fowlkes JB, Pipe JG, Adler RS, Buxton RB, Carson PL. Acoust Imaging. 1995;21:223–240. doi: 10.1118/1.597633. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Mol Cell Biol. 1994;14:1680–8. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Mol Cell Biol. 1996;16:5623–33. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Jones KC, Hunter T. Mol Cell Biol. 1998;18:2571–85. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniadecki NJ, Anguelouch A, Yang MT, Lamb CM, Liu Z, Kirschner SB, Liu Y, Reich DH, Chen CS. Proc Natl Acad Sci U S A. 2007;104:14553–8. doi: 10.1073/pnas.0611613104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi C, Nakayama K, Kubota M. Phys Med Biol. 2000;45:1511–20. doi: 10.1088/0031-9155/45/6/308. [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Vaque JP, Gutkind JS. Oncogene. 2007;26:3240–53. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Ursin G, Hovanessian-Larsen L, Parisky YR, Pike MC, Wu AH. Breast Cancer Res. 2005;7:R605–8. doi: 10.1186/bcr1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Wang HB, Dembo M, Hanks SK, Wang Y. Proc Natl Acad Sci U S A. 2001a;98:11295–300. doi: 10.1073/pnas.201201198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JG, Miyazu M, Matsushita E, Sokabe M, Naruse K. Biochem Biophys Res Commun. 2001b;288:356–61. doi: 10.1006/bbrc.2001.5775. [DOI] [PubMed] [Google Scholar]
- Wang JG, Miyazu M, Xiang P, Li SN, Sokabe M, Naruse K. Life Sci. 2005;76:2817–25. doi: 10.1016/j.lfs.2004.10.050. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. J Cell Biol. 1997;137:231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ML, George LK, Grant GD, Perou CM. Nat Rev Cancer. 2006;6:99–106. doi: 10.1038/nrc1802. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski P, Der CJ, Keely PJ. Journal of Cell Biology. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, Chen HC, Nowlen JK, Taylor SJ, Shalloway D, Guan JL. Mol Biol Cell. 1994;5:413–21. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.