

Abstract
MAPkinase signalling is essential for cell growth, differentiation and cell physiology. G proteins and tyrosine kinase receptors each modulate MAPkinase signalling through distinct pathways. We report here that RGS14 is an integrator of G protein and MAPKinase signalling pathways. RGS14 contains a GPR/GoLoco (GL) domain that forms a stable complex with inactive Giα1/3-GDP, and a tandem (R1, R2) Ras binding domain (RBD). We find that RGS14 binds and regulates the subcellular localization and activities of H-Ras and Raf kinases in cells. Activated H-Ras binds RGS14 at the R1 RBD to form a stable complex at cell membranes. RGS14 also co-localizes with and forms a complex with Raf kinases in cells. The regulatory region of Raf-1 binds the RBD region of RGS14, and H-Ras and Raf each facilitate one another’s binding to RGS14. RGS14 selectively inhibits PDGF-, but not EGF- or serum-stimulated Erk phosphorylation. This inhibition is dependent on H-Ras binding to RGS14 and is reversed by co-expression of Giα1, which binds and recruits RGS14 to the plasma membrane. Giα1 binding to RGS14 inhibits Raf binding, indicating that Giα1 and Raf binding to RGS14 are mutually exclusive. Taken together, these findings indicate that RGS14 is a newly appreciated integrator of G protein and Ras/Raf signalling pathways.
Keywords: RGS14, RGS proteins, GoLoco proteins, GPR proteins, scaffolds, H-Ras, Raf kinases, MAPkiase signalling
1. Introduction
Heterotrimeric G proteins (G proteins) are molecular switches that regulate a broad range of signalling events important for many aspects of cell and organ physiology. The regulators of G protein signalling (RGS proteins) inactivate Gα-GTP subunits to negatively regulate G protein signalling. In conventional signalling models [1], G proteins link cell surface receptors of neurotransmitters and hormones to intracellular signalling events, and RGS proteins limit the lifetime of those signalling events. RGS proteins are defined by a shared 130 amino acid RGS domain which binds Gα-GTP to act as a GTPase activating protein (GAP) and inhibit G protein signalling [2–4]. Family members are classified into six or more distinct subgroups based on amino acid sequence identities and functional similarities. Many are relatively small (20–30 kDa) simple proteins with unremarkable features outside of the RGS domain, while others are larger (60–160 kDa) more “complex” proteins with additional domains that bind other protein partners to confer distinct signalling functions and/or act as novel integrators of G protein signalling.
RGS14 is a highly unusual complex family member that contains a canonical RGS domain, a tandem (R1 and R2) Ras/Rap binding domain (RBD) [5, 6], and a GoLoco/GPR (GL) motif. RGS14 was originally identified as a novel binding partner for the Ras-like GTPases, Rap1/2 [7]. The full-length sequence [8] encodes a ~62 kDa protein most closely related to RGS12 (85 kDa) and RGS10 (22 kDa). We and others show that RGS14 interacts selectively with the Gi/o subfamily of G proteins to regulate their guanine nucleotide binding/hydrolysis activity and signalling functions [9–13]. The RGS domain binds directly to activated Gi/oα-GTP to confer non-selective GAP activity towards Giα and Goα. By contrast, we find that the GoLoco/GPR (GL) motif binds specifically to inactive Giα1- GDP and Giα3-GDP, but not Giα2 or Goα, to act as an inhibitor of GDP dissociation (GDI) much like Gβγ subunits [10, 12, 13]. Furthermore, we have shown that RGS14 is actively recruited from the cytosol to the plasma membrane by inactive Giα1-GDP or Giα3-GDP [14]. This recruitment is mediated by the GL domain of RGS14, and Giα1/RGS14 form a stable complex at the plasma membrane in cells. Other known Gα/GL protein complexes are thought to be involved with unconventional G protein signalling pathways [15–17] that rely on the combined action of non-receptor guanine nucleotide exchange factors (GEFs) and GL proteins in place of Gβγ subunits to regulate and mediate Gα signalling. These pathways have been shown to be important for cell division, neuronal development and synaptic plasticity in both lower and higher eukaryote [16, 18]. Thus, RGS14 may serve new functions that can be attributed to the action of its poorly understood GL domain distinct from those contributed by its RGS GAP domain.
Both G proteins and tyrosine kinase growth factor receptors activate MAPkinase signalling by distinct pathways [19–21]. MAPkinase signalling is important for many aspects of cell physiology including cell growth, survival, and differentiation as well as neuronal development and synaptic plasticity [22]. Since RGS14 tightly binds inactive forms of Giα1/3 and also contains tandem (R1 and R2) Ras/Rap binding domains (RBD), we investigated whether RGS14 could integrate the actions of Giα, Ras and Raf kinases to regulate MAP kinase signalling. We report that RGS14 binds activated H-Ras and its effectors Raf-1, B-Raf and A-Raf to regulate PDGF-stimulated Erk phosphorylation, and that this activity of RGS14 is regulated by its interactions with Giα1. RGS14 is recruited by H-Ras to form a stable complex at the plasma membrane, and it binds both H-Ras and/or Raf kinases either alone or together in cells. Expression of RGS14 inhibits PDGF-stimulated Erk phosphorylation and this inhibition is dependent on H-Ras binding, but is reversed by Giα1 which also blocks Raf binding to RGS14. Our findings demonstrate that RGS14 serves as a novel scaffold to integrate Giα1 and Ras/Raf/ MAPkinase signalling events through the action of its GL domain.
2. Experimental procedures
2.1 Plasmids and antibodies
The RGS14 cDNA used in this study was derived from rat (Genbank accession number of U92279) as previously described [10]. Truncated Flag-Raf-1 (ΔRaf-1) and 3xHA-B-Raf were kindly supplied by Dr. Haian Fu (Emory University). 3xHA-A-Raf cDNA was kindly supplied by Dr. Deborah Anderson (Saskatchewan Cancer Agency). HA-Raf-1 was kindly supplied by Dr. Ulf R. Rapp (Würzburg University). Glu–Glu epitope (EE) tagged recombinant Giα1/pcDNA3.1 plasmids, H-Ras 3xHA-tagged (N-terminus) plasmids and their constitutively active/inactive mutants were purchased from UMR cDNA Resource Center (Rolla, Missouri). The expression plasmids encoding RGS14 full-length, RGS14 deletion mutants coding for amino acids 1–213, 213–544, 213–490 and 444–544 cloned in-frame into the pcDNA3.1 (Invitrogen) were prepared as described previously [14]. Primer oligonucleotides encoding the 8 amino acid Flag tag (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) was used to generate N-terminally Flag-tagged RGS14 (Flag-RGS14). Site directed mutagenesis of RGS14 was performed by Quickchange Site Directed Mutagenesis kit (Stratagene). For the E92A, N93A double mutation placed into RGS14, the codon GAG was replaced by GCG, and the codon AAT was replaced by GCT; for the R333L mutation placed into RGS14, the codon AGA was replaced by CTT; for the H406A mutation, CAC were replaced by GCA. To express XFP-tagged Ric8A proteins in mammalian cells, the NheI/NotI YFP- and CFP-Ric8A DNA fragments described in Thomas, et al were subcloned into pcDNA3.1(+) (Invitrogen) [23].
Anti-Flag M2 antibody affinity gel was purchased from Sigma. Anti-Flag antibody, Alexa488-conjugated goat anti-rabbit and Alexa546-conjugated goat anti-mouse antibodies were purchased from Invitrogen. Anti-EE antibody was purchased from BD Biosciences. Monoclonal anti-HA horseradish peroxidase (HRP) conjugate antibody and monoclonal anti-HA TRITC (Rhodamine) conjugate antibody were purchased from Sigma. The following antibodies were purchased from Santa Cruz Biotechnology: anti-B-Raf (F-7) mouse monoclonal antibody (sc-5284), anti-Raf-1 (C-12) rabbit polyclonal antibody (sc-133), anti-Raf-1 (E-10) mouse monoclonal antibody (sc-7267), and anti-Giα1(R-4) mouse monoclonal antibody (sc-13533).
2.2 Confocal microscopy and fluorescence imaging
For cell imaging, HeLa cells were fixed for 10 min at room temperature with the following buffer: 20 mM PIPES pH 7.0, 1 mM MgCl2, 0.5 mM EGTA, 1 mM glutaraldehyde, 1 µg/ml aprotinin, 2 mM taxol, 0.1% Triton X-100, 2% paraformaldehyde. Fixed cells were subsequently blocked for 60 min at room temperature with PBS containing 10% goat serum and 1 mg/ml bovine serum albumin and incubated with a 1:1000 dilution of primary antibody, rabbit anti-Flag (Sigma), mouse anti-EE (BD Scientific), or Rhodamine-conjugated mouse anti-HA antibody (Sigma), for over night at 4°C. Cells were washed 3 times with PBS and then stained with 1:200 dilutions of Rhodamine-conjugated goat anti-mouse and FITC-conjugated goat anti-rabbit antibodies (Jackson Immuno-Research Laboratories) or Alexa553-conjugated goat anti-rabbit and Alexa633-conjugated goat anti-mouse antibodies (Invitrogen) for 1 h at room temperature. Cells were mounted with Vectashield mounting medium (Vector Laboratories). Images were collected on an Olympus IX51 inverted fluorescence microscope (Olympus) using a 100× oil immersion objective. Immunofluorescence analyses also were carried out using a LSM510 confocal laser scanning microscope (Zeiss). Images were acquired using an 63× oil immersion objective and processed using the Zeiss LSM image browser (version 2.801123) and Adobe Photoshop 7.0 (Adobe Systems).
2.3 Cell transfection and Ni-NTA pull-down assays of cell lysates
HeLa cells were obtained from the American Type Culture Collection. Cell transfections were performed according to protocols described in our previous work [14]. RGS14 pull-down assays were performed as described [10, 14, 24] with modifications. Cells were transiently transfected with selected recombinant plasmids according to the methods described above. Transfected cells were lysed in buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM EGTA, 10 mM MgCl2, 50 µg/ml Aprotinin, 100 µg/ml Leupeptin, 1 µM phenylmethylsulfonyl fluoride (PMSF) and 1% TritonX-100. Each lysate was incubated with 10 µg of either full-length Trx-H6-RGS14 or His6-Giα purified proteins for 2 hours. The complexes were incubated an additional 1 h with 100 µl of Ni-NTA for 60 min. The Ni-NTA beads were centrifuged and then washed five times for Raf-1 and seven times (for B-Raf) each with 1 ml ice cold washing buffer. Protein complex were eluted into 100 µl Laemmli buffer. Samples were separated by 11% SDS-PAGE and transferred to nitrocellulose membranes (Millipore Corp., Bedford, MA) and immunoblotted with selected antibodies for visualization.
2.4. Anti-Flag M2 antibody affinity gel immunoprecipitation and immunoblots
HeLa cells were seeded on 10 cm dishes and cDNA transfected using Lipofectamine 2000 (Invitrogen). For immunoprecipitation of expressed proteins, transfected cells were washed three times in ice cold PBS and lysed in buffer as described above in Ni-NTA pull-down assay. The lysate was cleared by centrifugation at 50,000 g for 30 min at 4°C. Protein concentrations were determined using the Bradford reagent (Bio-RAD). Lysates were then mixed with 50 µg Flag M2 gel (Sigma) directed against the Flag-tagged fusion protein. The mixed complexes were incubated for 2 h at 4°C with continuous rotation. Flag immunocomplexes were washed three times for Raf-1 and six times for B-Raf in ice-cold TBS buffer. Immunoprecipitates were suspended in a Laemmli sample buffer followed by boiling for 5 min. The protein samples were resolved by 11% SDS-PAGE, transferred to a nitrocellulous membrane (Bio-RAD) and probed with appropriate antibodies, e.g. HRP labeled monoclonal anti-Flag antibody (Sigma), HRP labeled monoclonal anti-HA antibody (Sigma), or polyclonal anti-Raf-1 antibody (Santa Cruz Biotechnology) followed by appropriate secondary antibody. Detection of immunolabelled proteins was accomplished by using the ECL-Plus chemiluminescent system (GE Healthcare).
2.5 Phospho-p44/42 Erk assay and statistical analysis
HeLa cells were seeded on 12-well plates and cDNA transfected with Lipofectamine 2000 (Invitrogen) as described above. After 24 h, transfected cells were switched into serum free DMEM for 6 h. Cells then were stimulated with 25 ng/ml PDGF (Chemicon), 100 ng/ml EGF (Sigma) or fetal calf serum, FCS (20% final) for the times described in the text. The reactions were stopped by transferring cells to ice, and immediately lysed by removing media and the addition of 400 µl/well Laemmli sample buffer. Recovered samples were then boiled for 5 min. The sample mixture was electrophoresed on 11% SDS-PAGE and transferred to nitrocellulose membranes and probed for both active and total Erk using anti-phospho-p44/42 Erk polyclonal antibody (Cell Signalling) and anti-p44/42 Erk polyclonal antibody (Cell Signalling), respectively. Detection of immunolabelled proteins was accomplished using the ECL-Plus chemiluminescent system. Quantification of proteins was achieved by densitometry using a Scion Imaging system. Numbers derived using this analysis from multiple experiments were pooled and subjected to statistical analysis (paired t-test).
3. Results
3.1 RGS14 binds to and co-localizes with activated H-Ras in cells
RGS14 was first identified as a binding partner for the Ras-like GTPase, Rap1/2 [7]. Ras and Rap each specifically bind RBD domains and RGS14 contains tandem (R1 and R2) RBDs [7, 25]. We tested if RGS14 could bind H-Ras. RGS14 (Flag-RGS14) was transfected together with different forms of Ras (HA-Ras) in HeLa cells. We observed that RGS14 bound to and co-immunoprecipitated with constitutively active (G/V mutant) H-Ras, but only weakly to wild type H-Ras and not at all to inactive (S/N mutant) H-Ras in HeLa cell lystes (Fig. 1A). We postulate that the weak binding of wild type H-Ras reflects a small population of active H-Ras-GTP present in cell lysates. We also examined the influence of H-Ras on RGS14 subcellular localization (Fig. 1B). When expressed alone in HeLa cells, RGS14-GFP localizes to the cytosol and H-Ras to membranes, in particular the plasma membrane. Active H-Ras(GV) localized most strongly to membranes, whereas inactive H-Ras(SN) failed to localize at membranes. When RGS14 is co-expressed with different forms of H-Ras, active H-Ras(GV) recruited RGS14 to the membrane. These findings strongly suggest that active H-Ras is a newly described binding partner for RGS14.
Figure 1. RGS14 binds and co-localizes with activated H-Ras in cells.
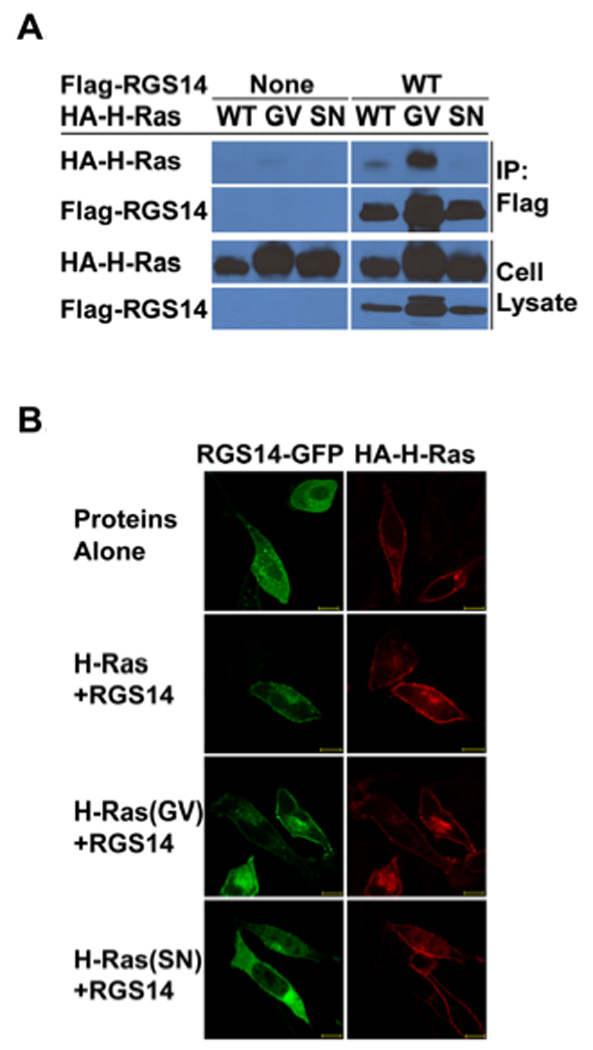
(A) RGS14 co-immunoprecipitates (co-IP) with active H-Ras from HeLa cell lysates. HeLa cells were co-transfected with Flag-tagged (Flag-RGS14) together with individual HA-tagged H-Ras (HA-H-Ras) isoforms including the activated (G/V) mutant isoform, or the inactive (S/N) mutant isoform, respectively. Whole-cell extracts were subjected to immunoprecipitation followed by immunoblot with the indicated antibodies. Cell extracts transfected with individual H-Ras isoforms alone were set as controls. The cell lysate shown in the lanes represent 5% of the whole cell extracts used for IP. (B) RGS14 co-localizes with active H-Ras(GV), but not inactive H-Ras(SN) on the plasma membrane. HeLa cells were transfected with HA-H-Ras, RGS14-GFP or RGS14-GFP together with wild type HA-H-Ras, active HA-H-Ras(G/V) or inactive HA-H-Ras(S/N), respectively. After transfection, cells were fixed, and HA-H-Ras isoforms were immunostained with Rhodamine-conjugated anti-HA antibody. Proteins were visualized by confocal microscopy as described in Experimental Procedures. RGS14 was detected by intrinsic fluorescence of GFP using confocal microscopy. Scale bars equal 10 µm. Results are representative of at least three separate experiments for each condition.
3.2. The first (R1) RBD domain of RGS14 is critical for H-Ras binding
We next examined which region of RGS14 is responsible for binding H-Ras. RGS14 contains two contiguous Ras/Rap-binding domains (RBDs), R1 and R2 (Fig. 2A). Based on previous studies, RBD domains present in Raf kinases and other proteins serve as specific binding sites for activated Ras and Rap isoforms, and a conserved arginine residue within all RBDs is critical for this interaction [9, 26]. RGS14 contains an analogous arginine within the R1 RBD (R336) whereas a histidine occupies the analogous residue in the R2 RBD (H409) [9]. Disrupting R336 inhibits Rap2 binding whereas disrupting this H406 does not [9, 27, 28]. To investigate H-Ras interaction with RGS14, we introduced these same single substitution mutations (R333L or H406A) into the R1 and R2 RBD domains, respectively, of Flag-tagged RGS14 (Fig. 2A and 2B). When each of these RGS14 mutants was co-expressed with either wild type H-Ras, active H-Ras(GV) or inactive H-Ras(SN), we found that H-Ras was inhibited from binding to the R333L mutant but not the H406A mutant of RGS14 (Fig. 2B). This also was reflected in RGS14 subcellular localization (Fig. 2C). When co-expressed together in HeLa cells, the R333L mutant of RGS14 failed to co-localize with active H-Ras(GV) whereas the H406A mutant co-localized at membranes with active H-Ras(GV) in a pattern similar to wild type RGS14 (compare Fig. 2C to Fig. 1B). These findings indicate that active H-Ras(GV) binds the R1, but not the R2 RBD of RGS14.
Figure 2. H-Ras binds the first (R1) RBD of RGS14.
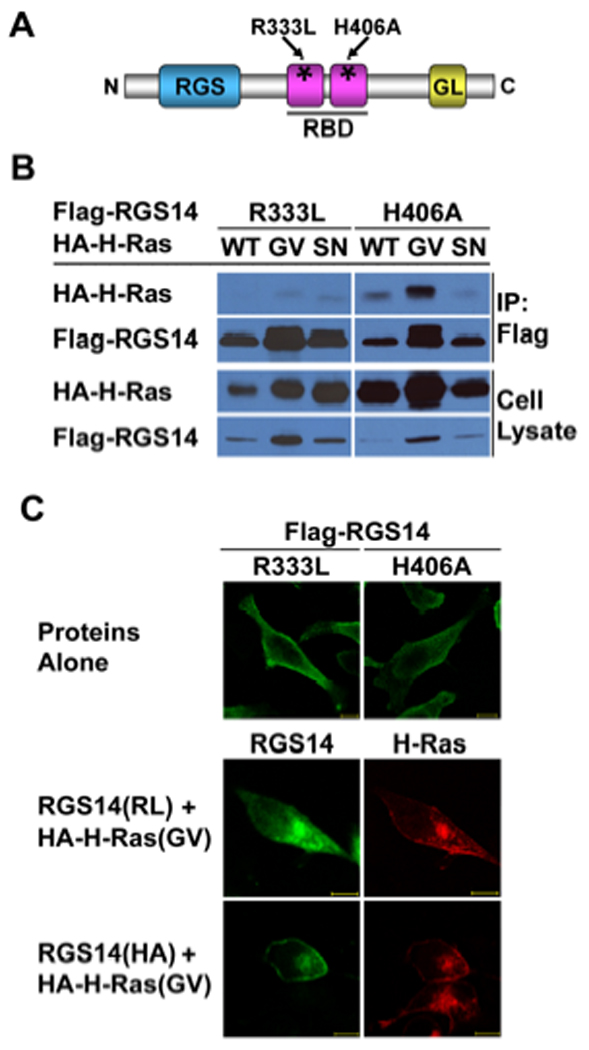
(A) Schematic diagram illustrating RGS14 constructs including point mutants of R333L within the R1 RBD, and H406A within the R2 RBD, as described. (B) Disruption of the R1 RBD, but not the R2 RBD, prevents H-Ras binding. HeLa cells were co-transfected with Flag-tagged RGS14 (Flag-RGS14) mutants together with individual HA-tagged Ras (HA-H-Ras) isoforms including the active (G/V) mutant or the inactive (S/N) mutant isoforms, respectively. Whole-cell extracts were subjected to anti-Flag immunoprecipitation (IP) followed by immunoblot with the indicated antibodies. The cell lysate shown in the lanes represent 5% of the whole cell extracts used for IP. (C) Disruption of the R1 RBD, but not the R2 RBD, alters co-localization of active H-Ras(GV) with RGS14 in HeLa cells. HeLa cells were co-transfected with HA-H-Ras(GV) and either the Flag-RGS14 R333L mutant or H406 mutant, constructs. After transfection, cells were fixed, and HA-H-Ras isoforms were immunostained with anti-HA-Rhodamine antibody as described. Flag-RGS14 was immunostained by anti-Flag rabbit polyclonal antibody followed by FITC labeled goat anti-rabbit secondary antibody. Proteins were visualized by confocal microscopy as described. Scale bars equal 10 µm. Results are representative of at least three separate experiments for each condition.
3.3. RGS14 binds to and co-localizes with Raf kinases in cells
The Raf kinases (Raf-1, B-Raf and A-Raf) are well described effectors of H-Ras. Therefore, we examined if RGS14 could also interact with one or more of the Raf kinases. Initial studies tested if purified recombinant His-RGS14 could bind any of the isoforms of Raf kinases (HA-tagged Raf-1 or B-Raf) when expressed in HeLa cell lysates. We found that expressed B-Raf and Raf-1 each bound purified His-RGS14 and were recovered by Ni-NTA resin (Fig. 3A). Similar results were observed when Flag-RGS14 was co-expressed with HA-B-Raf and HA-Raf-1 in HeLa cells. B-Raf and Raf-1 both were recovered following immuno-precipitation of RGS14 using anti-Flag resin (Fig. 3B), but the proteins did not bind non-specifically to the resin. RGS14-GFP also co-localized with either B-Raf or Raf-1 in both the cytosol and membranes of HeLa cells as visualized by confocal microscopy (Fig. 3C). We also found that RGS14 binds to and co-localizes with A-Raf in HeLa cells (Supplemental Figure 1). These findings suggest that Raf kinases are newly identified binding partners of RGS14, or components of RGS14-multi-protein complexes.
Figure 3. RGS14 binds Raf kinases.
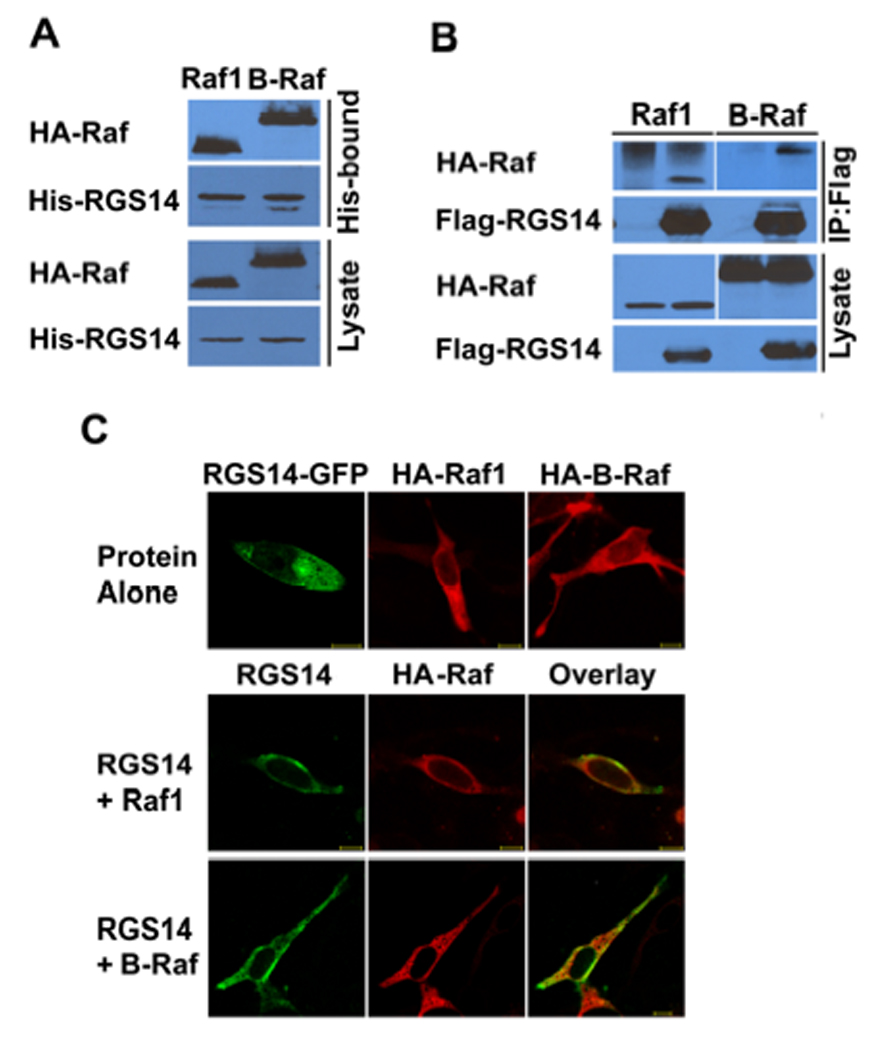
(A) Protein interaction trap (pull-down) assays to detect Raf complexed with purified RGS14. Cell lysates expressing either HA-Raf-1 or HA-B-Raf were incubated with purified Trx-H6-RGS14 and, following recovery by Ni-NTA, proteins were detected by anti-HA (Raf-1 or B-Raf) or anti-His (RGS14) antibody, respectively. (B) RGS14 co-immunoprecipitates (IP) with Raf kinases from cell lysates when expressed in cells. HeLa cells were co-transfected with Flag-tagged (Flag-RGS14) together with individual HA-tagged Raf kinase (HA-Raf-1 or HA-B-Raf) isoforms, respectively. Whole-cell extracts were subjected to immunoprecipitation followed by immunoblot with the indicated antibodies. The cell lysate shown in the lanes represents 5% of the whole cell extracts used for IP. (C) RGS14 and Raf kinases co-localize in the cytosol of cells. RGS14-GFP and Rafs (HA-Braf or HA-Raf-1) were co-transfected into HeLa cells. After transfection, cells were fixed, and Rafs were immunostained with anti-HA antibody (Raf-1 or B-Raf) as described. RGS14-GFP was detected by autofluorescence. Proteins were visualized by confocal microscopy as described. Scale bars equal 10 µm. Results are representative of at least three separate experiments for each condition.
3.4. The N-terminal regulatory region of Raf kinases binds to the RBD region of RGS14
To elucidate the binding regions between Raf and RGS14, we generated four truncated forms of Flag-RGS14 (Fig. 4A) including: 1) the isolated RGS domain (amino acids 1–213), 2) the isolated RBD domains (213–490), 3) the RBD/GoLoco domains (213–544), or 4) the isolated GoLoco domain (444–544). Each of the truncated forms of RGS14 was co-expressed together with either HA-B-Raf or HARaf-1 in HeLa cells, and cell lysates were subjected to anti-flag immuno-precipitation (Fig. 4A). We found that only those fragments containing the RBD domains were able to bind either Raf-1 or B-Raf, but not those fragments containing only the RGS domain or the GPR/GoLoco domain, thereby demonstrating that the tandem RBD domain region is essential for Raf binding. In contrast to what was observed with H-Ras binding to RGS14, introduction of the R333L or the H406A single point mutations into the RBDs failed to alter Raf binding to RGS14 (data not shown).
Figure 4. The N-terminal regulatory region of Raf kinases binds to the RBD region of RGS14.
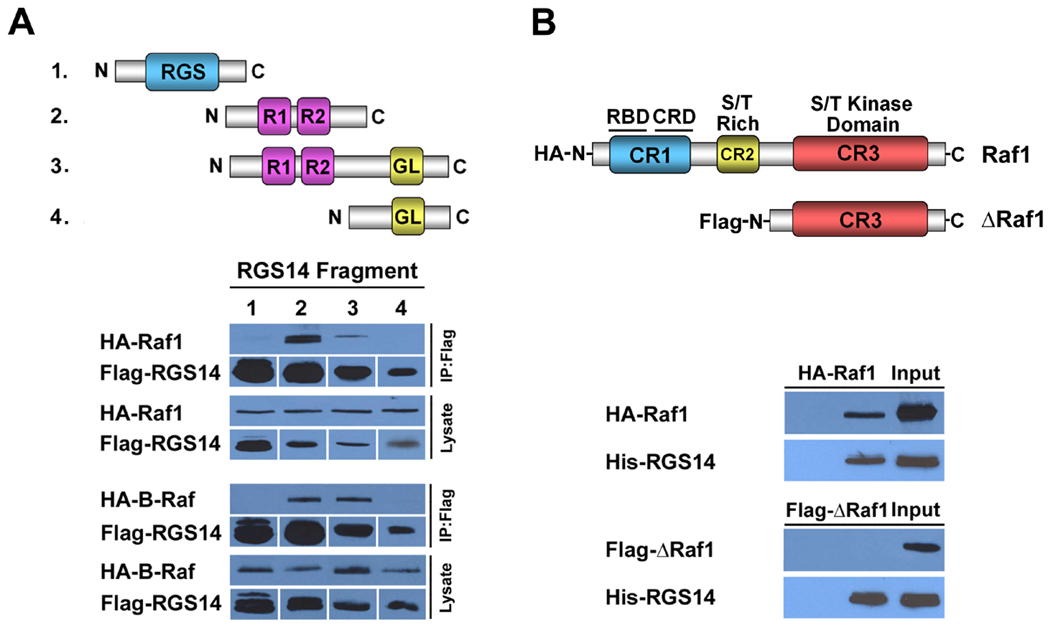
(A) The RBD region of RGS14 is required for Raf binding. Schematic diagram illustrating truncated Flag-RGS14 fragments used to co-immunoprecipitate with Raf kinases, as described in Experimental Procedures. RGS14 RBD domains are required for Raf binding. For immunoblots (below), HeLa cells were co-transfected with constructs expressing individual truncated Flag-RGS14 fragments together with HA-tagged Raf constructs (HA-Raf-1 or HA-B-Raf), respectively. Whole-cell extracts were subjected to immunoprecipitation followed by immunoblot analysis with the indicated antibodies. Only RGS14 fragments containing the RBD regions are able to bind either Raf-1 or B-Raf. Cell lysates shown in the lanes represent 5% of the whole cell extracts used for IP. (B) The N-terminal regulatory region of Raf-1 is required for RGS14 binding. Schematic diagram illustrating full-length Raf-1 and a truncated Raf-1 fragment missing the regulatory domains (CR1, CR2) but containing a functional kinase domain. HeLa cell lysates containing either HA-Raf-1 or HAB- Raf were mixed with purified Trx-H6-RGS14 and subjected to protein interaction trap assays (Ni- NTA pull-down). Recovered proteins were subjected to immunoblot analysis with anti-Raf-1 antibody and anti-Flag (to detect RGS14) antibody, respectively. Results are representative of at least three separate experiments for each condition.
Raf kinases share a conserved overall structural homology with three conserved domains designated as CR1, CR2 and CR3 (Fig. 4B). The CR1 region, located within the N-terminal region, contains a single RBD domain and a cysteine-rich zinc-binding domain (CRD). The CR2 region, located between CR1 and CR3, contains a serine/threonine rich domain, whereas the CR3, located in the C-terminal region, contains the serine/threonine (S/T) protein kinase catalytic domain. The CR1 and CR2 regions are important for the activation and regulation of Raf kinase activity and have been defined as essential sites for protein–protein interaction [29]. The N-terminal fragment region of Raf-1 (l-257) is able to homodimerize, whereas the C-terminal Raf fragment, BXB-Raf (l-25/306–648), does not [19]. We generated a truncated form of the Raf-1 (ΔRaf-1) missing the CR1 and CR2 regulatory regions but containing the CR3/kinase domain. When full-length Raf-1 and ΔRaf-1 were compared for their capacity to bind purified His-RGS14, we found that Raf-1, but not ΔRaf-1 bound RGS14, and was recovered using the Ni-NTA resin (Fig. 4B).
3.5. H-Ras and Raf-1 facilitate one another’s binding to RGS14 to form a heterotrimeric complex
Several scaffolding proteins have been identified that are important for the spatiotemporal organization and context-specific signalling of the different MAPkinase signalling pathways [30]. One hallmark of scaffolds is their capacity to facilitate binding and assemble functionally related protein partners. Through the tandem RBD domains, RGS14 can bind active H-Ras and Raf kinase independently. We examined if Flag-RGS14 could behave as a scaffold to bind both HA-H-Ras(GV) and HA-Raf-1 simultaneously. All three proteins were co-expressed in HeLa cells and cell lysates were subjected to anti-Flag immunoprecipitation. We found that H-Ras and Raf-1 both co-precipitated with RGS14 (Fig. 5A). A complicating factor in these experiments is that active H-Ras(GV), Raf-1 and RGS14 each bind one another independently, so confirming that H-Ras(GV) and Raf-1 are binding to the same RGS14 molecule in cells is technically difficult. To address this question we expressed each protein (Flag-RGS14, H-Ras(GV) and Raf-1) independently in different sets of cells. Cell lysates of each protein were prepared separately (Fig. 5B), and the amounts of expressed protein were quantified. The cell lysates were then mixed in various combinations to control and equalize the amount of each protein present in the samples. Proteins were then subjected to anti-Flag immunoprecipitation and H-Ras and Raf-1 bound to recovered Flag-RGS14 were detected (Fig. 5B). We observed that detectable amounts of H-Ras and Raf-1 each bound RGS14 independently, as before. However, when lysates of each protein were all mixed together, we found that considerably more active H-Ras and Raf-1 bound to RGS14. These findings are consistent with the notion that RGS14 is a scaffold that assembles H-Ras and Raf-1 to form a stable RGS14/H-Ras/Raf-1 ternary complex.
Figure 5. H-Ras and Raf facilitate one another’s binding to RGS14 to form a heterotrimeric complex.
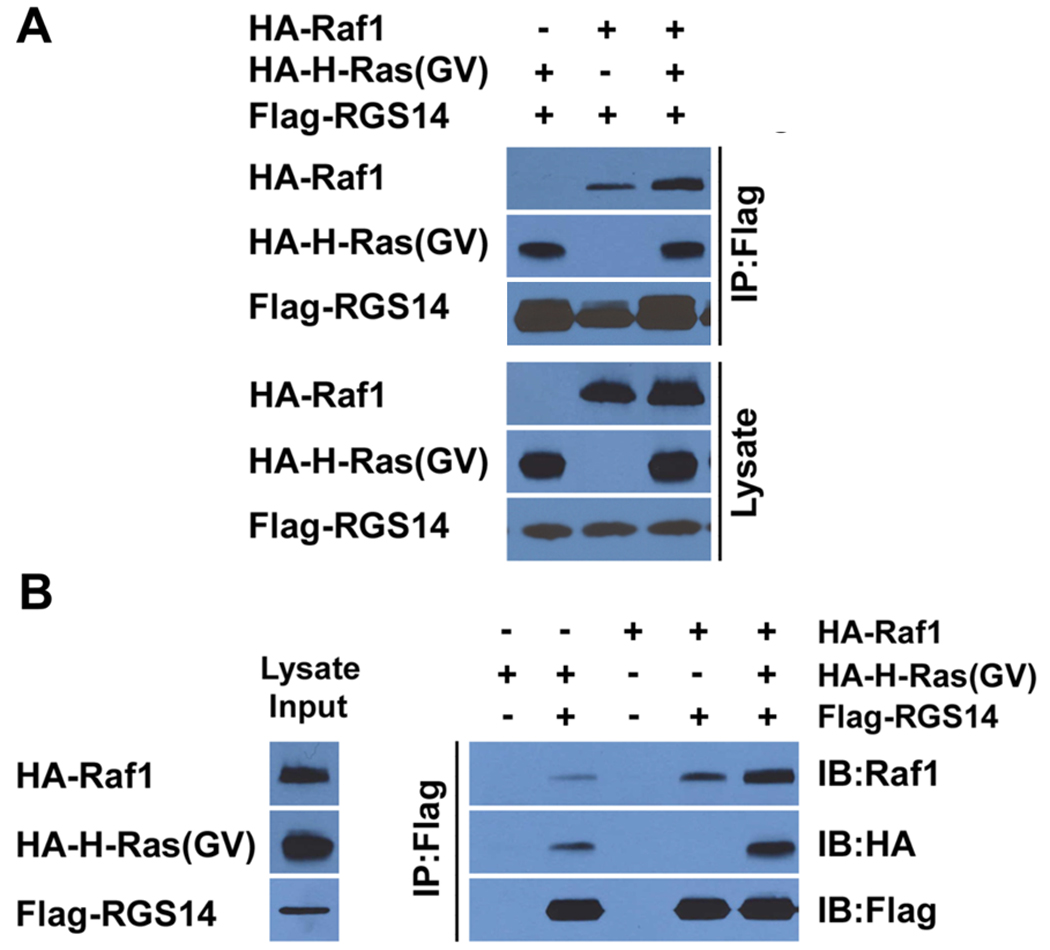
(A) RGS14 forms a heterotrimeric protein complex with Raf kinase and active H-Ras. HeLa cells were co-transfected with Flag-RGS14 together with either HA-Raf-1 kinase or active H-Ras(GV), or with both HA-Raf and HA-Ras(GV) together, respectively. Whole-cell extracts were subjected to immunoprecipitation (IP) and recovered proteins detected by immunoblot analysis using anti-HA antibodies to detect H-Ras(GV), anti-Raf-1 antibodies to detect Raf-1, or with anti-Flag to detect RGS14. Cell lysates shown in the lower panel represent 5% of the whole cell extracts used for IP. Results are representative of at least three separate experiments with similar results. (B) H-Ras and Raf-1 facilitate one another’s binding to RGS14. HA-Raf-1, HA-Ras(GV) and Flag-RGS14 were each expressed independently in different sets of HeLa cells. The relative amounts of expressed protein were determined in each extract, and appropriate volumes of extract containing equal amounts of expressed protein were mixed as described in experimental procedures. Resulting protein complexes were subjected to immunoprecipitation with anti-Flag resin, followed by immunoblot analysis with the indicated antibodies above. The cell lysates shown in the left lane represent 5% of the whole cell extracts used for IP.
3.6. RGS14 selectively inhibits PDGF-stimulated ERK phosphorylation
Various extracellular factors activate H-Ras and Raf kinases to stimulate MAP kinase signalling pathways that culminate with the phosphorylation and activation of Erk1/2. These include, but are not limited to, various growth factors (e.g. PDGF and EGF) and other mitotic factors present in blood serum. Since RGS14 binds both H-Ras and Raf kinases, we examined if RGS14 can modulate H-Ras/Raf-mediated Erk phosphorylation. We observe that HeLa cells express cell surface receptors for PDGF, EGF and serum factors that, when activated, stimulate Erk phosphorylation (Fig. 6). When cells were transfected with an RGS14 construct, we find that RGS14 protein expression markedly inhibited PDGF-stimulated Erk phosphorylation (Fig. 6A). In contrast, RGS14 did not alter Erk phosphorylation stimulated by either EGF (Fig. 6B) or 20% serum (Fig. 6C). These findings suggest that RGS14 binding to H-Ras and Raf kinases, (as shown above), serves to inhibit PDGF-stimulated MAPkinase signalling.
Figure 6. RGS14 inhibits PDGF-stimulated p42/44 Erk (Erk1/2) phosphorylation in HeLa cells.

HeLa cells expressing either empty vector (control) or a Flag-RGS14 construct were treated with either with (A) PDGF-AB, 20 ng/ml; (B), EGF, 100 ng/ml; or (C) fetal calf serum, 20 % final. Cells were treated for 10 min and compared to unstimulated cells as control. Protein extracts (100 ug) were prepared and subjected to immunoblot analysis using anti-phosphoErk p42/44 antibody (Erk1/2), anti-Flag (for RGS14) and anti-β-COP (loading control). Erk1/2 phosphorylation results from three to five separate experiments for each condition were quantitated by densitometry analysis and the results pooled and averaged (+/− SEM), as described in Experimental Procedures. Immunoblot results are shown from a single representative experiment used for densitometry. Differences in control and stimulated conditions were compared and subjected to statistical analysis (paired t-test). RGS14 inhibited PDGF-stimulated Erk1/2 phosphorylation significantly (*P<0.01).
3.7. RGS14-mediated inhibition of PDGF signalling depends on H-Ras binding and is reversed by Giα1
We next examined the importance of H-Ras binding on RGS14 inhibition of PDGF signalling. As shown earlier (Fig. 2), the R333L mutation placed into the R1 RBD domain prevents H-Ras binding to RGS14. As shown in Figure 7A, RGS14 inhibits PDGF-stimulated Erk phosphorylation as before. However, in stark contrast, we observe that the R333L mutation of RGS14 failed to inhibit PDGF-mediated Erk phosphorylation in HeLa cells (Fig. 7A), indicating that RGS14 inhibition of PDGF signalling depends on H-Ras binding. We have previously shown that RGS14 directly binds inactive Giα1-GDP via its GPR/GoLoco domain, and that Giα1 recruits RGS14 to form a stable complex at the plasma membrane [14]. Therefore, we next examined the influence of Giα1 binding on RGS14 inhibition of PDGF signalling (Fig. 7B). We observed that co-expression of Giα1 reversed RGS14 inhibition of PDGF signalling, whereas Giα1 alone had no effect of PDGF-stimulated Erk phosphorylation. Taken together, these findings indicate that RGS14 inhibition of PDGF signalling depends on H-Ras binding and that this inhibition is reversed by Giα1 binding to RGS14.
Figure 7. RGS14 inhibition of PDGF-signaling depends on H-Ras binding and is reversed by Giα1.
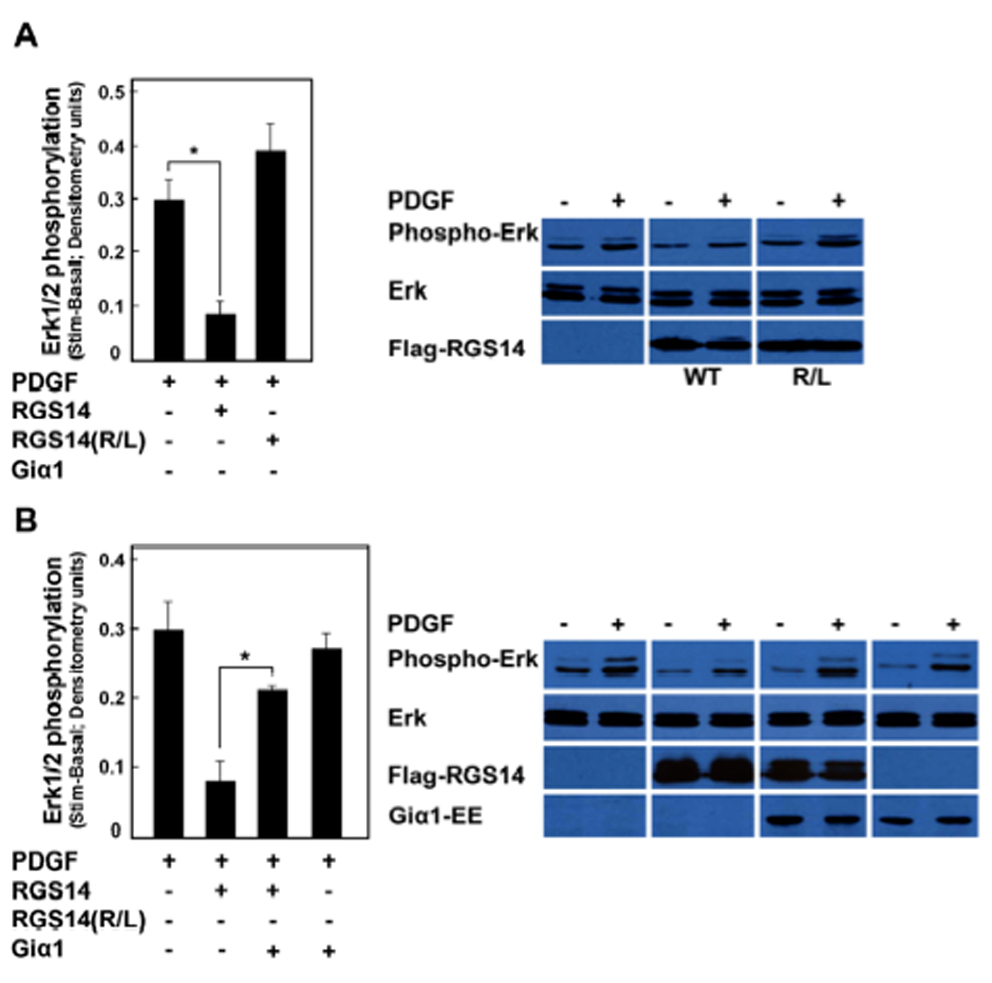
(A) RGS14 inhibition of PDGF-signaling depends on H-Ras binding. HeLa cells transfected with either empty vector (control), Flag-RGS14 or with the R333L mutant of Flag-RGS14 were stimulated with PDGF-AB (25 ng/ml) for 10 min. Phosphorylation of Erk1/2 in PDGF-AB was detected in cell lysates by immunoblot analysis using the anti-phosphoErk42/44 antibody. Results from three separate experiments were quantified by densitometry analysis and the pooled results averaged (+/− SEM) as described in Experimental Procedures. Immunoblot results are shown from a single representative experiment used for densitometry. Differences in unstimulated basal and stimulated conditions were compared and subjected to statistical analysis (paired t-test). RGS14 significantly inhibited PDGF-stimulated Erk1/2 phosphorylation (*P<0.05) but RGS14(R333L) did not. (B) RGS14 inhibition of PDGF-signalling is reversed by Giα1. HeLa cells transfected with either empty vector (control), Flag-RGS14, Flag-RGS14 and Giα1-EE together, or with Giα1-EE alone, were stimulated with PDGF-AB (25 ng/ml) for 10 min. Phosphorylation of Erk1/2 in PDGF-AB was detected in cell lysates by immunoblot analysis using the anti-phosphoErk p42/44 antibody. Results from three separate experiments were quantified by densitometry analysis and the pooled results averaged (+/− SEM) as described in Experimental Procedures. Immunoblot results are shown from a single representative experiment used for densitometry. Differences in unstimulated basal and stimulated conditions were compared and subjected to statistical analysis (paired t-test). RGS14 inhibition of PDGF-stimulated Erk1/2 phosphorylation was significantly reversed by co-expression of Giα1 (*P<0.01), whereas Giα1 alone had no effect on PDGF signalling.
3.8. Giα1 binding to RGS14 inhibits Raf binding to RGS14
We next sought to understand the basis for Giα1 reversal of RGS14 inhibition of PDGF signalling. HeLa cells were transfected with Flag-RGS14 alone or in the presence of Raf-1, Giα1 and/or H-Ras(GV) (Fig. 8). Cell lysates were subjected to anti-Flag immuno-precipitation and proteins bound to RGS14 were examined. As before, we observe that Raf-1, Giα1 and H-Ras(GV) each bind to RGS14 independently. However, in each case where Giα1 and Raf-1 are both present, Giα1 binds RGS14 and Raf-1 does not. In contrast, Giα1 binding does not affect H-Ras binding. This experiment was repeated in exactly the same way except with B-Raf in place of Raf-1, and similar results were obtained (data not shown). That is, Giα1 reduced the amount of B-Raf binding to RGS14. These findings suggest that Giα1 binding and Raf binding to RGS14 are mutually exclusive.
Figure 8. Giα1 inhibits Raf kinase binding to RGS14.
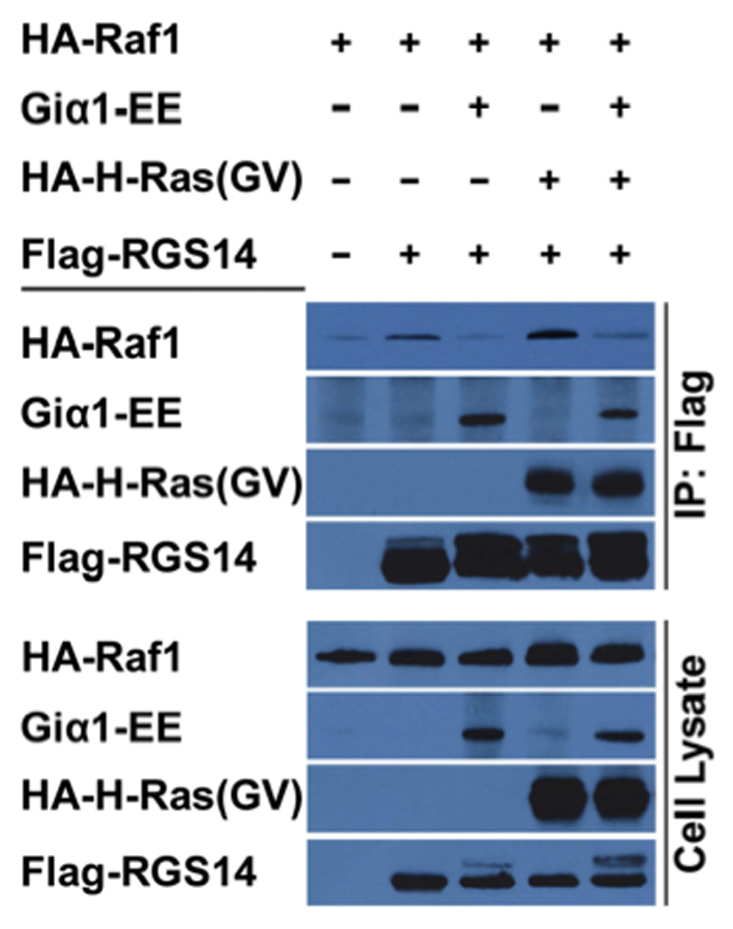
HeLa cells were transfected with HA-Raf-1 alone or in the presence of either Flag-RGS14, Flag-RGS14 plus Giα1-EE, RGS14 plus HA-H-Ras(GV) or Flag-RGS14 plus H-Ras(GV) and Giα1-EE. Cells were transfected as indicated by the labels above the lanes in the gel. Cell extracts were prepared and subjected to anti-Flag immunoprecipitation (IP), and recovered proteins were detected by immunoblot analysis using anti-HA (H-Ras and Raf-1), anti-EE (Giα1) or anti-Flag (RGS14). The cell lysates shown in the lanes represent 5% of the whole cell extracts used for IP. Results are representative of at least three separate experiments for each condition.
3.9. Giα1 indirectly recruits Raf kinases to the plasma membrane
Since Giα1 inhibits Raf binding to RGS14 in cells, we examined Giα1 interactions with Raf kinases. Initial studies examined if Giα1 binds to Raf kinases. HeLa cell lysates expressing either Raf-1 or B-Raf were mixed with purified His6-Giα1. When Giα1 was recovered by Ni-NTA pull-down and examined for the presence of bound Raf, we found that neither Raf-1 nor B-Raf bound to purified Giα1 (Fig. 9A). However, we also observed that expression of Giα1 recruited either B-Raf or Raf-1 to the plasma membrane in HeLa cells (Fig. 9B). When co-expressed together with RGS14 and either B-Raf or Raf-1, Giα1 recruited both proteins to the plasma membrane (Fig. 9C). In parallel studies, we also found that Giα1 recruited A-Raf to the plasma membrane (Supplemental Figure 2). Together, these findings suggest that Giα1 does not directly regulate Raf activity, but somehow contributes to the co-ordinate translocation of the signalling complex to the plasma membrane.
Figure 9. Giα1 does not bind Raf kinases, but does recruit Raf kinases to the plasma membrane in cells.
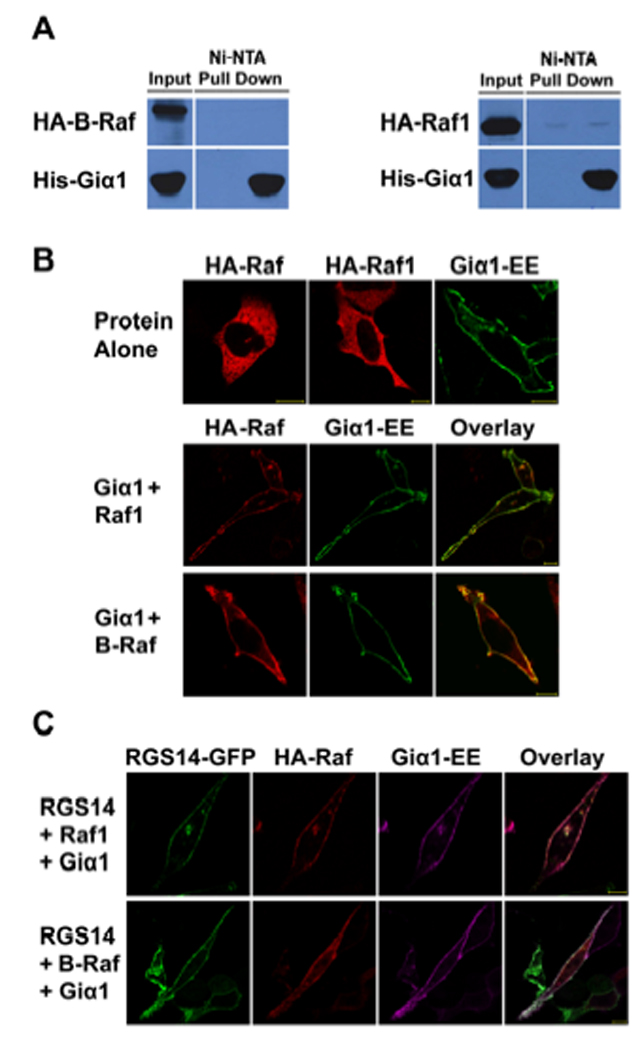
(A) Giα1 does not bind Raf kinases. Purified His6-Giα1 was incubated with cell extracts expressing either HA-Raf-1 or HA-B-Raf kinase. Samples were subjected to protein interaction trap assays (Ni-NTA pull-down) and recovered proteins were detected by immunoblot analysis. (B) Giα1 and Raf kinases co-localize at the plasma membrane. EE-tagged Giα1, Raf-1 or B-Raf were transfected into HeLa cells alone or Giα1-EE and each Raf kinase were co-transfected together. After transfection, cells were fixed, and Rafs were detected by immunostaining with anti-HA antibody and Giα1 was detected using the anti-EE antibody, as described. (C) Giα1 recruits RGS14 and Raf kinases to the plasma membrane. HeLa cells were transfected with RGS14-GFP, Giα1-EE and either HA-Raf-1 or HA-B-Raf., respectively. Rafs and Giα1 were detected as described above. RGS14-GFP was detected by autofluorescence of GFP. Proteins were visualized by confocal microscopy as described. Scale bars equal 10µm. Results are representative of at least three separate experiments for each condition.
4. Discussion
RGS14 is a highly unusual signalling protein that contains two distinct Gα interaction sites (RGS domain and GL domain) and tandem (R1 and R2) RBD domains that are known in other proteins to bind Ras and Rap GTPases. Indeed, RGS14 was first identified as a novel Rap1/2 binding partner [7]. We and others have shown previously that the RGS domain of RGS14 is a promiscuous and nonselective GAP for active forms of Giα and Goα, and that the GL domain is a selective GDI for Giα, but not Goα [7, 10–12, 27]. We subsequently reported that the GL motif of RGS14 selectively binds inactive Giα1-GDP and Giα3-GDP, but not Giα2, and that both of these Giα isoforms strongly recruit RGS14 to the plasma membrane to form a tight complex with the GL domain to influence the dynamic subcellular localization of RGS14 [14]. Here we expand those findings to show that RGS14 binds activated H-Ras and its Raf kinase effectors (Raf-1, B-Raf and A-Raf) to modulate their signalling. H-Ras and Raf each bind RGS14 independently, but the two proteins appear to facilitate one another’s binding to RGS14. Alternatively RGS14 may facilitate formation of the H-Ras/Raf complex which, in turn, binds more efficiently to RGS14. Active H-Ras selectively binds the R1 RBD domain of RGS14 and the Raf kinases also bind within the RBD region. We find that expression of RGS14 markedly inhibits PDGF-stimulated activation of Erk1/2 and that this inhibition is dependent on H-Ras binding but is reversed by co-expression of Giα1. In follow-up studies to understand this better, we found that Giα1 binding inhibited Raf binding to RGS14, suggesting that the binding of Giα1 and Raf are mutually exclusive. These findings demonstrate that RGS14 regulates the activity of multiple GTPases with distinct functions, and are consistent with the idea that RGS14 is a newly appreciated integrator of G protein (Giα) and Ras/Raf signalling.
RGS14 is one of only two known proteins (RGS12 the other) that contains three different binding domains for distinct GTPases, and it sits at the intersection of three families of signalling proteins that contain either an RGS domain, a GL domain(s) or an RBD domain(s). Recent studies indicate that proteins containing GL domains (GL proteins) modulate unconventional G protein signalling pathways and events that do not involve cell surface GPCRs [15, 17, 18]. Much of the previous work on RGS14 focuses on its presumed role as an RGS protein that modulates GPCR/G protein signalling. However, based on our findings here and our understanding of other proteins that contain GL domains, we propose that RGS14 is a newly appreciated integrator of unconventional G protein signalling and MAPkinase signalling. Our reported findings provide the framework of a model to describe how these functionally opposing domains work together to bind and modulate the functions of inactive and active Gα, and Ras/Raf/MAP kinase signalling.
Our proposed model highlights the GL domain as the first point of contact between Gα and RGS14, which differs from conventional models that highlight the RGS domain in this role [2, 4]. We envision RGS14 to function primarily as a GL protein that also contains an RGS domain. We have proposed that RGS14 is complexed with subsets of Giα1-GDP or Giα3-GDP via its GL domain and these complexes are devoid of Gβγ [14]. Our findings presented here are consistent with additional features of this model. We postulate that a non-receptor GEF such as Ric8A and/or possibly others [17, 31] stimulates nucleotide exchange and GTP binding to Giα which, in turn, promotes dissociation of RGS14 because the GL domain does not bind Gα-GTP. This is consistent with our recent observation that Ric8A interacts with RGS14 to promote Giα1 dissociation (C. Vellano et al, unpublished observation). Once free from Giα1, RGS14 is available to bind active H-Ras and Raf kinase to inhibit MAPKinase signalling, as we show here. In this model, the lifetime of the RGS14-Ras/Raf complex is limited by the RGS domain, which could restore Giα-GDP and promote reformation of Giα-GDP/GL-RGS14 complex, and consequential dissociation of Raf and H-Ras. Consistent with this idea, we find that Giα1 and Raf-1 binding to RGS14 is mutually exclusive. An arrangement that incorporates the RGS domain into the same protein restricts the RGS domain pairing to the Gα pre-selected by the GL domain (Giα1 or Giα3). This configuration imparts spatially restricted RGS/Gα selectivity and eliminates the necessity for intrinsic RGS/Gα selectivity. This is consistent with our earlier observation that the RGS domain is a non-selective GAP for Gi/oα [10]. We speculate that the RGS domain activity is regulated and unavailable as a GAP until the correct conformation is achieved. Consistent with this idea, we observe that both the GL domain [24] and the N-terminal region containing the RGS domain of RGS14 are phosphorylated (F.J. Shu, D.P. Cowan et al, unpublished observation), which could be important mechanisms for regulating these protein interactions.
Such a model exhibits many mechanistic parallels with established models of conventional GPCR/Gα signalling (1) except that a non-receptor GEF substitutes for a GPCR and the GL motif of RGS14 substitutes for Gβγ [23, 31, 32]. Of note, similar models have been proposed for proteins known to regulate cell division (but unrelated to MAPkinase signalling) in lower eukaryotes including worms and flies [32–34]. However, distinct GL and RGS proteins are thought to act in these systems whereas, in the case of RGS14 shown here, the RGS activity is already built into the GL protein and pre-positioned nearby to terminate RGS14 inhibition of Ras/Raf signalling. While some components of the working model are speculative, most are consistent with our observed findings and provide a framework for further experiments. Our previous work shows that RGS14 can modulate conventional GPCR/Gα (e.g. M2ChoR/Giα) signalling in membranes, and the model presented here does not rule out a parallel role for RGS14 in conventional GPCR/Gα signalling, as we have proposed [10, 35]. RGS14 may serve a (poorly understood) role in unconventional G protein signalling pathways involving non-receptor (GPCR) GEFs alone, or these in concerted action with GPCRs.
RGS14’s closest relative, RGS12, also was shown to bind H-Ras and B-Raf to inhibit PDGF Erk phosphorylation, though no Giα regulation of this activity was reported. RGS12 inhibition of PDGF signalling may be mediated by direct binding to the PDGF receptor [36] suggesting that RGS14 and RGS12 serve distinct functions. Consistent with this idea, RGS14 and RGS12 share both overlapping and distinct sequence and predicted domains structures (63% identity; 70% similarity), and have very different patterns of subcellular distribution. RGS12 localizes to punctate structures in the nucleus and cytosol [37, 38], while RGS14 is distributed diffusely throughout the cytosol where it shuttles in/out of the nucleus and also accumulates at centrosomes and the plasma membrane with Giα1/3 [11, 28].
The exact mechanism by which RGS14 inhibits PDGF-stimulated MAPkinase signalling is unclear. We propose that RGS14 could serve as a molecular scaffold that sequesters active H-Ras and Raf from their signalling pathways to passively inhibit signalling. We favor this idea which is consistent with our proposed model. Alternatively, RGS14 could directly alter H-Ras or Raf-1 activity, perhaps by stimulating Ras GTPase activity and/or inhibiting Raf kinase activity. H-Ras binds specifically to the R1 RBD, and previous work has shown that the RBD domains of other proteins simply serve as binding sites for active Ras-GTP or Rap-GTP, but do not directly alter GTP binding to or GTP hydrolysis activity of these GTPases [5, 6]. Consistent with these reports, we previously showed the RGS14 failed to alter Rap GTP binding/GTPase activity [10, 24]. This would suggest that RGS14’s effect on MAPkinase signalling is likely that of a passive scaffold that sequesters H-Ras and Raf. Alternatively RGS14 may serve to redirect Ras/Raf kinase activity away from MEK/Erk towards a different substrate and pathway. In this regard, RGS14 may act in a fashion similar to the well-described scaffold 14:3:3, which also binds Raf kinases to passively modulate their signalling activity. Further studies are required to determine if RGS14 redirects Raf kinase activity or if it competes with 14:3:3 for Raf binding.
Our previous work has shown that RGS14 is most highly expressed in neurons of adult brain [10], most notably within the hippocampus (Sarah E. Lee, et al, unpublished observation, and also http://www.brain-map.org). GL proteins studied thus far have been shown to be important for cell division, but also for neuronal development and synaptic plasticity in both lower and higher eukaryotes [16, 18]. Several studies have implicated a possible role for RGS14 in cell division [28, 39, 40]. However, since RGS14 is highly enriched in mitotic incompetent neurons of adult brain, it must serve functions distinct from (or in addition to) those relating to cell division. Within hippocampal neurons, MAPkinase signalling pathways play a crucial role in regulating various aspects of neuronal differentiation and synaptic plasticity [22]. It is possible that RGS14 serves an important, though as yet poorly defined role in regulating these processes. Other signalling proteins that share similar properties with RGS14 as scaffolds, GL proteins, and/or as regulators of MAPkinase signalling have been shown to play important roles in synaptic plasticity. For example, the mammalian partner of insceutable (mPins, aka LGN or AGS5) protein and RGS14 both contain GL/GPR motifs, are modulated by Ric8A [31], and are stabilized by Giα-GDP binding at the plasma membrane and centrosomes. In addition, mPins/LGN/AGS5 is reported to regulate opposing pulling forces during cell division of mitotic competent non-neuronal cells [41]. However, mPins/LGN/AGS5 also is expressed in mitotic incompetent adult hippocampal neurons (as is RGS14) and is enriched in synaptic membranes where it associates with PSD-95 and MAGUK scaffolding proteins in a Giα1-dependent manner to influence their trafficking and NMDA receptor surface expression [42]. Of note, we observe here (Fig. 9) that Giα1 does not bind Raf-1, yet is capable of recruiting this kinase to the plasma membrane along with RGS14, suggesting the involvement of a larger multi-protein signalling complex like PSD-95 or MAGUK. This also is consistent with a recent report showing that the FRMPD1 protein serves as a scaffold to regulate the subcellular localization and interactions of the GPR/GL-protein AGS3 and Giα3 [43]. Other scaffolding proteins play a key role in synaptic plasticity. Loss of Shank, a key scaffolding component of the postsynaptic density (PSD) of hippocampal neurons, results in mice that exhibit smaller dendritic spines, decreased synaptic transmission, but enhanced learning and memory [44]. Also, loss of the adaptor protein N-Shc/Shc-C, which directly links NMDA and BDNF-receptors to MAPkinase signalling in hippocampal neurons, yields mice that exhibit enhanced spatial learning and memory [45]. These reports support the idea that RGS14 may serve functions complimentary to those of other scaffolding proteins in neuronal synaptic plasticity.
5. Conclusion
We report that RGS14 binds activated H-Ras and Raf-1 kinases, both independently and together in cells. RGS14 inhibits PDGF stimulated MAPkinase activity in a H-Ras dependent manner. This inhibition of signaling and Raf-1 binding to RGS14 is reversed by Giα1, suggesting that Giα1 binding and Raf-1 binding to RGS14 are mutually exclusive. Giα1 expression in cells also recruits Raf-1 to the plasma membrane suggesting a larger Gi-regulated signalling complex. Together, our findings here suggest that RGS14 serves as a newly appreciated scaffold that integrate G protein and H-Ras/Raf/ MAP kinase signalling events.
Supplementary Material
Acknowledgements
*We thank Dr. Ulf R. Rapp (Wϋrzburg University), Dr. Haian Fu (Emory University) and Dr. Deborah Anderson (Saskatchewan Cancer Agency) for kindly sharing cDNA constructs used in these studies, as described in the Experimental Procedures section. We also thank Jon Waters for technical assistance in generating some mutant constructs. This work is supported by grants from the National Institutes of Health awarded to J.R.H. (R01NS37112 and R01NS049195).
Abbreviations
- RGS
regulators of G protein signalling
- RBD
Ras/Rap binding domain
- GPCR
G protein coupled receptor
- MAPkinase
Mitogen-activated protein kinases
- GPR/GoLoco (GL)
G protein regulatory motif
- GEF
guanine exchange factor
- GDI
guanine nucleotide dissociation inhibitor
- GΑP
GTPase activating protein
- NuMA
nuclear mitotic apparatus protein
- Ric-8A
resistance to inhibitors of cholinesterase 8A
- AGS
activator of G-protein signalling
- PSD
postsynaptic density
- NMDA
N-methyl-D-aspartic acid
- LGN
lateral geniculate nucleus, and Leu-Gly-Asn rich protein (aka mPINs or AGS5)
- MAGUK
membrane-associated guanylate kinase
- Frmpd1
FERM and PDZ domain containing 1
- BDNF
brain-derived growth factor
- PDGF
Platelet-derived growth factor
- EGF
Epidermal growth factor
- GFP
green fluorescent protein
- CFP
cyan fluorescent protein
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hepler JR, Gilman AG. Trends Biochem. Sci. 1992;17(10):383–387. doi: 10.1016/0968-0004(92)90005-t. [DOI] [PubMed] [Google Scholar]
- 2.Ross EM, Wilkie TM. Annu. Rev. Biochem. 2000;69(1):795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 3.De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG. Annu. Rev. Pharmacol. Toxicol. 2000;40(1):235–271. doi: 10.1146/annurev.pharmtox.40.1.235. [DOI] [PubMed] [Google Scholar]
- 4.Hollinger S, Hepler JR. Pharmacol. Rev. 2002;54(3):527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 5.Kiel C, Wohlgemuth S, Rousseau F, Schymkowitz J, Ferkinghoff-Borg J, Wittinghofer F, Serrano L. J. Mol. Bio. 2005;348(3):759–775. doi: 10.1016/j.jmb.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 6.Wohlgemuth S, Kiel C, Kramer A, Serrano L, Wittinghofer F, Herrmann C. J. Mol. Biol. 2005;348(3):741–758. doi: 10.1016/j.jmb.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 7.Traver S, Bidot C, Spassky N, Baltauss T, De Tand MF, Thomas JL, Zalc B, Janoueix-Lerosey I, Gunzburg JD. Biochem. J. 2000;350(1):19–29. [PMC free article] [PubMed] [Google Scholar]
- 8.Snow BE, Antonio L, Suggs S, Gutstein HB, Siderovski DP. Biochem. Biophys. Res. Commun. 1997;233(3):770–777. doi: 10.1006/bbrc.1997.6537. [DOI] [PubMed] [Google Scholar]
- 9.Traver S, Splingard A, Gaudriault G, De Gunzburg J. Biochem. J. 2004;379(3):627–632. doi: 10.1042/BJ20031889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollinger S, Taylor JB, Goldman EH, Hepler JR. J. Neurochem. 2001;79(5):941–949. doi: 10.1046/j.1471-4159.2001.00629.x. [DOI] [PubMed] [Google Scholar]
- 11.Cho H, Kozasa T, Takekoshi K, De Gunzburg J, Kehrl JH. Mol. Pharmacol. 2000;58(3):569–576. doi: 10.1124/mol.58.3.569. [DOI] [PubMed] [Google Scholar]
- 12.Kimple RJ, De Vries L, Tronchere H, Behe CI, Morris RA, Farquhar MG, Siderovski DP. J. Biol. Chem. 2001;276(31):29275–29281. doi: 10.1074/jbc.M103208200. [DOI] [PubMed] [Google Scholar]
- 13.Mittal V, Linder ME. J. Biol. Chem. 2004;279(45):46772–46778. doi: 10.1074/jbc.M407409200. [DOI] [PubMed] [Google Scholar]
- 14.Shu FJ, Ramineni S, Amyot W, Hepler JR. Cell. Signal. 2007;19(1):163–176. doi: 10.1016/j.cellsig.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Wilkie TM, Kinch L. Current Biology. 2005;15(20):R843–R854. doi: 10.1016/j.cub.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Hampoelz B, Knoblich JA. Cell. 2004;119(4):453–456. doi: 10.1016/j.cell.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 17.Blumer JB, Cismowski MJ, Sato M, Lanier SM. Trends Pharmacol. Sci. 2005;26(9):470–476. doi: 10.1016/j.tips.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Sato M, Blumer JB, Simon V, Lanier SM. Annu. Rev. Pharmacol. Toxicol. 2006;46:151–187. doi: 10.1146/annurev.pharmtox.46.120604.141115. [DOI] [PubMed] [Google Scholar]
- 19.Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, Zhang X-F. Recent. Prog. Horm. Res. 2001;56(1):127–156. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- 20.Goldsmith ZG, Dhanasekaran DN. Oncogene. 2007;26(22):3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- 21.Gutkind JS. J Biol Chem. 1998;273(4):1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- 22.Adams JP, Sweatt JD. Annu. Rev. Pharmacol. Toxicol. 2002;42(1):135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 23.Thomas CJ, Tall GG, Adhikari A, Sprang SR. J. Biol. Chem. 2008 doi: 10.1074/jbc.M802422200. M802422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollinger S, Ramineni S, Hepler JR. Biochemistry. 2003;42(3):811–819. doi: 10.1021/bi026664y. [DOI] [PubMed] [Google Scholar]
- 25.Herrmann C, Horn G, Spaargaren M, Wittinghofer A. J. Biol. Chem. 1996;271(12):6794–6800. doi: 10.1074/jbc.271.12.6794. [DOI] [PubMed] [Google Scholar]
- 26.Fabian JR, Vojtek AB, Cooper JA, Morrison DK. Proc. Natl. Acad. Sci. U.S.A. 1994;91(13):5982–5986. doi: 10.1073/pnas.91.13.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mittal V, Linder ME. Biochem. J. 2006;394(Pt 1):309–315. doi: 10.1042/BJ20051086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho H, Kim D, Kehrl JH. J. Biol. Chem. 2005;280(1):805–814. doi: 10.1074/jbc.M408163200. [DOI] [PubMed] [Google Scholar]
- 29.Yuryev A, Wennogle LP. Genomics. 2003;81(2):112–125. doi: 10.1016/s0888-7543(02)00008-3. [DOI] [PubMed] [Google Scholar]
- 30.Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Oncogene. 2007;26(22):3185–3202. doi: 10.1038/sj.onc.1210411. [DOI] [PubMed] [Google Scholar]
- 31.Tall GG, Gilman AG. Proc. Natl. Acad. Sci. U.S.A. 2005;102(46):16584–16589. doi: 10.1073/pnas.0508306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellaiche Y, Gotta M. Curr. Opin. Cell. Biol. 2005;17(6):658–663. doi: 10.1016/j.ceb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Hampoelz B, Hoeller O, Bowman SK, Dunican D, Knoblich JA. Nat. Cell Bio. 2005;7(11):1099–1105. doi: 10.1038/ncb1318. [DOI] [PubMed] [Google Scholar]
- 34.Hongyan W, Kian Hong N, Hongliang Q, Siderovski DP, Chia W, Fengwei Y. Nat. Cell Bio. 2005;7(11):1091–1098. doi: 10.1038/ncb1317. [DOI] [PubMed] [Google Scholar]
- 35.Hepler JR, Cladman W, Ramineni S, Hollinger S, Chidiac P. Biochemistry. 2005;44(14):5495–5502. doi: 10.1021/bi048359d. [DOI] [PubMed] [Google Scholar]
- 36.Sambi BS, Hains MD, Waters CM, Connell MC, Willard FS, Kimple AJ, Pyne S, Siderovski DP, Pyne NJ. Cell. Signal. 2006;18(7):971–981. doi: 10.1016/j.cellsig.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Chatterjee TK, Fisher R. A. J. Biol. Chem. 2000;275(38):29660–29671. doi: 10.1074/jbc.M000330200. [DOI] [PubMed] [Google Scholar]
- 38.Chatterjee TK, Fisher R. A. Mol. Cell Biol. 2002;22(12):4334–4345. doi: 10.1128/MCB.22.12.4334-4345.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho H, Kehrl JH. J. Cell Biol. 2007;178(2):245–255. doi: 10.1083/jcb.200604114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin-McCaffrey L, Willard FS, Oliveira-dos-Santos AJ, Natale DRC, Snow BE, Kimple RJ, Pajak A, Watson AJ, Dagnino L, Penninger JM, Siderovski DP, D'Souza SJA. Dev. Cell. 2004;7(5):763–769. doi: 10.1016/j.devcel.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 41.Du Q, Macara IG. Cell. 2004;119(4):503–516. doi: 10.1016/j.cell.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 42.Sans N, Wang PY, Du Q, Petralia RS, Wang YX, Nakka S, Blumer JB, Macara IG, Wenthold R. J. Nat. Cell. Biol. 2005;7(12):1179–1190. doi: 10.1038/ncb1325. [DOI] [PubMed] [Google Scholar]
- 43.An N, Blumer JB, Bernard ML, Lanier SM. J. Biol. Chem. 2008 doi: 10.1074/jbc.M803497200. M803497200:[Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang CS, Shi SH, Ule J, Ruggiu M, Barker LA, Darnell RB, Jan YN, Jan LY. Cell. 2005;123(1):105–118. doi: 10.1016/j.cell.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 45.Miyamoto Y, Chen L, Sato M, Sokabe M, Nabeshima T, Pawson T, Sakai R, Mori N. J. Neurosci. 2005;25(7):1826–1835. doi: 10.1523/JNEUROSCI.3030-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.