

Abstract
Upregulation of expression of the close homolog of adhesion molecule L1 (CHL1) by reactive astrocytes in the glial scar reduces axonal regeneration and inhibits functional recovery after spinal cord injury (SCI). Here we investigate the molecular mechanisms underlying upregulation of CHL1 expression by analyzing the signal transduction pathways in vitro. We show that astrogliosis stimulated by bacterial lipopolysaccharide (LPS) upregulates CHL1 expression in primary cultures of mouse cerebral astrocytes, coinciding with elevated protein synthesis and translocation of protein kinase δ (PKCδ) from cytosol to the membrane fraction. Blocking PKCδ activity pharmacologically and genetically attenuates LPS-induced elevation of CHL1 protein expression through a phosphatidylinositol 3-kinase (PI3K) dependent pathway. LPS induces extracellular signal-regulated kinases (ERK1/2) phosphorylation through PKCδ and blockade of ERK1/2 activation abolishes upregulation of CHL1 expression. LPS-triggered upregulation of CHL1 expression mediated through translocation of nuclear factor κB (NF-κB) to the nucleus is blocked by a specific NF-κB inhibitor and by inhibition of PI3K, PKCδ, and ERK1/2 activities, implicating NF-κB as a downstream target for upregulation of CHL1 expression. Furthermore, the LPS-mediated upregulation of CHL1 expression by reactive astrocytes is inhibitory for hippocampal neurite outgrowth in cocultures. Although the LPS-triggered NO-guanylate cyclase-cGMP pathway upregulates GFAP expression in cultured astrocytes, we did not observe this pathway to mediate LPS-induced upregulation of CHL1 expression. Our results indicate that elevated CHL1 expression by reactive astrocytes requires activation of PI3K/PKCδ-dependent pathways and suggest that reduction of PI3K/PKCδ activity represents a therapeutic target to downregulate CHL1 expression and thus benefit axonal regeneration after SCI.
Keywords: astrocytes, lipopolysaccharide, close homolog of L1, PI3K, PKCδ, ERK1/2, NF-κB, NO
INTRODUCTION
Adhesive receptors at the cell surface play important roles in cell communication not only during ontogenetic development, but also in the adult during modulation of synaptic efficacy and regeneration during or after acute or chronic injury of the central and peripheral nervous systems (Loers and Schachner 2007; Maness and Schachner 2007). Knowledge of signal transduction mechanisms are thus important in probing for a recognition molecule's ability to influence cellular responses to stimuli from its immediate environment and in integrating signal transduction events elicited within a cell by other cell surface receptors. Also, synthesis and degradation of a recognition molecule elicited, for instance, by neurotrophins or pro- and anti-inflammatory cytokines are important for understanding and, consequently, modulating the communication between cells. This knowledge serves to improve the beneficial targeting of recognition molecule expression in cases where these molecules exert adverse effects on regeneration after an acute lesion.
We have become interested in the functional role of the immunoglobulin superfamily adhesion molecule CHL1, a close homolog of the adhesion molecule L1 (Chaisuksunt et al. 2000a; Chen et al. 1999; Dong et al. 2002; Hillenbrand et al. 1999; Holm et al. 1996; Leshchyns'ka et al. 2006; Morellini et al. 2007; Nikonenko et al. 2006; Pratte et al. 2003; Rolf et al. 2003; Zhang et al. 2000) in spinal cord regeneration since it is implicated in reduced regeneration in the compression-lesioned spinal cord of the adult mouse (Jakovcevski et al. 2007). This effect was unexpected since CHL1 had previously been shown to promote neurite outgrowth, neuronal survival and migration in vitro (Buhusi et al. 2003; Chen et al. 1999). In CHL1 deficient mice, functional recovery, as assessed by locomotor rating and video-based motion analyses, was improved compared to wild-type littermate control mice. This improved functional recovery was associated with enhanced monoaminergic reinnervation of the caudal part of the spinal cord and an altered pattern of post-traumatic synaptic rearrangements at cell bodies of spinal cord motoneurons. Restricted recovery of wild-type mice was likely related to early and persistent (up to 2 months after the lesion) upregulation of CHL1 expression by glial fibrillary acidic protein-positive astrocytes at the lesion site. To understand the apparently adverse effects of CHL1 on axonal regrowth in vivo, in vitro experiments were carried out to analyze whether the presence of CHL1 at the cell surface of reactive astrocytes or at the cell surface of neurons mediated this effect. To this aim, homogenotypic and heterogenotypic co-cultures of neurons and astrocytes isolated from CHL1-deficient and wild-type control littermates were assessed for neurite outgrowth. Neurite outgrowth was only reduced when CHL1 was simultaneously expressed on both cell types. This inhibitory effect on neurite outgrowth was considered to be due to a homophilic CHL1-CHL1 interaction, implicating CHL1 as a glial scar component in restriction of post-traumatic axonal regrowth and remodeling of spinal circuits.
Based on these observations we investigated whether upregulation of the cytokine FGF-2 after central nervous system trauma (Mocchetti et al. 1996; Zai et al. 2005) would serve as a link between enhanced CHL1 expression and reduced regeneration after optic nerve crush (Rolf et al. 2003) as well as spinal cord injury (Jakovcevski et al. 2007). CHL1 expression was indeed enhanced in a dose- and time-dependent manner by activation of known FGF receptor-dependent signaling pathways, involving MAP kinase, Ca2+-calmodulin-dependent kinase II and phosphoinositol 3-dependent kinase (PI3K). Not only in vitro, but also in vivo, when injected into the unlesioned spinal cord, FGF-2 enhanced the expression of CHL1 in parallel with the previously described enhancement of glial fibrillary acidic protein expression (Eclancher et al. 1996; Liu et al. 2005). Also, in vitro assays confirmed that FGF-2 enhances CHL1-mediated migration and proliferation of astrocytes as indicated by its more potent effects on wild-type astrocytes than CHL1-deficient astrocytes (Jakovcevski et al. 2007).
In this study, we were interested in whether pro-inflammatory mechanisms would influence CHL1 expression by astrocytes. Elucidation of signal transduction pathways evoked by pro-inflammatory agents would be important in view of the possibility to reduce CHL1 expression by astrocytes, thus curbing one of the inhibitory factors affecting regeneration after spinal cord injury in acute and chronic neurodegenerative diseases of adult mammals. To this end, we investigated the effects of bacterial lipopolysaccharide (LPS) on CHL1 expression in primary cultures of astrocytes and demonstrated that the PI3K/PKCδ-dependent ERK1/2 pathway mediates upregulation of CHL1 expression by reactive astrocytes. Our findings indicate that targeting PI3K/PKCδ/MAP kinase pathways may serve as a strategy to attenuate CHL1 expression by the glial scar, thus enhancing functional recovery after spinal cord injury (SCI).
MATERIALS AND METHODS
Reagents and Antibodies
Lipopolysaccharide (LPS, Escherichia coli), L-N6-(1-iminoethyl)-lysine hydrochloride (L-NIL; an inhibitor of iNOS), PTIO (an NO scavenger), NS 2028 (an inhibitor of guanylate cyclase), S-nitroso glutathione (GSNO, an NO donor), 3-morpholinosydnonimine (SIN-1; an NO donor), S-nitroso-N-acetylpenicillamine (SNAP, an NO donor), forskolin, poly-D-lysine (PDL), 100x protease inhibitor cocktail and 100x phosphatase inhibitor cocktail I & II were purchased from Sigma (St. Louis, MO). Mouse recombinant tumor necrosis factor-α (TNFα), phorbol-12-myristate-13-acetate (PMA), Ro-31-8220 (a broad-spectrum PKC inhibitor), Gö6983 (a broad-spectrum PKC inhibitor), Rottlerin (a specific PKCδ inhibitor), PKCβII/EGFR inhibitor, Gö6976 (a specific PKCα, PKCβ inhibitor), LY294002 (PI3K antagonist), Wortmannin (PI3K antagonist), PI3Kα inhibitor IV, PI3Kβ inhibitor VI (TGX-221), PI3Kγ inhibitor, PD98059 (ERK antagonist), U0126 (MEK antagonist), and H89 (PKA inhibitor) were obtained from Calbiochem (San Diego, CA). IKK-NBD peptide (NF-κB inhibitor), 8-bromo-cGMP (a cell-permeable cGMP analog), and MY-5445 (cGMP phosphodiesterase inhibitor) were provided by Biomol (Plymouth Meeting, PA). Recombinant mouse interferon-γ (IFNγ) was purchased from R&D Systems (Minneapolis, MN).
The following antibodies were used: goat anti-CHL1 antibody (1:500, R&D Systems), antibodies against various PKC isoforms (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-Akt (Ser473) (1:1000, Cell Signaling Technology, Beverly, MA), anti-phospho-ERK1/2 and ERK1/2 (1:500, Cell Signaling Technology), anti-NF-κB p65 subunit and anti-NF-κB p50 subunit (1:2000, Biomol), rabbit anti-neuronal class III β-tubulin (Tuj1, 1:500, Covance, Berkeley, CA), and anti-GAPDH (1:1000, Chemicon, Temecula, CA).
Primary Cultures of Astrocytes and Microglia
Primary astrocytes and microglia were cultured from the cerebral cortices of 1- to 2-day-old C57BL/6 mice as described (Jakovcevski et al. 2007). In brief, cerebra were dissected, chopped, triturated, and plated on tissue culture flasks that had been coated with poly-D-lysine (PDL, 50 μg/ml). The cells were grown in Dulbecco's Modified Eagle's Medium/F12 (DMEM/F12, Invitrogen, Piscataway, NJ, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen), 1% Pen/Strep at 37°C with 5% CO2. When the cells had grown to confluence, the flasks were shaken at 200 rpm for 3 h at 37°C to isolate microglia. On culture days 8 to 10, the cell cultures were continuously shaken for 24 h at 200 rpm to remove oligodendroglia. Then, the adherent astrocytes were subcultured in multiwell-plates or dishes. When cells had reached subconfluence, the culture medium was replaced with serum-free DMEM/F12 overnight before treatments. When microglia had reached confluence, LPS (30 ng/ml) or IFNγ (60 ng/ml) were applied to the dish for 12 h. Cells were then washed twice with pre-warmed PBS, and the culture medium was replaced. After a two-day culture period, the medium was collected and filtered to be used as conditioned culture medium.
Experimental Treatment of Astrocyte Cultures
Astrocytes were treated with 1.0 μg/ml LPS for different time periods or for 24 h with different concentrations of LPS. To examine the effects of PKC, PI3K, ERK1/2, nuclear factor κB (NF-κB), nitric oxide (NO), inducible nitric oxide synthase (iNOS), or guanylate cyclase (GC) antagonists/inhibitors, astrocytes were pre-treated with the indicated reagents for 1 h, followed by 1.0 μg/ml of LPS treatment at the indicated times. To investigate whether the NO-GC-cGMP pathway participates in LPS-induced CHL1 protein expression, GSNO, MY-5445, or 8-bromo-cGMP was applied to the cultures at the indicated times and concentrations.
Cell Viability and Cell Proliferation
Astrocytes were plated into PDL pre-coated 96-well plates (20,000 cells/well in 200 μl DMEM) with or without LPS treatment at the indicated times and concentrations. In order to obtain a reference curve, a series of dilutions of known cell numbers were prepared and plated into 96-well-plates under the same condition for 2 h to allow the cell attach (Suppl. Fig. 1). Mitochondrial activity was measured using the MTT assay kit (Sigma) according to the manufacturer's instructions. Cell viability and cell proliferation in treated cultures is presented graphically as the percentage of live cells in relation to live cells in the vehicle-treated, control condition (Mosmann 1983).
Whole-cell Lysate Preparation
Astrocytes were treated at 37°C with selected compounds as described above. The cells were collected in radioimmunoprecipitation assay (RIPA) buffer (Sigma) supplemented with 1 mM phenylmethylsulfonyl fluoride, 1x protease inhibitor cocktail and phosphatase inhibitor cocktail I & II. After brief sonication with a Sonic Dismembranator (Fisher Scientific), whole-cell lysates were prepared by centrifugation at 20,600xg for 5 min at 4°C.
siRNA Analyses
siRNAs designed to knock down the endogenous expression of PKCα, PKCβ, PKCδ, and siRNA for the negative control were purchased from Ambion (Austin, TX). Transfections of siRNAs were performed by use of siPORTNeoFX reagent according to the procedures recommended by the manufacturer (Ambion) (Liao et al. 2008). Briefly, the siPORTNeoFX reagent was diluted in Opti-MEM I reduced-serum medium (Invitrogen) and incubated at room temperature for 10 min, thereafter being mixed with the indicated siRNA (50 nM) diluted in Opti-MEM I reduced-serum medium, followed by incubation at room temperature for 10 min. The complexes formed were dispensed into 24-well cell culture plates and overlaid with cells at a density of 1.6×105 cells/ml. At 24 h post-transfection, cells were replaced with fresh medium and grown for an additional 60 h before harvest. LPS was added to the cells 24 h prior to termination.
Staining of Mitochondria
Astrocytes grown on coverslips were treated with Rottlerin 100 nM for 24 h before being fixed in 4% paraformaldehyde (PFA). Following fixation, the cells were washed in PBS for three times and then incubated with the MitoTracker Deep Red 633 FM probes (Invitrogen) 400 nM for 20 min. After rinsing the cells with fresh PBS, fluorescence (deep red) was visualized using a Zeiss LSM 5 confocal laser-scanning microscope.
Cytosolic and Membrane Fractionation
The preparation of cytosolic and membrane fractions was performed as described previously with some modifications (Shah et al. 2003). Briefly, serum-starved astrocytes were treated with 1.0 μg/ml LPS or 200 nM PMA for the time periods indicated, washed twice with ice-old PBS and kept frozen at −80°C until processing. Frozen cells were rapidly thawed and harvested in homogenization buffer containing 25 mM Tris·HCl, pH 7.4, 2 mM EDTA, 10 mM β-mercaptoethanol, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1x protease inhibitor cocktail and 1x phosphatase inhibitor cocktail I & II. After incubation on ice for 10 min, the cells were briefly sonicated. Nuclei and unbroken cells were pelleted by centrifugation at 500xg for 5 min at 4°C, and the supernatant was centrifuged at 100,000xg for 30 min at 4°C in an Optima™ TLX Ultracentrifuge to yield the cytosolic fraction. The pellet fractions were resuspended in ice-cold homogenization buffer containing 1% Triton X-100 for 30 min, sonicated, and centrifuged at 100,000xg for 20 min at 4°C. The resulting supernatant was taken as the membrane fraction.
Cytosolic and Nuclear Extraction
Astrocytes grown in 10 cm tissue culture dishes were treated with 1.0 μg/ml LPS for the indicated times. The cytosolic and nuclear fractions from treated and untreated cells were isolated with the NE-PER nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL) following the manufacturer's instructions.
Cocultures of Hippocampal Neurons and Astrocytes
Primary cultures of cerebral cortical astrocytes were prepared as described above, plated on glass coverslips coated with PDL (100 μg/ml). Confluent cultures of astrocytes were treated with or without 1.0 μg/ml LPS in the absence of serum for 3 days and dissociated hippocampal neurons were plated on the monolayers (Jakovcevski et al. 2007). To exclude an effect of LPS on neurons, the astrocytes were washed twice with pre-warmed PBS before coculture. To avoid extensive neurite outgrowth that hinders subsequent analyses, cultures were fixed only 6 h after plating and stained with anti-Tuj1 antibody to identify neurons. For the study of specificity of CHL1 on neurite outgrowth, a goat anti-CHL1 monoclonal antibody directed against the extracellular domain of the molecule was added at a final concentration of 100 μg/ml on 1 h before LPS administration to astrocytes. After 3 days of incubation, the astrocytes were washed twice with pre-warmed PBS and then cocultured with freshly dissociated hippocampal neurons for 6 h. Immunofluorescence was visualized using a Zeiss LSM 5 confocal laser-scanning microscope with a 40× objective. At least 10 random and non-overlapping microscopic fields were captured for each coverslip. Lengths and numbers of neurites were measured using NIH ImageJ software.
Immunoblot Analysis
Equal amounts of protein were electrophoretically separated on 4 to 12% NuPAGE Novex Bis-Tris gradient gels (Invitrogen) and then transferred to nitrocellulose membranes (Invitrogen). After blocking with 8% nonfat milk for 1 h at room temperature, blots were incubated overnight at 4°C with primary antibodies and probed with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Piscataway, NJ) for 1 h at room temperature. The immunocomplexes were then visualized with the enhanced chemiluminescence detection system (GE Healthcare). Blots were quantified by band densitometry of scanned films using the Gel-Pro Analyzer program (Media Cybernetics, Inc., Rockville, MD).
Immunocytochemistry
Astrocytes grown on coverslips were treated with or without LPS 1.0 μg/ml for 3 days. The culture medium was removed and replaced with pre-warmed, serum-free MEM followed by adding the same volume of fresh 8% PFA (final concentration, 4% PFA). Cells were fixed for 10 min at room temperature, washed three times with PBS, and processed for immunocytochemical staining using various antibodies. Fixed cells were blocked in 10% normal donkey serum with or without 0.2% Triton X-100 at room temperature for 1 h and then incubated with a rabbit antibody against Tuj1 and goat antibody against CHL1 overnight at 4°C. After washing three times in PBS, Cy3-conjugated donkey anti-goat (1:400, Jackson ImmunoResearch, West Grove, PA) and Alexa 488-conjugated donkey anti-rabbit (1:400, Jackson ImmunoResearch) were applied for 1 h in the dark at room temperature. Cell nuclei were labeled with TO-PRO-3 (1:1000, Invitrogen). Immunofluorescence was visualized by using a Zeiss LSM 5 confocal laser-scanning microscope.
Statistical Analysis
All experiments were repeated at least three times using independent culture preparations. All numerical data are presented as group mean values with standard errors of the mean (SEM). Statistical analysis was conducted by one-way analysis of variance followed by paired comparisons using Student's t test with Bonferroni corrections. Significance threshold value was 0.05.
RESULTS
Astrocyte Activation Induced by LPS Upregulates CHL1 Protein Expression
Bacterial LPS is a prototype pro-inflammatory stimulator of astrogliosis and enhances expression of the gliosis indicator glial fibrillary acid protein (GFAP) in cultures of mouse astrocytes (Brahmachari et al. 2006). To investigate CHL1 expression in reactive astrocytes, primary cultures of mouse astrocytes were treated with 1.0 μg/ml LPS for 6–72 h. Under control conditions in the absence of LPS, CHL1 was expressed in astrocytes at a low basal level, but exposure of these cells to LPS significantly upregulated CHL1 protein expression. LPS enhanced CHL1 expression in a time- and dose-dependent manner (Fig. 1A,B). We also found that incubation of culture astrocytes with LPS (1 μg/ml) for 2 days enhanced GFAP protein levels by 59% compared to the normal astrocytes (data not shown). Cell viability assays indicated that LPS did not induce cell death at any of the different times and concentrations tested (Fig. 1C,D).
Fig. 1.

LPS upregulates CHL1 protein expression in primary cultures of mouse astrocytes. A. Time-dependence and B. dose-dependence of CHL1 expression upon treatment with LPS. The representative immunoblots of culture lysates show protein levels of CHL1 (top panel). GAPDH (bottom panel) was used as a loading control. Relative levels of CHL1 expression are shown as a percentage of the non-treated control group (control (Ctrl) set to 100%). *P<0.05 versus control group without LPS. C–D. Cell viability was examined by the MTT assay. N=5. E–F. The expression of CHL1 was determined and quantified in the cytosolic and membrane fractions of cultured astrocytes after LPS treatment. *P<0.05 versus control group without LPS. G. The astrocytes with (right) or without (left) treatment with 1.0 μg/ml LPS for 2 days were stained with goat anti-CHL1 antibody and visualized under a Zeiss LSM 5 confocal laser-scanning microscope. Scale bar = 50 μm .
To determine the subcellular distribution of CHL1 expression, we prepared the cytosolic and membrane fractions of cultured astrocytes after LPS treatment. We found that CHL1 was significantly upregulated in the membrane fraction, beginning at 6 h after 1.0 μg/ml LPS treatment which is earlier than observed in the total cell lysate. The latter is homogenized from whole cells which cause signals to be diluted in the membrane fraction. High CHL1 expression persisted for 1–3 days post-treatment (Fig. 1E,F). There was no significant change in CHL1 levels in the membrane fraction at early time points (1, 3, 4 h) (data not shown). Cell surface expression of CHL1 protein was detectably enhanced at the cell surface after 2 days treatment (Fig. 1G). Besides, hypertrophic morphology of astrocytes was seen after 2 days incubation with LPS (Fig. 1G), indicating that LPS induces a reactive phenotype of cultured astrocytes obtained from early postnatal brain.
To address the question as to whether increased CHL1 expression is directly or indirectly related to cell proliferation, we measured proliferation of cultured astrocytes in the absence and presence of LPS using the MTT assay kit. After 4 days of incubation with 0.5 and 1.0 μg/ml LPS, the percentages of proliferating cells among total cell numbers were, respectively, 97±6.5% (n=3) and 102±7.2% (n=3), compared with the untreated control group (set to 100%). Therefore, higher LPS-induced expression levels of CHL1 reflect higher CHL1 protein expression in individual cells, independent of their state of proliferation.
LPS has been reported to stimulate astrocytes in vitro and in vivo into an astrogliotic state that is associated with secretion of several pro-inflammatory cytokines (Drescher and Whittum-Hudson 1996; Giri et al. 2004). We thus examined the effects of TNFα and IFNγ on CHL1 expression. Exposure of cultures to TNFα (1–500 ng/ml) or IFNγ (1–200 ng/ml) for 24 h did not induce detectable changes in CHL1 expression (data not shown). Furthermore, conditioned media from cultured microglia pretreated with LPS or IFNγ for 24 h did not enhance astrocytic CHL1 expression (data not shown). These observations indicate that elevated expression of CHL1 induced by LPS is not mediated via the pro-inflammatory cytokines TNFα and IFNγ.
PKCδ Mediates LPS-induced Upregulation of CHL1 Expression
It has been shown that LPS causes increased phosphorylation of PKCδ (Shen et al. 2005) and treatment with PKC inhibitors abolishes LPS-induced signaling activities in glial cells (Akundi et al. 2005; Kim et al. 2005). These findings suggested that the PKC signal transduction pathway contributes to the stimulatory changes induced by LPS. To investigate whether PKC is involved in LPS-induced CHL1 protein expression, we first down-regulated PKC by long term treatment with phorbol ester (PMA, 0.5–1.0 μM for 24 h) which causes proteolytic degradation of all PKC isozymes of the conventional and novel subfamilies (Shah et al. 2003). Depletion of PKC reduced LPS-induced upregulation of CHL1 expression (Fig. 2A). In the absence of LPS, however, depletion of PKC by PMA downregulated CHL1 expression (Fig. 2A, right panel), suggesting that activation of PKC is also involved in regulating CHL1 expression independent of LPS signaling. Since PKC isoforms have been classified as conventional (α, βI, βII, γ), novel (δ, ε, θ, η) and atypical (ξ) PKCs based on their regulatory properties (Dempsey et al. 2000; Toker 1998), we next examined the expression of these PKC isoforms by cultured astrocytes. Quantitative immunoblot analyses indicated the presence of PKCα, βI, βII, δ, ε, and η (Fig. 2B). Stimulation with PMA for 13 h at 2 μM resulted in down-regulation of all PKC isozymes (data not shown), indicating that these isoforms are all PMA-sensitive. Our cultures of astrocytes did not express PKCγ and θ (Fig. 2B).
Fig. 2.

LPS-induced upregulation of CHL1 expression in cultured astrocytes is mediated by PKC. A. PKC depletion by PMA treatment (1.0 μM for 13 h) abolishes upregulation of CHL1 by LPS (1.0 μg/ml for 24 h) (left panel) and also downregulates endogenous CHL1 expression (right panel). B. Immunoblot analysis of endogenous expression of PKC isoforms. C. CHL1 upregulation by LPS was regulated by PKC inhibitors. *P<0.05 versus control group without LPS; #P<0.05 versus LPS alone group. D. Cell viability was examined by the MTT assay. E. Mitochondria were stained by the MitoTracker Deep Red 633 FM probes and visualized under Zeiss LSM 5 confocal laser-scanning microscope. Scale bar = 20 μm (shown in Fig. 2E right panel). F. CHL1 protein expression in cultured astrocytes transfected with PKCα, PKCβ, PKCδ siRNA, or negative control siRNA (NTC) incubated with or without LPS (1.0 μg/ml) for 24 h. Relative levels of CHL1 expression are expressed as percentage of vehicle treatment (left panel). Expression levels of PKCα, PKCβI/II, and PKCδ in astrocytes transfected with siRNA of PKCα, PKCβ, PKCδ, and NTC were analyzed by Western blot analyses (right panel). *P<0.05 versus NTC without LPS treatment; #P<0.05 versus NTC with LPS treatment.
To further test the involvement of PKC isoforms, astrocyte cultures were pretreated with different inhibitors of PKC isozymes prior to incubation with LPS. The broad-spectrum PKC inhibitors Ro318220 and Go6983 abolished LPS-induced expression of CHL1 protein (Fig. 2C), suggesting that PKC activation is important for upregulation of CHL1 expression. Morveover, increased CHL1 expression was abolished by incubation of cultures with the PKCδ specific inhibitor Rottlerin at the low dosages of 10–50 nM (Fig. 2C), whereas the conventional PKC antagonist Gö6976 or the inhibitor of PKCβII/EGFR had no effect (Fig. 2C). The critical participation of PKCδ in the upregulation of CHL1 expression by LPS was further confirmed by siRNA analyses in which LPS-induced CHL1 upregulation was significantly reduced upon siRNA suppression of PKCδ expression (Fig. 2F). Repression of PKCδ also markedly reduced the basal CHL1 expression by approximately 40% in the absence of LPS. In contrast, no such effect was observed after knockdown of the expression of conventional PKC (Fig. 2F). Collectively, these results demonstrate that LPS-induced upregulation of CHL1 protein expression is dependent on specific activation of a novel PKCδ.
Rottlerin inhibits PKCδ kinase activity at a ki of 3–6 μM. ki values for PKCα,β,γ and PKCε,η,ζ are 30–42 μM, 80–100 μM, respectively (Gschwendt et al. 1994; Way et al. 2000). To rule out toxic effects of the PKCδ inhibitor Rottlerin under our serum-free treatment conditions, we performed a cell viability assay. After a 24 h exposure of cultures to Rottlerin, the viability assay showed that Rottlerin (50–500 nM) did not induce cell death compared with the cells without treatment (Fig. 2D). However, increasing Rottlerin concentration to 1 μM caused approximately 30% of all cells to die under serum-free cultured conditions, whereas in the presence of 10% FBS, cell viability assays showed that Rottlerin 1 μM did not induce cell death (data not shown). Mitochondria, an indicator of cell viability, were identified by MitoTracker Deep Red633 FM. We performed another test for cell viability. There were no significant differences in the number or morphology of the mitochondria between control or Rottlerin-treated cells (Fig. 2E), indicating that under our treatment conditions Rottlerin does not affect mitochondria.
LPS Induces Different Expression Patterns of PKC Isoforms and Transient Translocation of PKCδ
To investigate which PKC isozyme was induced by LPS stimulation, we assessed the effect of LPS on the expression of the six PKC isozymes expressed by cultured astrocytes. Exposure of cultures to 1.0 μg/ml LPS for 0.5–24 h did not induce detectable changes in protein levels of several PKC family members, including α, βII, and η (Fig. 3A). PKCβI expression was slightly increased 0.5–1 h after exposure to LPS, and PKCε was modestly elevated at 12–24 h after exposure to LPS (Fig. 3A). It is noteworthy that expression levels of several PKC isoforms did not change in a linear fashion as a function of time during incubation with LPS (Fig. 3A). The most robust observation was that LPS increased protein expression of PKCδ in a time-dependent manner (Fig. 3A,B). Moreover, this LPS-dependent upregulation of PKCδ expression was reduced by the PKCδ inhibitor Rottlerin (Fig. 3C). These observations indicate that PKCδ influences LPS-induced upregulation of CHL1 expression.
Fig. 3.

LPS induces expression of different PKC isoforms in cultured astrocytes. A. Time course of the effect of LPS treatment on PKC isoform expression. Control (Ctrl) refers to incubation without LPS for 24 h. This pertains also to the other figures and concomitant legends. GAPDH was used as loading control. B. Expression levels of PKCδ after LPS treatment were determined. C. Cells preincubated for 1 h with the indicated concentrations of Rottlerin were treated with LPS (1.0 μg/ml) for 24 h. PKCδ expression in cell lysates was quantified by immunoblot analysis.
PKC translocation from cytosol to the cell membrane is a measure of enzyme activation. To determine which PKC isoform is involved in LPS-stimulated signaling, we prepared subcellular fractions and measured expression of PKC isoforms in the cytosolic and membrane fractions of cultured astrocytes after LPS treatment. First, in agreement with previous observations (Shah et al. 2003; Slepko et al. 1999), we found that exposure of astrocyte cultures to 200 nM PMA for 10 min resulted in translocation of several PKC isoforms to the cell membrane, indicating that our subcellular fraction action procedure was adequate (Fig. 4B). Next, to explore whether LPS affects PKC translocation to the membrane fraction and thus activation, astrocyte cultures were treated with 1.0 μg/ml LPS for 30 min to 24 h. The results showed that PKCδ was generally the most reliable isoform to translocate from cytosol to the cell membrane in 30 min (Fig. 4A). Also, this translocation was abolished by the PKCδ inhibitor Rottlerin (Fig. 4C). In addition, we tested PKC translocation after short-term application of LPS. Again, translocation of only PKCδ to the membrane fraction was found at 15 min after exposure to LPS (Fig. 4D). In agreement with our observations in whole-cell lysates (Fig. 3A), PKCδ expression was increased in both cytosolic and membrane fractions (Fig. 4A). These observations implicate LPS-induced PKCδ activation in upregulation of CHL1 expression.
Fig. 4.

LPS induces transient translocation of PKCδ from cytosol to membrane in cultured astrocytes. A. Cells were treated with 1.0 μg/ml LPS for the indicated times, and cytosol and membrane fractions were prepared. B. Cytosolic and membrane fractions from PMA-treated (200 nM) or untreated cells were subjected to immunoblot analysis with antibodies to different PKC isoforms. C. Fractions were subjected to immunoblot analysis for PKCδ expression. D. Cells were treated with or without LPS for the indicated times prior to analysis of PKCδ.
PI3Kβ and δ Are Involved in LPS-induced Upregulation of CHL1 Expression
PI3K has been shown to be involved in LPS-induced signaling in glial cells (Pahan et al. 1999). To address the question of whether PI3K contributes to LPS-stimulated upregulation of CHL1 expression, blockade of PI3K activity by the inhibitor LY294002 or Wortmannin was tested. Both LY294002 and Wortmannin caused a dose-dependent reduction of LPS-induced CHL1 expression (Fig. 5A,B), indicating that PI3K upregulates CHL1 expression in astrocytes.
Fig. 5.

Inhibition of PI3K reduces LPS-induced CHL1 and PKCδ expression in cultured astrocytes. A–E. Expression of CHL1 protein was determined by immunoblot analysis in cells pretreated with LY294002, Wortmannin, or inhibitors for PI3Kα, β, γ for 1 h, followed by incubation with or without 1.0 μg/ml LPS for 24 h. *P<0.05 versus LPS alone group. F. Time-dependence of Akt phosphorylation (Ser473) in the subcellular fractions upon treatment with 1.0 μg/ml LPS. *P<0.05 versus control group without LPS treatment. G. Whole-cell lysates were subjected to immunoblot analysis for PKCδ. H. Cytosolic and membrane fractions were analyzed by immunoblot for PKCδ in astrocytes treated with LY294002 (10 μM) or Wortmannin (30 nM) for 1 h before incubation with 1.0 μg/ml LPS for 30 min.
PI3Ks are members of a family of lipid kinases that has been divided into three distinct classes (Class I, II, and III) based on their primary structure, mode of regulation and substrate specificity (Vanhaesebroeck et al. 2001). The activation of Class I PI3Ks is one of the most important signal transduction pathways used by cell surface receptors to control intracellular events (Hawkins et al. 2006). The Class I enzymes are further divided into Class IA (α, β, δ) and B (γ) subgroups based on their distinct p110 catalytic subunits and modes of regulation (Hawkins et al. 2006). It has been reported that PI3Kδ expression is restricted to leukocytes (Chantry et al. 1997). To identify which type of PI3Ks is involved in the upregulation of CHL1 in astrocytes, we applied specific inhibitors of PI3Ks isozymes prior to incubation with LPS. The inhibitors specific for PI3Kβ (TGX-221) and PI3Kγ significantly blocked upregulation of CHL1 expression (Fig. 5D,E), whereas the PI3Kα inhibitor had no effect (Fig. 5C). We also measured phosphorylation of protein kinase B (PKB, also known as Akt) at Ser473 which is an indirect assessment of PI3K activity. We found that LPS induced Akt phosphorylation in both cytosolic and membrane fractions of cultured astrocytes (Fig. 5F). Collectively, these results demonstrate that activation of PI3K, especially PI3Kβ and γ, is involved in LPS-mediated upregulation of CHL1.
PKC isozymes are downstream targets for PI3K through phosphoinositide-dependent kinase-1 (PDK-1) under diverse physiological conditions (Toker 2000). We confirm here that inhibition of PI3K signaling by pretreatment of astrocytes with 10 μM LY294002 or 30 nM Wortmannin abolishes LPS-induced upregulation of PKCδ expression in whole-cell lysates (Fig. 5G) and translocation to the cell membrane (Fig. 5H), indicating that PI3K acts upstream of PKCδ in regulation of CHL1 expression in LPS-activated astrocytes.
ERK1/2 Activation Regulates LPS-induced CHL1 Expression
ERK1/2 are ubiquitous enzymes implicated in LPS-induced signaling in many cell types (Akundi et al. 2005; Lai et al. 2003; Lee et al. 2003). Abnormal regulation of ERK1/2 activity has been observed in several models of CNS injury and inflammation (Atkins et al. 2007; Pannu et al. 2004; Park et al. 2004; Zhao et al. 2007). We here evaluated the role of ERK1/2 in the LPS-triggered signaling leading to upregulation of CHL1 expression by astrocytes. Cells were pretreated with PD98059 (a specific ERK1/2 antagonist) or U0126 (a specific inhibitor of MEK) for 1 h, followed by treatment with 1.0 μg/ml LPS for 24 h. Western blot analyses showed that inhibition of ERK1/2 inhibits LPS-induced upregulation of CHL1 expression (Fig. 6A,B). Furthermore, LPS induced phosphorylation of ERK1/2 in a time-dependent manner with a peak at 3 h after stimulation with LPS (Fig. 6C). This effect was blocked by PD98059 in a dose-dependent manner (Fig. 6D). These findings indicate that inhibition of EKR1/2 activity by LPS downregulates CHL1 expression.
Fig. 6.

ERK1/2 activation enhances LPS-induced CHL1 expression in cultured astrocytes. A–B. LPS-induced upregulation of CHL1 expression is abolished by MEK (U0126) and ERK1/2 (PD98059) inhibitors. C. Time-dependence of ERK1/2 phosphorylation upon treatment with 1.0 μg/ml LPS. D–E. Phosphorylation of ERK1/2 induced by 1.0 μg/ml LPS for 3 h was abolished by inhibitors of phosphorylation of ERK1/2 (PD98059) or PKCδ (Rottlerin).
PKC is an upstream regulator of ERK1/2 activity in glial cells (Hsieh et al. 2007; Lee et al. 2003). In our study, we confirmed that PKC activation caused by treatment with the PKC activator PMA results in phosphorylation of ERK1/2 in cultured astrocytes (data not shown). In addition, LPS-stimulated translocation of PKCδ to the membrane fraction was inhibited by the PKCδ-specific inhibitor Rottlerin (Fig. 4C), but not by the inhibitor of ERK1/2 activation PD98059 (data not shown). Together with the results that LPS-induced phosphorylation of ERK1/2 was attenuated by Rottlerin (Fig. 6E), we propose that PKCδ is an upstream component of ERK1/2 signaling in regulating LPS-induced CHL1 expression.
LPS-induced Upregulation of CHL1 Expression Depends on PI3K/PKCδ-mediated Nuclear Translocation of NF-κB
The transcription factor NF-κB is critical for the induction of several pro-inflammatory cytokines in astrocytes (Kozuka et al. 2007). According to the TRANSFAC database predictions on the CHL1 promoter sequence from −1679 to + 421, the promoter has three putative NF-κB binding sites at −1385 to −1375; −1290 to −1280; and −1061 to −1051. To investigate whether activation of NF-κB is involved in LPS-induced upregulation of CHL1 expression in astrocytes, the specific NF-κB inhibitor, IKK-NBD (the NF-κB essential modifier-binding domain) peptide, was used, which inhibits NF-κB activation without altering the basal activation of NF-κB (Dasgupta et al. 2004; May et al. 2000). Pretreatment of astrocytes with 5–10 μM IKK-NBD peptide inhibited the LPS-induced upregulation of CHL1 expression (Fig. 7A), suggesting that LPS increases CHL1 expression via NF-κB activation.
Fig. 7.

Nuclear translocation of NFκB is involved in the LPS-induced increase of CHL1 expression in cultured astrocytes. A. Cells preincubated with the indicated concentrations of IKK-NBD peptide for 1 h were stimulated with 1.0 μg/ml LPS for 3 h. Cell lysates were subjected to immunoblot analysis of CHL1 expression. B–C. Equal amounts of proteins from control and stimulated cells were subjected to immunoblot analysis, and phosphorylation of NFκB isoforms was determined in the cytosolic and nuclear fractions. D. Cells were preincubated with Rottlerin (50 nM), LY294002 (10 μM), and PD98059 (20 μM) for 1 h before LPS treatment for 3 h. Phosphorylation of p65 NFκB in the nuclear fraction was subjected to immunoblot analysis.
NF-κB complexes are composed mainly of the p50 and p65 subunits that translocate to the nucleus in response to stimulation with LPS and pro-inflammatory cytokines (Ghosh and Karin 2002; Giri et al. 2004). Thus, the nuclear translocation of NF-κB serves as an indicator of transcriptional activation. To determine whether NF-κB translocation to the nucleus is involved in LPS-induced upregulation of CHL1 expression, we prepared cytosolic and nuclear fractions from LPS-treated or untreated astrocytes. Translocation of NF-κB was determined by immunoblot analysis using anti-p65 and anti-p50 NF-κB antibodies. As shown in Fig. 7B,C, 1.0 μg/ml LPS stimulated translocation of p65 NF-κB and p50 NF-κB to the nucleus in a time-dependent manner. Furthermore, nuclear translocation of p65 NF-κB was blocked by pretreatment with Rottlerin, LY294002, and PD98059 (Fig. 7D), indicating that NF-κB activation was abolished by inhibition of PI3K, PKCδ, and ERK1/2 activities. These results indicate that NF-κB is a downstream target for upregulation of CHL1 expression in response to LPS-induced astrocyte activation.
The NO-guanylate Cyclase-cGMP Pathway Does Not Participate in LPS-induced Upregulation of CHL1 Expression
NO has been implicated in the pathogenesis of stroke and other degenerative neurological diseases, such as demyelinating conditions and ischemic or traumatic brain and spinal cord injury (Akama et al. 1998; Brosnan et al. 1994; Hawkins et al. 1998; Mitrovic et al. 1994; Murphy 2000; Nakahara et al. 2002). Recent studies have demonstrated that the NO-GC-cGMP pathway participates in the activation of astrocytes stimulated by LPS and by pro-inflammatory cytokines, such as IFNγ (Brahmachari et al. 2006). We, therefore, investigated whether CHL1 expression levels are regulated by the iNOS-NO-GC-cGMP pathway. First, we found that exposure of astrocyte cultures to 10–50 μM L-NIL (an inhibitor of iNOS) or 50–100 μM PTIO (a scavenger of NO) for 24 h did not change basal levels of CHL1 protein expression (data not shown). Next, to explore whether iNOS-NO affects LPS-induced upregulation of CHL1 expression, astrocyte cultures were pretreated with 10–50 μM L-NIL or 50–100 μM PTIO for 1 h, followed by exposure to 1.0 μg/ml LPS for 24 h. The results showed that pretreatment of L-NIL or PTIO did not alter LPS-induced upregulation of CHL1 expression (Fig. 8 A,B). Furthermore, we examined the effect of GSNO, an NO donor, on expression of CHL1. Immunoblot analysis showed that release of NO by GSNO also did not change CHL1 expression (Fig. 8 C,D), in agreement with our observations that other NO donors, such as 3-morpholinosydnonimine (SIN-1) or S-nitroso-N-acetylpenicillamine (SNAP) did not change CHL1 expression (data not shown). These data indicate that iNOS-NO signaling is not involved in LPS-induced upregulation of CHL1 expression.
Fig. 8.

The NO-GC-cGMP pathway is not involved in LPS-induced CHL1 expression in cultured astrocytes. A–B. Cells were pretreated with the iNOS inhibitor (L-NIL) and the NO scavenger PTIO at the indicated concentrations for 1 h prior to incubation with or without 1.0 μg/ml LPS for 24 h. Cell lysates were subjected to immunoblot analysis for CHL1 expression. C. Time-dependence and D. dose-dependence of CHL1 expression upon treatment with the NO donor GSNO. E. Cells pretreated with different concentrations of the guanylate cyclase inhibitor NS-2028 NS for 1 h were incubated with 1.0 μg/ml LPS for 24 h. Cell lysates were subjected to immunoblot analysis for CHL1 expression. F–G. Cells were treated with different concentrations of 8-Br-cGMP or MY-5445 for 24 h, and CHL1 expression in cell lysates was subjected to immunoblot analysis. N=5.
Guanylate cyclase (GC)-cGMP is intimately coupled to NO-induced downstream signaling events (Bredt 2003; Monfort et al. 2002). To further investigate whether CHL1 expression is regulated by the NO-GC-cGMP pathway, inhibitors for GC or cGMP and a cell-permeable cGMP derivative were applied to the astrocyte cultures. The results showed that pretreatment of NS-2028, a specific inhibitor of GC, did not prevent LPS-induced elevation of CHL1 expression (Fig. 8E). As expected, neither 8-Br-cGMP, a cell-permeable cGMP derivative, nor MY-5445, an inhibitor of cGMP phosphodiesterase affected CHL1 protein levels in astrocytes (Fig. 8F,G). These observations show that LPS-induced upregulation of CHL1 expression in astrocytes is not mediated via the NO-GC-cGMP pathway.
Neurite Outgrowth on LPS-treated Astrocytes was Inhibited by LPS-mediated Upregulation of CHL1 Expression
Our previous study has shown that CHL1 participates in the inhibition of axonal regeneration in the adult glial scar after spinal cord injury (Jakovcevski et al. 2007). Thus, we were interested in whether overexpression of CHL1 by astrocytes would disturb neurite outgrowth in vitro. The longest neurite, total neurite length per neuron, and the numbers of neurites per neuron were measured. We found that neurites on LPS-stimulated astrocytes were 30% shorter and contained 29% fewer neurites than neurons on control non-treated astrocytes (Fig. 9A–C). Further, to test that the inhibitory effect was due to of CHL1 overexpression mediated by LPS, an antibody against the extracellular domain of CHL1 was used. The presence of the anti-CHL1 antibody during LPS treatment significantly increased neurite numbers and lengths compared to LPS-treatment alone (Fig. 9). These observations indicate that it is the upregulation of CHL1 expression by LPS that renders activated astrocytes less supportive for neurite outgrowth.
Fig. 9.

Neurite outgrowth in cocultures of neurons and astrocytes. The length of the longest neurite (A), total neurite lengths (B), and number of neurites per neuron (C) were analyzed in hippocampal neuons seeded on top of astrocytes that had or had not been treated with LPS or antibody against CHL1. Data represent mean ± SEM values for n > 200 neurons from 2 independent experiments. *P<0.05 versus control group without LPS; #P<0.05 versus LPS alone group.
DISCUSSION
Inhibition of axonal regeneration after spinal cord injury has been attributed in part to the non-permissive environment of the glial scar, which reduces axonal growth. Although resident microglia and oligodendrocytes as well as invading macrophages and Schwann cells contribute to the astroglial scar, reactive astrocyte responses appear to predominate in glial scar formation. CHL1 shows enhanced expression in reactive astrocytes both in vivo and in vitro (Jakovcevski et al. 2007; Rolf et al. 2003) and is considered a glial scar molecule that restricts post-traumatic axonal growth and inhibits the functional recovery after compression lesion of the mouse spinal cord (Jakovcevski et al. 2007). Therefore, understanding the mechanisms that regulate CHL1 expression in astrocytes may clarify the molecular mechanisms leading to upregulation of CHL1 expression in reactive astrocytes and, furthermore, may provide therapeutic strategies to downregulate CHL1 levels for therapeutic benefit. In the present study we show that pro- inflammatory LPS leads to upregulation of CHL1 expression in cultures of mouse astrocytes via signaling pathways as schematized in Figure 10. Findings from this study identify PI3K/PKCδ as key signaling mediators in regulating CHL1 expression in astrocytes, and demonstrate that the induction of CHL1 expression by LPS is involved in the non-permissive environment of reactive astrocytes for neurites outgrowth in vitro.
Fig. 10.
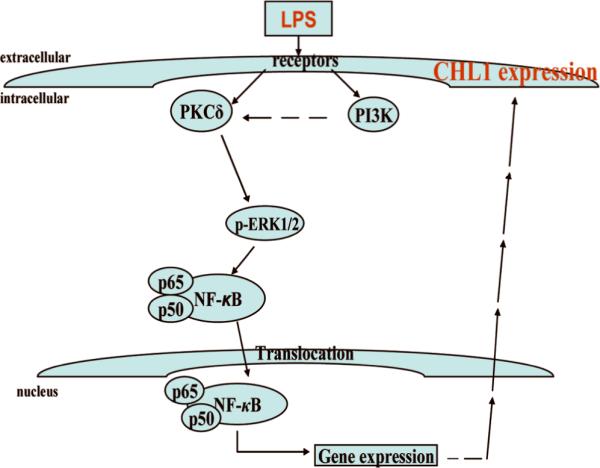
Schematic presentation of the signaling pathway for LPS-induced PI3K/PKCδ-dependent CHL1 expression in cultured astrocytes from early postnatal mouse cerebral cortex.
The response of adult astrocytes to injury is referred to as reactive astrogliosis which is characterized by enhanced GFAP expression, cellular hypertrophy and proliferation (Gadea et al. 2008). Cultured astrocytes from the developing early postnatal brain (Brahmachari et al. 2006) as well as from the adult rat spinal cord (Codeluppi et al. 2009) demonstrate phenotypic attributes of reactive gliosis after stimulation with agents such as LPS, interferon-γ, endothelin-1, HIV- 1gp120, amyloid β (Aβ)1–42, or epidermal growth factor. The cultured astrocytes treated with LPS from the present study also showed cellular hypertrophy and increased levels of GFAP, characteristics of reactive astrocytes, thus phenotyping at least to a considerable extent the features of astrogliotic cells in vivo.
PKC family members mediate a variety of cell functions, including cell differentiation, proliferation, and apoptosis (Newton 1995). PKCδ is a key member of the novel PKC family which does not require Ca2+ for activation but is activated by diacylglycerol (DAG) (Gschwendt et al. 1994). The present study has provided evidence that CHL1 expression is regulated by PKCδ in astrocytes. (1) PKCδ is highly expressed in cultured astrocytes being upregulated as well as activated by LPS through a PI3K-dependent pathway; (2) suppression of PKCδ activity by inhibitors or by blocked siRNA-mediation down-regulated CHL1 expression by LPS; (3) depletion of PKCs including PKCδ by long-term stimulation of PMA abolished LPS-triggered upregulation of CHL1 expression; (4) blocking the activation of downstream targets, such as ERK and NF-κB, inhibited LPS-induced elevation of CHL1 through PI3K/PKCδ-dependent mechanisms. Collectively, these finding indicate that PI3K/PKCδ mediates upregulation of CHL1 expression in the reactive astrocytes in vitro.
Conventional PKCs can be activated by myelin-associated inhibitors of axonal growth and by chondroitin sulfated proteoglycan, and inhibition of PKC causes robust axonal regeneration (Sivasankaran et al. 2004). In our study, LPS-induced upregulation of CHL1 expression by cultured astrocytes was not blocked by inhibitors or gene knockdown of conventional PKC isozymes. Moreover, astrocyte activation by LPS did not elicit translocation or persistent protein synthesis of these PKCs. Our recent Western blot results, however, demonstrate that rat spinal cord contusion induces robust and persistent PKCδ expression by 1 week and later after injury, but not conventional PKCs (including PKCα, βI, βII) (unpublished data).
Upregulation of CHL1 expression has been observed both in vitro and in vivo to depend on the action of FGF-2 (Jakovcevski et al. 2007; Rolf et al. 2003). However, LPS acts independently of FGF-2 since we could show that FGF-2 enhances astrocyte proliferation (Jakovcevski et al. 2007), but LPS does not enhance proliferation in the same time of exposure to the cultures (this study). Thus, LPS may act through the toll-like receptor-4 (TLR-4) as a specific receptor for LPS which has been shown to be active in astrocytes in several culture systems (Brahmachari et al. 2006; Pannu et al. 2004) although toll-like receptors have not been described in astrocytes by another study (Lehnardt et al. 2003). As previously described for microglia, the specific PKCδ inhibitor Rottlerin can modulate TLR-4 induced signaling (Kim et al. 2005). Although we cannot exclude that LPS-induced upregulation of CHL1 expression in cultures of astrocytes is due to the presence of microglia, we consider it unlikely. First, we found that CHL1 is not expressed in cultured microglia from mouse cerebral cortex tested by immunocytochemistry and Western blot analysis (data not shown), which is consistent with our previous observations in vivo (Jakovcevski et al. 2007). CHL1 is also not expressed in macrophages of rat injured spinal cord (unpublished data). Also, when cultures of astrocytes prepared under the present conditions were co-cultured at a ratio of 50 to 1 of astrocytes to microglia (Brahmachari et al. 2006), a similar expression pattern of LPS-induced upregulation of CHL1 expression was found in the microglia-depleted cultures of astrocytes as in the co-cultures with microglia. These observations indicate that upregulation of CHL1 in activated astrocytes is independent of microglia/macrophages. Furthermore, the pro-inflammatory cytokines TNFα and IFNγ did not induce detectable changes in CHL1 expression. Together with our observation that conditioned medium from cultured microglia pre-treated with LPS or IFNγ did not change astrocytic CHL1 expression, we conclude that LPS-triggered elevation of CHL1 expression is not mediated by the pro-inflammatory cytokines TNFα and IFNγ. However, this does not exclude that LPS may act as a general pro-inflammatory inducer (Atkins et al. 2007; Park et al. 2004; Vallieres et al. 2006).
It has been shown that activation of NF-κB mediates the expression of many pro-inflammatory molecules such as TNFα and IL-6. Furthermore, activation also induces expression of GFAP in astrocytes (Brahmachari et al. 2006; Pahan et al. 2000; Pahan et al. 1999). Consistently, we observed that LPS induced an increase in CHL1 expression in our astrocyte cultures via an NF-κB-dependent mechanism. Previous studies have indicated that PI3K/PKCδ/MAP kinases are important mediators in regulation of NF-κB–dependent gene expression such as iNOS, cyclooxygenase-2, IL-8, ICAM-1 (Hsieh et al. 2007; Pahan et al. 1999; Vallee et al. 2004). We found that nuclear translocation of NF-κB induced by LPS was blocked by antagonists of PI3K, PKCδ, and ERK1/2, indicating that NF-κB serves as a downstream target for LPS-triggered CHL1 upregulation. However, the MAP kinase pathway not only influences the activity of transcription factor NF-κB, but also of other transcription factors, such as CREB (Ejarque-Ortiz et al. 2007; Simi et al. 2005).
LPS leads to activation of inducible nitric-oxide synthase (iNOS) and production of NO in primary cultured astrocytes (Brahmachari et al. 2006; Pahan et al. 1997). It has been demonstrated that NO plays an important role in regulating GFAP expression in astrocytes through the GC-cGMP-PKG pathway (Brahmachari et al. 2006). However, in our study, we did not observe this pathway to be involved in LPS-triggered CHL1 upregulation. Our conclusion is based on the following: First, the increase of CHL1 expression stimulated by LPS was not reduced by the application of an iNOS inhibitor or NO scavenger. Second, NO donors such as GSNO, SIN-1 or SNAP did not increase CHL1 expression in astrocytes. Third, a specific inhibitor of GC did not prevent LPS-triggered CHL1 elevation. Fourth, a cGMP derivative or an inhibitor of cGMP phosphodiesterase did not change CHL1 protein levels. We thus conclude that LPS-triggered upregulation of CHL1 expression in cultured mouse astrocytes does not depend on the iNOS-NO-GC-cGMP signaling mechanism.
In summary, we have provided some mechanistic details on the regulation of CHL1 expression by LPS in mouse cerebral cortical astrocyte as schematized in Fig 10. Our study shows that LPS-induced upregulation of CHL1 expression is mediated through the PI3K/PKCδ/MAP kinase pathway independent of an iNOS-NO-GC-cGMP signaling mechanism. The consequence of CHL1 upregulation in reactive astrocytes perturb neurite outgrowth in vitro, as shown in the present studies, and is associated with inhibition of regeneration in vivo (Jakovcevski et al. 2007). Further experiments are needed to dissect in more detail the signal transduction pathways upstream and downstream of the MAP kinase pathway in astrocytes so that MAP kinase function in LPS-induced upregulation of CHL1 expression can be influenced in a targeted manner with the aim to influence the aversive upregulation of CHL1 expression after spinal cord injury and possibly other injuries of the central nervous system for therapeutic benefit.
Supplementary Material
Acknowledgments
Melitta Schachner is a New Jersey Professor of Spinal Cord Research.
Supported by NIH RO1 NS 035647 (JRW).
REFERENCES
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci U S A. 1998;95(10):5795–800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akundi RS, Candelario-Jalil E, Hess S, Hull M, Lieb K, Gebicke-Haerter PJ, Fiebich BL. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia. 2005;51(3):199–208. doi: 10.1002/glia.20198. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Oliva AA, Jr., Alonso OF, Chen S, Bramlett HM, Hu BR, Dietrich WD. Hypothermia treatment potentiates ERK1/2 activation after traumatic brain injury. Eur J Neurosci. 2007;26(4):810–9. doi: 10.1111/j.1460-9568.2007.05720.x. [DOI] [PubMed] [Google Scholar]
- Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26(18):4930–9. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS. Nitric oxide signaling specificity--the heart of the problem. J Cell Sci. 2003;116(Pt 1):9–15. doi: 10.1242/jcs.00183. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Reactive nitrogen intermediates in human neuropathology: an overview. Dev Neurosci. 1994;16(3–4):152–61. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- Buhusi M, Midkiff BR, Gates AM, Richter M, Schachner M, Maness PF. Close homolog of L1 is an enhancer of integrin-mediated cell migration. J Biol Chem. 2003;278(27):25024–31. doi: 10.1074/jbc.M303084200. [DOI] [PubMed] [Google Scholar]
- Chaisuksunt V, Campbell G, Zhang Y, Schachner M, Lieberman AR, Anderson PN. The cell recognition molecule CHL1 is strongly upregulated by injured and regenerating thalamic neurons. J Comp Neurol. 2000a;425(3):382–92. doi: 10.1002/1096-9861(20000925)425:3<382::aid-cne4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, Cooper JA, Hoekstra MF. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236–41. doi: 10.1074/jbc.272.31.19236. [DOI] [PubMed] [Google Scholar]
- Chen S, Mantei N, Dong L, Schachner M. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. J Neurobiol. 1999;38(3):428–39. doi: 10.1002/(sici)1097-4695(19990215)38:3<428::aid-neu10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Codeluppi S, Svensson CI, Hefferan MP, Valencia F, Silldorff MD, Oshiro M, Marsala M, Pasquale EB. The Rheb-mTOR pathway is upregulated in reactive astrocytes of the injured spinal cord. J Neurosci. 2009;29(4):1093–104. doi: 10.1523/JNEUROSCI.4103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K. Antineuroinflammatory effect of NF-kappaB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J Immunol. 2004;173(2):1344–54. doi: 10.4049/jimmunol.173.2.1344. [DOI] [PubMed] [Google Scholar]
- Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- Dong L, Chen S, Richter M, Schachner M. Single-chain variable fragment antibodies against the neural adhesion molecule CHL1 (close homolog of L1) enhance neurite outgrowth. J Neurosci Res. 2002;69(4):437–47. doi: 10.1002/jnr.10250. [DOI] [PubMed] [Google Scholar]
- Drescher KM, Whittum-Hudson JA. Modulation of immune-associated surface markers and cytokine production by murine retinal glial cells. J Neuroimmunol. 1996;64(1):71–81. doi: 10.1016/0165-5728(95)00156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eclancher F, Kehrli P, Labourdette G, Sensenbrenner M. Basic fibroblast growth factor (bFGF) injection activates the glial reaction in the injured adult rat brain. Brain Res. 1996;737(1–2):201–14. doi: 10.1016/0006-8993(96)00732-9. [DOI] [PubMed] [Google Scholar]
- Ejarque-Ortiz A, Medina MG, Tusell JM, Perez-Gonzalez AP, Serratosa J, Saura J. Upregulation of CCAAT/enhancer binding protein beta in activated astrocytes and microglia. Glia. 2007;55(2):178–88. doi: 10.1002/glia.20446. [DOI] [PubMed] [Google Scholar]
- Gadea A, Schinelli S, Gallo V. Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci. 2008;28(10):2394–408. doi: 10.1523/JNEUROSCI.5652-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J Immunol. 2004;173(8):5196–208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199(1):93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34(Pt 5):647–62. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res. 1998;118:155–72. doi: 10.1016/s0079-6123(08)63206-9. [DOI] [PubMed] [Google Scholar]
- Hillenbrand R, Molthagen M, Montag D, Schachner M. The close homologue of the neural adhesion molecule L1 (CHL1): patterns of expression and promotion of neurite outgrowth by heterophilic interactions. Eur J Neurosci. 1999;11(3):813–26. doi: 10.1046/j.1460-9568.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- Holm J, Hillenbrand R, Steuber V, Bartsch U, Moos M, Lubbert H, Montag D, Schachner M. Structural features of a close homologue of L1 (CHL1) in the mouse: a new member of the L1 family of neural recognition molecules. Eur J Neurosci. 1996;8(8):1613–29. doi: 10.1111/j.1460-9568.1996.tb01306.x. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Wang HH, Wu CY, Jou MJ, Yen MH, Parker P, Yang CM. BK-induced COX-2 expression via PKC-delta-dependent activation of p42/p44 MAPK and NF-kappaB in astrocytes. Cell Signal. 2007;19(2):330–40. doi: 10.1016/j.cellsig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Jakovcevski I, Wu J, Karl N, Leshchyns'ka I, Sytnyk V, Chen J, Irintchev A, Schachner M. Glial scar expression of CHL1, the close homolog of the adhesion molecule L1, limits recovery after spinal cord injury. J Neurosci. 2007;27(27):7222–33. doi: 10.1523/JNEUROSCI.0739-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DC, Kim SH, Jeong MW, Baek NI, Kim KT. Effect of rottlerin, a PKC-delta inhibitor, on TLR-4-dependent activation of murine microglia. Biochem Biophys Res Commun. 2005;337(1):110–5. doi: 10.1016/j.bbrc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Kozuka N, Kudo Y, Morita M. Multiple inhibitory pathways for lipopolysaccharide- and pro-inflammatory cytokine-induced nitric oxide production in cultured astrocytes. Neuroscience. 2007;144(3):911–9. doi: 10.1016/j.neuroscience.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM. Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Immunol. 2003;170(12):6244–9. doi: 10.4049/jimmunol.170.12.6244. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Shin CY, Yoo BK, Ryu JR, Choi EY, Cheong JH, Ryu JH, Ko KH. Induction of matrix metalloproteinase-9 (MMP-9) in lipopolysaccharide-stimulated primary astrocytes is mediated by extracellular signal-regulated protein kinase 1/2 (Erk1/2) Glia. 2003;41(1):15–24. doi: 10.1002/glia.10131. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100(14):8514–9. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchyns'ka I, Sytnyk V, Richter M, Andreyeva A, Puchkov D, Schachner M. The adhesion molecule CHL1 regulates uncoating of clathrin-coated synaptic vesicles. Neuron. 2006;52(6):1011–25. doi: 10.1016/j.neuron.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Liao M, Zhang Y, Dufau ML. Protein kinase Calpha-induced derepression of the human luteinizing hormone receptor gene transcription through ERK-mediated release of HDAC1/Sin3A repressor complex from Sp1 sites. Mol Endocrinol. 2008;22(6):1449–63. doi: 10.1210/me.2008-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lu B, Tu CQ, Chi LT, Wang GL, Pei FX. Effect of basic fibroblast growth factor on the expression of glial fibrillary acidic protein after tractive spinal cord injury in rats. Chin J Traumatol. 2005;8(2):117–20. [PubMed] [Google Scholar]
- Loers G, Schachner M. Recognition molecules and neural repair. J Neurochem. 2007;101(4):865–82. doi: 10.1111/j.1471-4159.2006.04409.x. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10(1):19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289(5484):1550–4. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merrill JE. Nitric oxide as a potential pathological mechanism in demyelination: its differential effects on primary glial cells in vitro. Neuroscience. 1994;61(3):575–85. doi: 10.1016/0306-4522(94)90435-9. [DOI] [PubMed] [Google Scholar]
- Mocchetti I, Rabin SJ, Colangelo AM, Whittemore SR, Wrathall JR. Increased basic fibroblast growth factor expression following contusive spinal cord injury. Exp Neurol. 1996;141(1):154–64. doi: 10.1006/exnr.1996.0149. [DOI] [PubMed] [Google Scholar]
- Monfort P, Munoz MD, Kosenko E, Felipo V. Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phosphodiesterase. J Neurosci. 2002;22(23):10116–22. doi: 10.1523/JNEUROSCI.22-23-10116.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morellini F, Lepsveridze E, Kahler B, Dityatev A, Schachner M. Reduced reactivity to novelty, impaired social behavior, and enhanced basal synaptic excitatory activity in perforant path projections to the dentate gyrus in young adult mice deficient in the neural cell adhesion molecule CHL1. Mol Cell Neurosci. 2007;34(2):121–36. doi: 10.1016/j.mcn.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29(1):1–13. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Nakahara S, Yone K, Setoguchi T, Yamaura I, Arishima Y, Yoshino S, Komiya S. Changes in nitric oxide and expression of nitric oxide synthase in spinal cord after acute traumatic injury in rats. J Neurotrauma. 2002;19(11):1467–74. doi: 10.1089/089771502320914697. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270(48):28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Nikonenko AG, Sun M, Lepsveridze E, Apostolova I, Petrova I, Irintchev A, Dityatev A, Schachner M. Enhanced perisomatic inhibition and impaired long-term potentiation in the CA1 region of juvenile CHL1-deficient mice. Eur J Neurosci. 2006;23(7):1839–52. doi: 10.1111/j.1460-9568.2006.04710.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. Expression of a dominant-negative mutant of p21(ras) inhibits induction of nitric oxide synthase and activation of nuclear factor-kappaB in primary astrocytes. J Neurochem. 2000;74(6):2288–95. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Namboodiri AM, Sheikh FG, Smith BT, Singh I. Increasing cAMP attenuates induction of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem. 1997;272(12):7786–91. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- Pahan K, Raymond JR, Singh I. Inhibition of phosphatidylinositol 3-kinase induces nitricoxide synthase in lipopolysaccharide- or cytokine-stimulated C6 glial cells. J Biol Chem. 1999;274(11):7528–36. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- Pannu R, Won JS, Khan M, Singh AK, Singh I. A novel role of lactosylceramide in the regulation of lipopolysaccharide/interferon-gamma-mediated inducible nitric oxide synthase gene expression: implications for neuroinflammatory diseases. J Neurosci. 2004;24(26):5942–54. doi: 10.1523/JNEUROSCI.1271-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EM, Joh TH, Volpe BT, Chu CK, Song G, Cho S. A neuroprotective role of extracellular signal-regulated kinase in N-acetyl-O-methyldopamine-treated hippocampal neurons after exposure to in vitro and in vivo ischemia. Neuroscience. 2004;123(1):147–54. doi: 10.1016/j.neuroscience.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Pratte M, Rougon G, Schachner M, Jamon M. Mice deficient for the close homologue of the neural adhesion cell L1 (CHL1) display alterations in emotional reactivity and motor coordination. Behav Brain Res. 2003;147(1–2):31–9. doi: 10.1016/s0166-4328(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Rolf B, Lang D, Hillenbrand R, Richter M, Schachner M, Bartsch U. Altered expression of CHL1 by glial cells in response to optic nerve injury and intravitreal application of fibroblast growth factor-2. J Neurosci Res. 2003;71(6):835–43. doi: 10.1002/jnr.10533. [DOI] [PubMed] [Google Scholar]
- Shah BH, Soh JW, Catt KJ. Dependence of gonadotropin-releasing hormone-induced neuronal MAPK signaling on epidermal growth factor receptor transactivation. J Biol Chem. 2003;278(5):2866–75. doi: 10.1074/jbc.M208783200. [DOI] [PubMed] [Google Scholar]
- Shen S, Yu S, Binek J, Chalimoniuk M, Zhang X, Lo SC, Hannink M, Wu J, Fritsche K, Donato R. Distinct signaling pathways for induction of type II NOS by IFNgamma and LPS in BV-2 microglial cells. Neurochem Int. 2005;47(4):298–307. doi: 10.1016/j.neuint.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Simi A, Edling Y, Ingelman-Sundberg M, Tindberg N. Activation of c-fos by lipopolysaccharide in glial cells via p38 mitogen-activated protein kinase-dependent activation of serum or cyclic AMP/calcium response element. J Neurochem. 2005;92(4):915–24. doi: 10.1111/j.1471-4159.2004.02938.x. [DOI] [PubMed] [Google Scholar]
- Sivasankaran R, Pei J, Wang KC, Zhang YP, Shields CB, Xu XM, He Z. PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat Neurosci. 2004;7(3):261–8. doi: 10.1038/nn1193. [DOI] [PubMed] [Google Scholar]
- Slepko N, Patrizio M, Levi G. Expression and translocation of protein kinase C isoforms in rat microglial and astroglial cultures. J Neurosci Res. 1999;57(1):33–8. doi: 10.1002/(SICI)1097-4547(19990701)57:1<33::AID-JNR4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Toker A. Signaling through protein kinase C. Front Biosci. 1998;3:D1134–47. doi: 10.2741/a350. [DOI] [PubMed] [Google Scholar]
- Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57(4):652–8. [PubMed] [Google Scholar]
- Vallee S, Laforest S, Fouchier F, Montero MP, Penel C, Champion S. Cytokine-induced upregulation of NF-kappaB, IL-8, and ICAM-1 is dependent on colonic cell polarity: implication for PKCdelta. Exp Cell Res. 2004;297(1):165–85. doi: 10.1016/j.yexcr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Vallieres N, Berard JL, David S, Lacroix S. Systemic injections of lipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia. 2006;53(1):103–13. doi: 10.1002/glia.20266. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci. 2000;21(5):181–7. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Yoo S, Wrathall JR. Increased growth factor expression and cell proliferation after contusive spinal cord injury. Brain Res. 2005;1052(2):147–55. doi: 10.1016/j.brainres.2005.05.071. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Roslan R, Lang D, Schachner M, Lieberman AR, Anderson PN. Expression of CHL1 and L1 by neurons and glia following sciatic nerve and dorsal root injury. Mol Cell Neurosci. 2000;16(1):71–86. doi: 10.1006/mcne.2000.0852. [DOI] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci. 2007;27(9):2357–68. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.