

Abstract
Prostate cancer (PC), a complex disease, can be relatively harmless or extremely aggressive. To identify candidate genes involved in causal pathways of aggressive PC, we implemented a systems biology approach by combining differential expression analysis and co-expression network analysis to evaluate transcriptional profiles using lymphoblastoid cell lines from 62 PC patients with aggressive phenotype (Gleason grade ≥ 8) and 63 PC patients with nonaggressive phenotype (Gleason grade ≤ 5). From 13935 mRNA genes and 273 microRNAs tested, we identified significant differences in 1100 mRNAs and 7 microRNAs with false discovery rate < 0.01. We also identified a co-expression module demonstrating significant association with the aggressive phenotype of PC (p=3.67×10−11). The module of interest was characterized by over-representation of cell cycle-related genes (false discovery rate = 3.50×10−50). From this module, we further defined 20 hub genes that were highly connected to other genes. Interestingly, five of the 7 differentially expressed microRNAs have been implicated in cell cycle regulation and two (miR-145 and miR-331-3p) are predicted to target three of the 20 hub genes. Ectopic expression of these two microRNAs reduced expression of target hub genes and subsequently resulted in cell growth inhibition and apoptosis. These results suggest that cell cycle is likely to be a molecular pathway causing aggressive phenotype of PC. Further characterization of cell cycle-related genes (particularly, the hub genes) and miRNAs that regulate these hub genes could facilitate identification of candidate genes responsible for the aggressive phenotype and lead to a better understanding of PC etiology and progression.
Keywords: gene network, microRNA, systems biology, lymphoblastoid cell line, prostate cancer
Introduction
Prostate cancer (PC) remains the most commonly diagnosed non-skin cancer in men in the United States. Approximately one in three men over the age of 50 shows histological evidence of PC. However, only about 10% will be diagnosed with clinically significant PC, implying that most PCs never progress to become life-threatening. So far, little is known about what makes some PCs biologically aggressive and more likely to progress to metastastic and potentially lethal disease. PC is a complex disease, believed to be caused by variations in a large number of genes and their complex interactions. Conventional approaches used to elucidate genetic risk factors and genetic mechanisms include family-based linkage analysis, pathway-based association study and genome-wide association study (GWAS). Among these approaches, GWAS has been very successful with over a dozen single nucleotide polymorphisms (SNPs) identified with elevated risk to PC (1). However, the observed associations have yet to be translated into a full understanding of the genes or genetic elements mediating disease susceptibility. Furthermore, few PC risk variants identified from GWAS have any association with clinical characteristics. This is not surprising because these risk SNPs are identified by comparing PC cases with controls. Studies using case-case design are clearly needed to identify associations of genetic variants with aggressive PC.
Traditionally, microarray-based transcriptional profiling analysis produces massive gene lists (usually based on p-value) without consideration of potential relationships among these genes. The gene-by-gene approach often lacks a coherent picture of disease-related pathological interactions. To facilitate candidate gene discovery, there is now an increasing interest in using a systems biology approach. This approach allows for a higher order interpretation of gene expression relationships and identifies modules of co-expressed genes that are functionally related, and eventually characterizes causal pathways and genetic variants. So far, studies using the approach have successfully identified disease-related transcriptional networks and genetic variants that contribute to the disease phenotypes (2–7). For example, an early study analyzed the gene expression profiles in large population-based adipose tissue cohorts and found a marked correlation between gene expression in adipose tissue and obesity-related traits. The systems biology approach identified a core network module that was causally associated with obesity (2). This study has recently been validated through characterization of transgenic and knockout mouse models of genes predicted to be causal for obesity phenotype (7).
Expression levels of many genes show abundant natural variation in species from yeast to human (8). Studies have demonstrated significant association of genetic polymorphisms with gene expression in a variety of human cell lines and tissues (9). In addition to genetic factors, however, microRNAs (miRNA) are emerging as key players in the regulation of gene expression. miRNAs are small non-coding RNAs that control the expression of protein-coding transcripts. Each miRNA has multiple target genes that are regulated at the post-transcriptional level. They have been implicated in various diseases, and may influence tumorigenesis by acting as oncogenes and tumor suppressors. For example, the miR-17/92 cluster cooperates with c-MYC to accelerate tumor development (10, 11). Germline variations in miRNAs and their target genes have been reported to have a profound effect not only on tumor progression but also an individual’s risk of developing cancer (12, 13). Hence, miRNAs are related to diverse cellular processes and regarded as important components of the gene regulatory network.
To identify the genes that contribute to the aggressive phenotype of PC, we implemented a systems biology approach and analyzed whole genome gene expression profiles in 125 lymphoblastoid cell lines (LCLs) derived from 62 aggressive and 63 non-aggressive PC patients. We identified a set of mRNA genes and miRNAs whose expression levels were associated with not only cell cycle regulation but also aggressive nature of PC. We then verified the functional role of two miRNAs using prostate cancer cell lines. These results suggested that the cell cycle-related biological process may be genetically dysregulated in PC patients and that miRNAs may be significantly involved in development of the aggressive phenotype.
Materials and Methods
Study Subjects
The patients were selected based on our ongoing clinic-based case-control study (14, 15). The characteristics of these patients were listed in Table 1. All subject in the study provided written informed consent. The study was approved by the Mayo Clinic IRB.
Table 1.
Clinical characteristics of prostate cancer patients
| Low Grade PC N=63 |
High Grade PC N=62 |
||
|---|---|---|---|
| Patient Characteristics: | |||
| Age, median (range) Age, quartiles |
65 (44–74) | 65 (44–74) | |
| 40 – 58 | 7 (11.1) | 7 (11.3) | |
| 59 – 64 | 22 (34.9) | 22 (35.5) | |
| 65 – 69 | 22 (34.9) | 22 (35.5) | |
| 70 – 84 | 12 (19) | 11 (17.7) | |
| PSA | |||
| < 4 | 10 (15.9) | 10 (16.1) | |
| 4 – 9.9 | 34 (54) | 32 (51.6) | |
| 10 – 19.9 | 12 (19) | 9 (14.5) | |
| ≥ 20 | 7 (11.1) | 11 (17.7) | |
| Unknown | 0 | 0 | |
| Pathologic Characteristics: | |||
| Nodal Status | |||
| Negative | 62 (98.4) | 51 (82.3) | |
| Positive | 1 (1.6) | 11 (17.7) | |
| Unknown | 0 | 0 | |
| Stage | |||
| 1 or 2 | 47 (74.6) | 16 (25.8) | |
| 3 or 4 | 15 (23.8) | 35 (56.5) | |
| Unknown | 1 (1.6) | 11 (17.7) | |
| Grade | |||
| 4 | 5 (7.9) | 0 | |
| 5 | 58 (92.1) | 0 | |
| 8 | 0 | 30 (48.4) | |
| 9 | 0 | 30 (48.4) | |
| 10 | 0 | 2 (3.2) | |
Cell lines and RNA extraction for profiling analysis
Peripheral blood lymphocytes were collected from 125 Caucasian men with median age of 65 years old (range 44–74) and transformed with Epstein-Bar virus to establish immortalized cell lines. The transformed cell lines were cultured in RPMI 1640 media supplemented with 15% fetal bovine serum, and 1% penicillin/streptomycin at 37°C in humidified incubators in an atmosphere of 5% CO2. Experimental series were set up by seeding 5-ml cultures in T25 flasks. Each culture was fed with 5ml of fresh media twice a week until the cell number reached ~106 in a T75 flask. The cells were harvested and suspended in 500 µl of RNA Stabilization reagent (RNAlater) and stored at −80°C for further processing. Total RNA was extracted from each cell culture using miRNeasy Mini Kit (QIAGEN) according to the manufacturer’s guidelines. This protocol effectively recovered both mRNA and miRNA. The integrity of these total RNAs was assessed using an Agilent 2100 Bioanalyzer.
mRNA and miRNA microarrays
Illumina human-6 V2 gene expression BeadChip and microRNA expression panel (based on miRbase release 9.0) were used for mRNAs and miRNA profiling analyses, respectively (Illumina, Inc., San Diego, CA). RNA aliquot of 200ng from each cell culture was labeled and hybridized to each array using standard Illumina protocols. BeadChips (mRNA) or sample array matrices (miRNA) were scanned on an Illumina BeadArray reader. For mRNA, 30 triplicate samples, 30 duplicate samples and 65 singleton samples were run for a total of 215 expression profiles. For miRNA, there were 84 duplicate samples and 6 quadruplicate samples for a total of 192 expression profiles. Based on principal component analysis, we removed 26 individual miRNA profiles due to substantial shifts away from a main cluster. However, replicates from each of the 26 individuals were still included in the analysis as they were in the main cluster. These expression profiles have been deposited in NCBI’s Gene Expression Omnibus (GEO) with accession number GSE14794.
Data processing
We processed 215 mRNA profiles from a total of 125 independent patients and 166 miRNA profiles from a total of 90 independent patients. For both mRNA and miRNA data, raw data from BeadStudio (Illumina, San Diego, CA) were first transformed using a variance stabilization transformation algorithm (16) and then normalized using quantile normalization. We averaged samples with replicates and excluded probes with median detection p value ≥ 0.01 (the p values were generated in BeadStudio software). This procedure reduced the number of mRNA probes from 48702 to 13935 and miRNA probes from 736 to 366. Among the 366 miRNAs, 273 in miRBase database1 version 9.1 were included in the study. The remaining 93 that were putative miRNAs identified in a RAKE analysis were excluded from further analysis.
Data analysis
The pathological grades (Gleason Score) <= 5 and >=8 were used to dichotomize samples into low grade (non-aggressive) and high grade (aggressive) groups. We applied a two sample t-test with multiple testing correction to identify genes and miRNAs that were significantly differentially expressed between the two Gleason grade groups. We defined q-value of false discovery rate (FDR) < 0.01 to be statistically significant. Pearson correlation coefficients were also calculated in order to compare results from the following network analysis.
To explore the phenotype-related genes and their interactions, we applied a systems biology approach using a weighted gene co-expression network analysis (WGCNA) (17–20). Unlike other gene co-expression networks using a binary variable to encode gene co-expression (connected=1, unconnected=0), the WGCNA converts co-expression measures into connection weights or topology overlap measures (TOM). Because the program was computationally intensive when running on large numbers of genes we simplified the computation by selecting a subset of genes for analysis. We selected the genes in two steps: first, we selected the genes that showed significant correlation with PC grade (FDR<0.01); from the rest of genes, we then selected the top 2000 most variable genes based on coefficient of variance. We inputted expression profiles of these selected genes to construct weighted gene co-expression modules using the WGCNA R package (18, 19, 21). We defined modules using static method by hierarchically clustering the genes using 1-TOM as the distance measure with a height cutoff = 0.95 and a minimum size (gene number) cut-off = 40 for the resulting dendrogram.
To identify which module is correlated with clinical phenotype, we first calculated module eigengene (ME; i.e., first principal component of the expression values across subjects) using all genes in each module. We then correlated the MEs to PC grade using the Pearson correlation. We determined intramodular connectivity for each gene by summing the connectivities of that gene with each other gene in that module. We used program VisANT (Integrative Visual Analysis Tool for Biological Networks and Pathways) (22) to construct gene-gene interaction (connections) networks.
Gene ontology analysis
To explore whether genes in each target group share a common biological function, we searched for over-representation in gene ontology (GO) categories. We used 13935 mRNA accession numbers as reference gene list. We inputted each group of genes into DAVID (The Database for Annotation, Visualization and Integrated Discovery) for GO term enrichment analysis. The DAVID is a program that checks for an enrichment of genes with specific GO, KEGG, and SwissProt terms (23).
Nucleofection of miRNA mimics in VCaP and LNCaP cells
We cultured LNCaP cells (24) in RPMI 1640 and VCap (25) cells in Dulbecco's modified Eagle's medium, respectively. Both cell lines were grown in the media containing 10% fetal bovine serum, 1% penicillin and streptomycin at 37°C with 5% CO2. Cells were nucleofected with double stranded synthetic microRNA mimics (syn-hsa-miR-145 miScript miRNA and syn-hsa-miR-331 miScript miRNA) and scrambled controls (Qiagen, Germantown, MD) using program T-09 (Lonza, Cologne, Germany). Nucleofection efficiency was monitored by nucleofecting the cells with 2.0 µg of pmaxGFP plasmid DNA in 6 well plates. Cells were visualized and tested at 48 hrs after nucleofection.
Cell viability assay and FACS
After nucleofection, cells were placed on 24 well plates. Media were changed twice after 10 hrs of plating and then once every 24 hrs. Cell viability of treated cells was examined using LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen, Eugene, OR) after 48 hrs treatment and visualized using a fluorescent microscope (100 x) after 15 min staining. FACS analysis was performed using a FACSCalibur Flow Cytometer (Becton Dikinson) following the method of Riccardi and Nicoletti (26).
qRT-PCR
Expression level of target genes were quantified at 48 hrs after treatment by qRT-PCR using the Lightcycler 480 SYBR Green I master mix (Roche, Indianapolis, IN) in an ABI 7500 real time PCR system. Primer sequences were listed in Supplementary Table 1. GAPDH expression level was used as normalization control. Relative expression values were calculated following the 2 −ΔΔ Ct method of Schmittgen and Livak (27) using values from 3 independent experiments.
Results
Correlation between transcripts and pathological grades
To identify transcripts whose expression traits were associated with aggressive phenotype of PC, we applied a two sample t-test using 13935 detectable gene expression profiles in 62 high grade and 63 low grade PC cases. Among all genes tested, we found significant association in 1100 genes (FDR <0.01). For the 125 PC cases, 90 (45 high-grade and 45 low-grade cases) were also available for miRNA profiling analysis. The two sample t-test using 273 detectable miRNA expression profiles identified significant association with PC grade in 7 miRNAs (FDR<0.01) (Supplementary Table 2). The 7 miRNAs included miR-222, miR-221, miR-331-3p, miR-16, miR-145, miR-9* and miR-551a. Because miR-9 and miR-9* are processed from the same precursor, we also observed an association of miR-9 with the PC grade (FDR=0.013). However, we did not find any association of miR-15a with PC grade (p=0.65) although miR-15a and miR-16 are located in the same miRNA cluster.
To functionally classify these 1100 significant genes, we used the online biological classification tool DAVID (23) and observed significant enrichment of these genes in multiple GO categories. The most significant enrichment was the GO category of cell cycle biological process with FDR=3.40×10−23. The other significant GO categories included DNA replication (FDR=1.60×10−13) and chromosome (FDR=2.10×10−13). In fact, all significant GO category clusters were related to cell cycle biological function (Supplementary Table 3).
In an effort to provide additional evidence to support our initial observation, we downloaded gene expression profiles from another study with benign prostate tissues (28). After obtaining the relevant clinical information, we re-analyzed the Affymetrix U95av2-based expression profiles derived from 5 benign prostate tissues in patients with aggressive phenotype (Gleason Score ≥8) and 4 benign prostate tissues in patients with non-aggressive phenotype (Gleason Score ≤5). Statistical analysis using t-test revealed significant difference in 1847 RNA probes (p<0.05). Interestingly, GO analysis of these differential genes showed that cell cycle regulation was the most significantly enriched GO category with p=2.97×10−5 (FDR=0.056) (Supplementary Table 3). We further analyzed these differentially expressed genes and found significant overlap between the benign tissues and the cell lines (p<0.01).
Gene co-expression networks and biological pathways
Because co-expressed genes are biologically related, grouping these highly connected genes by network analysis may shed light on underlying functional processes in a manner complementary to standard differential expression analyses. To ensure that phenotype-related genes were used to construct the network, we included the 1100 most significant genes with FDR <0.01 along with the top 2000 most variable genes (selected from remaining 12835 genes) determined by their coefficient of variance. The WGCNA analysis identified four modules of genes with high topological overlap (Figure 1). The modules were defined as a cluster of highly connected genes (nodes). Each major branch in the figure represented a color-coded module containing a group of highly correlated genes. The modules turquoise, brown, blue and yellow included 265, 106, 229 and 65 genes, respectively.
Figure 1. Gene co-expression network analysis.
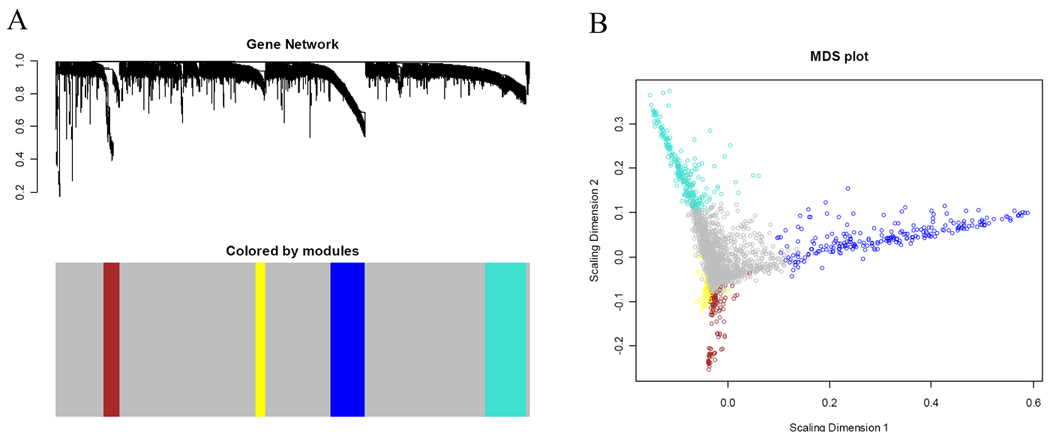
A. Branches (gene modules) of highly correlated genes by average linkage hierarchical clustering of 3100 genes. The colored bars directly corresponded to the module (color) designation for the clusters of genes. Grey denoted genes that were not part of any module. The remaining colors were used for the four modules. B. Multi-dimensional scaling plot of the entire gene expression network. Each dot represented a gene, where the color corresponded to the gene module. The distance between each dot indicated their topological overlap.
To examine if these modules were associated with aggressive PC, we correlated the module eigengene to the Gleason grade and found significant correlation of the PC grade only with the turquoise module (p=3.67×10−11). The other three modules did not show any correlation (all p>0.05). To biologically characterize those modules, we applied the DAVID tool (23) to classify these genes in each module and observed various level of GO category enrichment in all 4 modules (Table 2). Specifically, the PC grade-related turquoise module demonstrated significant enrichment in the biological process of cell cycle (FDR=3.50×10−50). The blue module showed over-representation in protein acetylation (FDR=8.21×10−7). The brown and yellow modules show a strong trend but not statistical significance (FDR>0.01) for GO category enrichment.
Table 2.
Module significance in aggressive PC and GO analysis
| Correlation with Pathological Grade |
DAVID GO Analysis | ||||||
|---|---|---|---|---|---|---|---|
| Modules | Total Gene Count |
r | p-value | Term | Gene Count |
p-value | FDR |
| Turquoise | 265 | 0.548 | 3.67E-11 | Cell Cycle | 97 | 1.86E-53 | 3.50E-50 |
| Brown | 106 | −0.08 | 0.377 | Nuclear Pore Complex Interacting |
3 | 2.46E-04 | 0.464 |
| Blue | 229 | 0.058 | 0.521 | Acetylation | 30 | 5.27E-10 | 8.21E-07 |
| Yellow | 65 | 0.106 | 0.241 | Membrane | 27 | 2.78E-04 | 0.422 |
Clinical trait-related hub genes
The importance of a gene is often dependent on how well it associates with other genes in a network. Studies suggest that more centralized genes in the network are more likely to be key drivers to proper cellular function than peripheral genes (nodes) (18). These centralized genes are called hub genes, implying that they are highly connected genes. Intramodular hub genes are defined based on their high correlation with the module eigengene, i.e. as a good representative of a module. We focused our analysis on genes in the turquoise module because of its relevance to clinical trait (Table 2). We used the WGCNA algorithm to calculate intramodular connectivity (connection strength of a given gene with other genes in a particular module). To visualize the relationship between gene significance and intramodular connectiviy, we plotted scaled connectivity on x-axis and gene significance (absolute correlation coefficient r value between gene expression and PC grade) on y axis. We observed significant positive correlation (r = 0.61, p = 7.1×10−19) (Figure 2A). The genes with higher connectivity tended to have stronger correlation with PC grade, suggesting a potentially important role of highly connected genes (hub genes) in the aggressive phenotype of PC.
Figure 2. Identification of clinical trait-related hub genes.
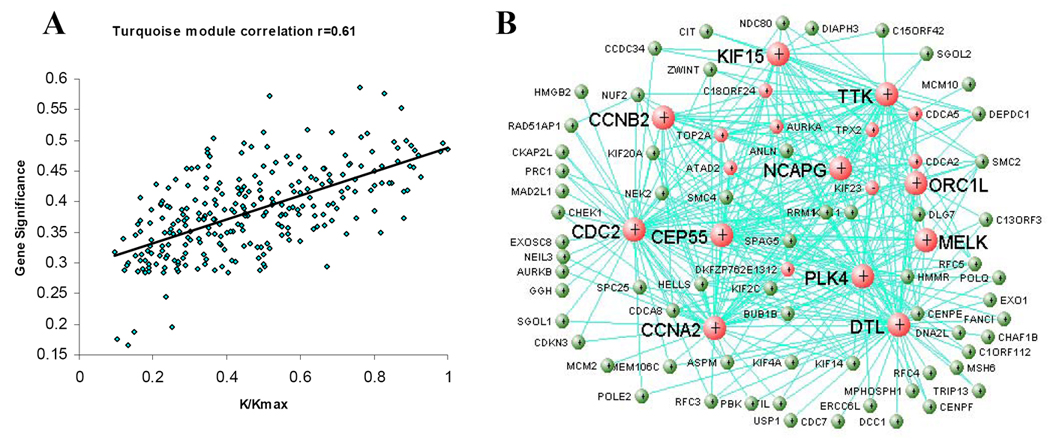
A. Scatterplot between gene significance (absolute r) (y-axis) and scaled intramodular connectivity (K/Kmax). Each point corresponded to a gene in the turquoise module. The intramodular connectivity was significantly correlated with gene significance (r = 0.61, p = 7.1 × 10−19). B. Visualization of gene-gene interaction within turquoise module. The connections were drawn using VisANT tool (ref 22). The genes with at least one connection when weighted cutoff value >=0.16 were shown. Each node represented a gene. Red nodes were hub genes. Bigger nodes indicated more connections.
To further visualize gene-gene interactions, we exported the WGCNA-generated connectivity information to the VisANT (22) and observed various degrees of gene-gene connections (interactions). We raised the weighted cutoff value to >=0.16 to identify hub genes with the strongest connections with other genes. The raised cutoff reduced the total number of connections per gene. Under this criterion, we observed 84 genes, each with at least one connection, and 20 genes, each with at least 10 connections (Figure 2B). We defined the 20 highly connected genes as hub genes. The genes CDC2 and DTL were the strongest, each with 55 connections, while CCNA2 had 50. More importantly, all 20 hub genes not only showed significant correlation with pathological grade but also have been implicated in cell cycle-related functions (Table 3).
Table 3.
Connectivity and gene significance of 20 selected hub genes
| Symbol | Gene Name | Accession Number |
Number of Connections |
Gene Significance* | |||
|---|---|---|---|---|---|---|---|
| r | p | FDR | Rank ** | ||||
| CDC2 | cell division cycle 2 | NM_001786 | 55 | 0.495 | 4.49E-09 | 8.92E-07 | 43 |
| DTL | denticleless homolog | NM_016448 | 55 | 0.485 | 1.00E-08 | 1.68E-06 | 51 |
| CCNA2 | cyclin A2 | NM_001237 | 50 | 0.482 | 1.30E-08 | 2.03E-06 | 54 |
| PLK4 | polo-like kinase 4 | NM_014264 | 48 | 0.441 | 2.67E-07 | 1.78E-05 | 128 |
| TTK | TTK protein kinase | NM_003318 | 40 | 0.475 | 2.26E-08 | 2.91E-06 | 66 |
| CEP55 | centrosomal protein 55kDa | NM_018131 | 35 | 0.419 | 1.16E-06 | 5.32E-05 | 186 |
| KIF15 | kinesin family member 15 | NM_020242 | 26 | 0.484 | 1.05E-08 | 1.71E-06 | 52 |
| CCNB2 | cyclin B2 | NM_004701 | 20 | 0.416 | 1.40E-06 | 6.11E-05 | 196 |
| ORC1L | origin recognition complex, subunit 1-like |
NM_004153 | 19 | 0.491 | 6.27E-09 | 1.16E-06 | 46 |
| MELK | maternal embryonic leucine zipper kinase |
NM_014791 | 17 | 0.500 | 2.82E-09 | 6.17E-07 | 39 |
| NCAPG | non-SMC condensin I complex, subunit G |
NM_022346 | 17 | 0.373 | 1.84E-05 | 4.36E-04 | 360 |
| HJURP | Holliday junction recognition protein |
NM_018410 | 14 | 0.487 | 8.30E-09 | 1.45E-06 | 49 |
| Ska1 | spindle and KT associated 1 | NM_145060 | 13 | 0.370 | 2.12E-05 | 4.81E-04 | 376 |
| TPX2 | TPX2, microtubule- associated, homolog |
NM_012112 | 13 | 0.335 | 1.36E-04 | 1.89E-03 | 613 |
| TOP2A | topoisomerase (DNA) II alpha 170kDa |
NM_001067 | 12 | 0.497 | 3.74E-09 | 7.78E-07 | 41 |
| CDCA5 | cell division cycle associated 5 |
NM_080668 | 12 | 0.431 | 5.31E-07 | 2.95E-05 | 154 |
| KIF23 | kinesin family member 23 | NM_004856 | 12 | 0.379 | 1.29E-05 | 3.28E-04 | 334 |
| CDCA2 | cell division cycle associated 2 |
NM_152562 | 11 | 0.392 | 6.11E-06 | 1.90E-04 | 274 |
| ATAD2 | ATPase family, AAA domain containing 2 |
NM_014109 | 11 | 0.373 | 1.86E-05 | 4.40E-04 | 361 |
| AURKA | aurora kinase A | NM_198436 | 10 | 0.379 | 1.34E-05 | 3.37E-04 | 340 |
Gene Significance represents statistical significance of Pearson correlations between a specific gene expression and pathological grade of PC.
Rank is based on FDR value among 13935 genes with most significant gene as 1.
Hub genes as miRNA targets
Because each miRNA may regulate multiple mRNA genes, we asked if the expression traits in hub genes were the result of regulatory effects from miRNAs. To explore this, we downloaded all miRNA target genes predicted by TargetScan (29–31). We focused our search on the 20 hub genes and the 7 differential miRNAs. We found that three of the 20 hub genes were the predicted targets for two differentially expressed miRNAs. The three hub genes CCNA2, CDCA5 and KIF23 were significantly up-regulated in aggressive PC (Table 3). The miR-145, significantly down-regulated in aggressive PC, was predicted to bind to 3’ UTR of the CCNA2. The miR-331-3p, also significantly down-regulated in aggressive PC, was predicted to target the genes CDCA5 and KIF23. More interestingly, we observed significant correlation in expression level for each of these miRNA-gene pairs. The miR-145:CCNA2 pair showed inverse correlation with p=1.48×10−4. The miR-331-3p:CDCA5 and miR-331-3p:KIF23 pairs demonstrated inverse correlation with p=2.25×10−4 and p=0.029, respectively.
Functional evaluation of miR-145 and miR-331-3p in vitro
To evaluate the potential regulatory roles of miR-145 and miR-331-3p, we ectopically expressed these miRNAs in prostate cancer cell lines LNCaP (24) and VCaP (25). We found that ectopic expression of the miR-145 reduced the CCNA2 level by 54% in VCaP cells and 45% in LNCaP cells. Ectopic expression of the miR-331 reduced the CDCA5 level by 44 % in VCaP and 48% in LNCaP cells, and the KIF23 level by 43 % in VCaP and 44 % in LNCaP cells (Figure 3A). To investigate the functional consequences of ectopic expression of these miRNAs, we examined cell viability using a flow cytometer. Gene transfer efficiency was monitored in GFP transfected control groups and ~ 80% of transfection was observed in both prostate cancer cell lines. We found significant cell growth arrest and apoptosis by the expression of these miRNAs. Specifically, the miR-145 and miR-331 ectopic expression induced 37% and 39% apoptosis in the VCaP cells; and 32% and 33% apoptosis in LNCaP cells respectively. In contrast, scrambled cells didn’t show any significant apoptosis (Figure 3B and 3C).
Figure 3. Biological effect of ectopic expression of miR-145 and miR-331 in prostate cancer cell lines.
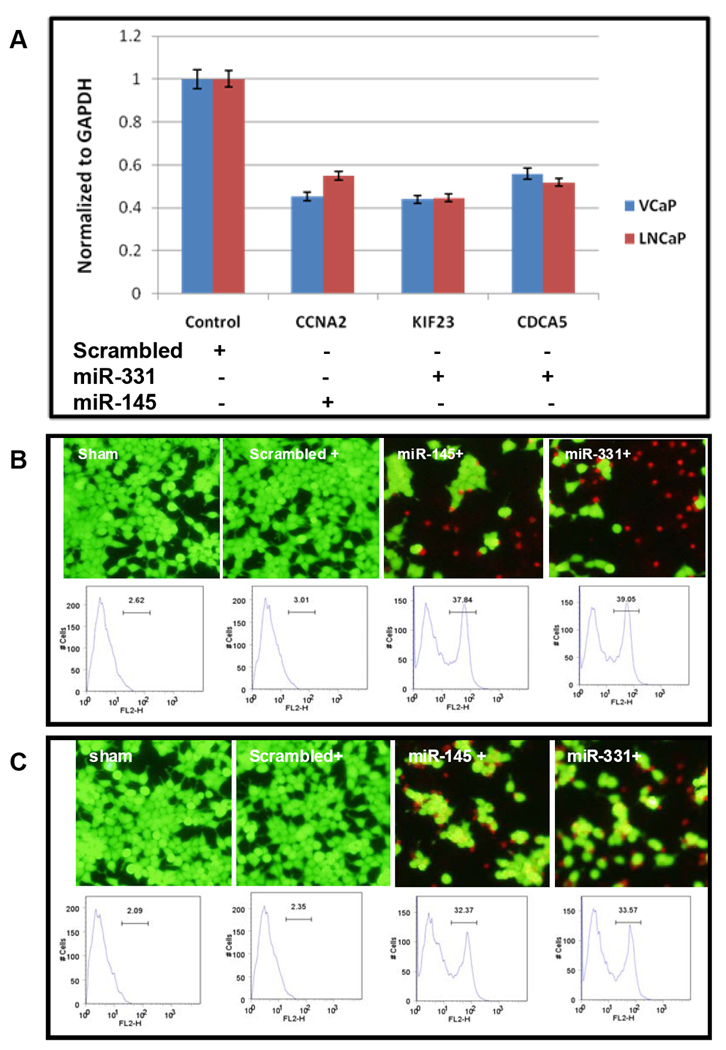
A. qRT-PCR was used to measure expression level of target genes using total RNA from nucleofected cells. Expression values were normalized to GAPDH. Expression levels of target genes were significantly reduced by ectopic expression of the two miRNAs. Cell viability was examined in VCaP cells (B) and LNCaP cells (C). Live and dead cells were stained in green and red, respectively. Percentage of apoptotic cell population measured by FACS was shown below each corresponding cell image.
Discussion
Clinical phenotypes of PC vary from an indolent disease requiring no treatment to one in which tumors metastasize and escape local therapy even when with early detection. Identification of candidate genes for aggressive PC has been a difficult task. In this study, we applied a systems biology approach to study the aggressive phenotype of PC. This approach utilized gene expression profiles and organized genes into modules based on co-expression. By examining expression profiles in 125 lymphoblastoid cell lines derived from PC patients, we observed four co-expression modules. Importantly, one of four modules not only enriched genes known to play critical roles in cell cycle regulation but also demonstrated significant correlation with aggressive phenotype of PC. These results, along with results from benign prostate tissues (Supplementary Table 2), strongly suggested that germline variations of cell cycle-related genes may be a major cause to aggressive PC.
Hub genes are believed to play major roles in a highly interacted network. In this study, we have defined 20 highly connected hub genes in an aggressive PC-associated module. Further data mining revealed significant involvement of these hub genes in the cell cycle regulation and the development of various tumors. For example, the gene CDC2 (connected to 55 other genes) is essential for G1/S and G2/M phase transitions of eukaryotic cell cycle. Aberrant activation of the CDC2 may contribute to tumorigenesis by promoting cell proliferation and survival (32). The gene DTL (55 connections) plays important roles in DNA synthesis, cell cycle progression, cytokinesis, proliferation, and differentiation (33). The DTL may regulate p53 polyubiquitination (34) and CDT1 proteolysis in response to DNA damage (35) and may also be essential for early G2/M checkpoint (36). Suppression of the DTL causes accumulation of G(2)/M cells, resulting in growth inhibition of cancer cells(37). The gene CCNA2 (50 connections) belongs to the highly conserved cyclin family. The gene is expressed in all tissues and binds/activates CDC2 kinases, and thus promotes both cell cycle G1/S and G2/M transitions. Overexpression of the gene was associated with high grade (38) and poor prognosis (39) in breast cancer. These data strongly suggest that dys-regulation of these cell cycle-related hub genes may be crucial for the development of aggressive phenotype of PC.
It is worthwhile to mention that none of the 20 hub genes were among the top gene list identified by differential gene expression analysis (Supplementary Table 2). The hub genes with the greatest and least statistical significance are MELK (FDR= 6.17×10−7) and TPX2 (FDR= 1.89×10−3), respectively. The MELK is ranked 39th and the TPX2 is ranked 613th in differential analysis (Table 3). Depending on the purpose of a study, a top gene list approach (based on differential expression p-value) will be more suitable for biomarker discovery because this type of study is directed at finding disease markers. However, for an understanding of etiology, simply selecting top differential genes identified by two sample t-test (or similar methods) may miss important genes. Therefore, a systems biology-based network analysis may provide an important alternative and more meaningful tool for candidate gene discovery.
miRNA has been emerged as a crucial regulator of gene expression. In this study, we identified 7 differentially expressed miRNAs, five of which have been implicated in regulation of cell cycle. For example, the top two miRNAs (miR-222/221) directly targeted cell growth suppressive cyclin-dependent kinase inhibitors p27 and p57 mRNAs, and reduce their protein levels (40, 41). Ectopic expression of the miR-222/221 also resulted in activation of CDK2 and facilitation of G1/S phase transition (42), which agreed with our present study: significant increases of the miR-222/221 (FDR<= 4.73×10−6) as well as the CDK2 (FDR=7.79×10−4) in aggressive PC. The target gene p27 (CDKN1B), however, only showed slightly decreased expression (mean=8.78 in high grade and 8.79 in low grade on log2 scale, p=0.79). The lack of significant decrease in the p27 may be explained by the fact that the miRNAs regulate the target gene at the posttranscriptional level. Another target gene p57 (CDKN1C) was undetectable in our lymphoblastoid cell lines and therefore was not included in the analysis.
Important role of the miR-222/221 in aggressive PC was recently confirmed by in vivo and in vitro studies. For example, in vivo overexpression of miR-221 was able to confer a high growth advantage to LNCaP-derived tumors in SCID mice while anti-miR-221/222 treatment in the highly aggressive PC3 cell line reduced tumor growth (43). Furthermore, up-regulation of these two miRNAs in PC-derived primary cell lines showed significant inverse correlation with the p27 expression. Additionally, both in vitro and in vivo results implicated that p21 and p27 had compensatory roles in advanced prostate cancer cells, and down-regulation of both these molecules essentially enhanced the aggressive phenotype (44). These results suggest that the miR-221/222 may contribute to the oncogenesis and progression of PC through p27(Kip1) down-regulation.
The other three miRNAs that affect cell cycle regulation include miR-16, miR-145 and miR-331. The miR-16 can trigger an accumulation of cells in G0/G1 by silencing multiple cell cycle genes simultaneously (45, 46) and negatively regulate two other targets HMGA1 and CAPRIN1 involved in cell proliferation (47). In our data set, we observed up-regulation of the miR-16 and down-regulation of the target genes HMGA1 and CAPRIN1. Particularly, expression difference of the HMGA1 was statistically significant (mean=7.83 in high grade and 7.90 in low grade, FDR=0.007). The miR-145 showed inhibition of tumor cell growth by direct silencing c-Myc (48). The MYC is an oncogenic, nuclear phosphoprotein that plays a key role in cell cycle progression, apoptosis and cellular transformation. Down-regulation of the miR-145 in aggressive PC was consistent with up-regulation of the MYC in the same sample set (mean=11.39 in high grade and 11.30 in low grade, p=0.04, FDR=0.10). Consequently, we observed significant up-regulation of Myc-regulated miRNAs (11) including miR-363 (FDR=0.016), miR-92a (FDR=0.022), miR20b (FDR=0.028) and miR-18b (FDR=0.030). Additionally, our previous study demonstrated that miR-331 was significantly associated with cell cycle-related genes (49). By ectopic expression of the miR-145 and miR-331-3p, the current study demonstrated significant reduction of corresponding target genes, inhibition of cell growth and accumulation of apoptotic cells (Figure 3). These findings suggest that differential expression of these miRNAs at germline level may dys-regulate target hub genes which could lead to an abnormal cell division and proliferation, and eventually developing an aggressive phenotype of PC.
Overall, this study used a systems biology approach to identify genes that are potentially involved in the aggressive phenotype of PC. This approach moves beyond single gene investigation to provide a systems level perspective on the potential relationships between members of a network. Our results strongly suggest that dys-regulation of cell cycle may significantly contribute to the deadly form of PC. These findings are important not only because we have discovered a candidate pathway and related hub genes but also because we have identified candidate miRNAs and their predicted target genes. Further studies are needed to determine genetic causes of expression alterations in both differentially expressed miRNAs and mRNA genes. Additional functional studies will determine whether variations in the selected hub genes and miRNAs are attributable to the aggressive nature of PC. These studies will facilitate candidate gene discovery and lead to better understanding of the aggressive phenotype of PC, a more clinically relevant form of the disease.
Supplementary Material
Acknowledgments
We also thank Drs. Lihua Li and Aaron Sarver for their comments and suggestions.
This study is supported from NIH grant CA126786 (L.W.), Mayo Clinic SPORE in prostate cancer, NIH CA91956 (L.W.) and Mayo Clinic Comprehensive Cancer Center Shared Resource grant (P30 CA15083). V.T is supported by grant funding from the Academic Health Center, University of Minnesota. Grant support from the Laboratory Medicine and Pathology, University of Minnesota to S.S is duly acknowledged.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Witte JS. Prostate cancer genomics: towards a new understanding. 2009;10(2):77–82. doi: 10.1038/nrg2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Zhu J, Lum PY, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452(7186):429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18(6–7):463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghazalpour A, Doss S, Zhang B, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2(8):e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci. 2008;28(6):1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ray M, Ruan J, Zhang W. Variations in the transcriptome of Alzheimer's disease reveal molecular networks involved in cardiovascular diseases. Genome Biol. 2008;9(10):R148. doi: 10.1186/gb-2008-9-10-r148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, Deignan JL, Qi H, et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41(4):415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452(7186):423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 9.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10(3):184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435(7043):828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 13.Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2008;105(20):7269–7274. doi: 10.1073/pnas.0802682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, McDonnell SK, Hebbring SJ, et al. Polymorphisms in mitochondrial genes and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17(12):3558–3566. doi: 10.1158/1055-9965.EPI-08-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, McDonnell SK, Slusser JP, et al. Two common chromosome 8q24 variants are associated with increased risk for prostate cancer. Cancer Res. 2007;67(7):2944–2950. doi: 10.1158/0008-5472.CAN-06-3186. [DOI] [PubMed] [Google Scholar]
- 16.Lin SM, Du P, Huber W, Kibbe WA. Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res. 2008;36(2):e11. doi: 10.1093/nar/gkm1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS computational biology. 2008;4(8):e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horvath S, Zhang B, Carlson M, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103(46):17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 20.Oldham MC, Konopka G, Iwamoto K, et al. Functional organization of the transcriptome in human brain. Nature neuroscience. 2008;11(11):1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24(5):719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 22.Hu Z, Snitkin ES, DeLisi C. VisANT: an integrative framework for networks in systems biology. Briefings in bioinformatics. 2008;9(4):317–325. doi: 10.1093/bib/bbn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Lovci MT, Kwon YS, Rosenfeld MG, Fu XD, Yeo GW. Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model. Proc Natl Acad Sci U S A. 2008;105(51):20179–20184. doi: 10.1073/pnas.0807121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korenchuk S, Lehr JE, L MC, et al. VCaP, a cell-based model system of human prostate cancer. In vivo (Athens, Greece) 2001;15(2):163–168. [PubMed] [Google Scholar]
- 26.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- 27.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 28.Singh D, Febbo PG, Ross K, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1(2):203–209. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 29.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 32.Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008;27(34):4733–4744. doi: 10.1038/onc.2008.104. [DOI] [PubMed] [Google Scholar]
- 33.Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle. 2006;5(22):2676–2687. doi: 10.4161/cc.5.22.3500. [DOI] [PubMed] [Google Scholar]
- 34.Banks D, Wu M, Higa LA, et al. L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle. 2006;5(15):1719–1729. doi: 10.4161/cc.5.15.3150. [DOI] [PubMed] [Google Scholar]
- 35.Higa LA, Banks D, Wu M, Kobayashi R, Sun H, Zhang H. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle. 2006;5(15):1675–1680. doi: 10.4161/cc.5.15.3149. [DOI] [PubMed] [Google Scholar]
- 36.Sansam CL, Shepard JL, Lai K, et al. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 2006;20(22):3117–3129. doi: 10.1101/gad.1482106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueki T, Nishidate T, Park JH, et al. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene. 2008;27(43):5672–5683. doi: 10.1038/onc.2008.186. [DOI] [PubMed] [Google Scholar]
- 38.Aaltonen K, Ahlin C, Amini RM, et al. Reliability of cyclin A assessment on tissue microarrays in breast cancer compared to conventional histological slides. Br J Cancer. 2006;94(11):1697–1702. doi: 10.1038/sj.bjc.6603147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husdal A, Bukholm G, Bukholm IR. The prognostic value and overexpression of cyclin A is correlated with gene amplification of both cyclin A and cyclin E in breast cancer patient. Cell Oncol. 2006;28(3):107–116. doi: 10.1155/2006/721919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galardi S, Mercatelli N, Giorda E, et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem. 2007;282(32):23716–23724. doi: 10.1074/jbc.M701805200. [DOI] [PubMed] [Google Scholar]
- 41.Medina R, Zaidi SK, Liu CG, et al. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68(8):2773–2780. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YK, Yu J, Han TS, et al. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37(5):1672–1681. doi: 10.1093/nar/gkp002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercatelli N, Coppola V, Bonci D, et al. The inhibition of the highly expressed miR-221 and miR-222 impairs the growth of prostate carcinoma xenografts in mice. PLoS ONE. 2008;3(12):e4029. doi: 10.1371/journal.pone.0004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy S, Singh RP, Agarwal C, Siriwardana S, Sclafani R, Agarwal R. Downregulation of both p21/Cip1 and p27/Kip1 produces a more aggressive prostate cancer phenotype. Cell Cycle. 2008;7(12):1828–1835. doi: 10.4161/cc.7.12.6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linsley PS, Schelter J, Burchard J, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol. 2007;27(6):2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Q, Fu H, Sun F, et al. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008;36(16):5391–5404. doi: 10.1093/nar/gkn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaddar T, Rouault JP, Chien WW, et al. Two new miR-16 targets: caprin-1 and HMGA1, proteins implicated in cell proliferation. Biol cell. 2009;101(9):511–524. doi: 10.1042/BC20080213. [DOI] [PubMed] [Google Scholar]
- 48.Sachdeva M, Zhu S, Wu F, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106(9):3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Oberg AL, Asmann YW, et al. Genome-wide transcriptional profiling reveals microRNA-correlated genes and biological processes in human lymphoblastoid cell lines. PLoS One. 2009;4(6):e5878. doi: 10.1371/journal.pone.0005878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.