

Abstract
Alzheimer’s disease (AD) is the most common progressive dementia and is pathologically characterized by brain deposition of amyloid-β (Aβ) peptide as senile plaques. Inflammatory and immune response pathways are chronically activated in AD patient brains at low levels, and likely play a role in disease progression. Like microglia, activated astrocytes produce numerous acute-phase reactants and proinflammatory molecules in the AD brain. One such molecule, S100B, is highly expressed by reactive astrocytes in close vicinity of β-amyloid deposits. We have previously shown that augmented and prolonged activation of astrocytes has a detrimental impact on neuronal survival. Furthermore, we have implicated astrocyte-derived S100B as a candidate molecule responsible for this deleterious effect. To evaluate a putative relationship between S100B and AD pathogenesis, we crossed transgenic mice overexpressing human S100B (TghuS100B mice) with the Tg2576 mouse model of AD, and examined AD-like pathology. Brain parenchymal and cerebral vascular β-amyloid deposits and Aβ levels were increased in bigenic Tg2576-huS100B mice. These effects were associated with increased cleavage of the β-C-terminal fragment of amyloid precursor protein (APP), elevation of the N-terminal APP cleavage product (soluble APPβ), and activation of β-site APP cleaving enzyme 1. In addition, double transgenic mice showed augmented reactive astrocytosis and microgliosis, high levels of S100 expression, and increased levels of proinflammatory cytokines as early as 7-9 months of age. These results provide evidence that (over)-expression of S100B acts to accelerate AD-like pathology, and suggest that inhibiting astrocytic activation by blocking S100B biosynthesis may be a promising therapeutic strategy to delay AD progression.
Keywords: S100B, astrocyte, amyloid-β, inflammatory response, Alzheimer’s transgenic mice
INTRODUCTION
Chronic activation of glial cells in and around β-amyloid plaques may be pathoetiologic in Alzheimer’s disease (AD) via production of numerous neurotoxic acute-phase reactants, proinflammatory cytokines, and immunostimulatory molecules (Akiyama et al., 2000; Griffin, et al., 1998b). However, not all forms of gliosis are deleterious, and this is underscored by well-documented dual roles of microglia in exacerbating or mitigating AD pathology depending on the nature of the stimulus (Akiyama et al., 2000; Bard et al., 2000; Griffin et al., 1998b; Schenk et al., 1999; Tan et al., 2002; Town et al., 2005, 2008). Similarly, mounting evidence suggests that a variety of biologically active molecules produced by activated astrocytes may play a dichotomous role in various brain pathologies, including AD (Mrak and Griffin, 2001; Nedergaard and Dirnagl, 2005; Ridet et al., 1997). Among these substances, S100B (the ββ form of S100 protein) plays central roles in Ca2+-dependent regulation of a variety of intracellular functions, and is neurotrophic at physiological levels (Donato, 1999). In the brain, S100B is the predominant isoform of the S100 family, where it is expressed by astrocytes and is thought to mediate glial activation (Mrak and Griffin, 2001; Ridet et al., 1997). However, S100B can also be detrimental, given that S100B 1) at micromolar concentrations produces neuronal damage by causing overexpression of inducible nitric oxide synthase and subsequent release of nitric oxide (Hu et al., 1997); 2) is up-regulated in a variety of neurodegenerative diseases including AD (Mrak and Griffin, 2001); 3) associates with neuritic β-amyloid plaques in a transgenic mouse model of AD (Sheng et al., 2000); 4) stimulates glial activation through a cytokine cycle; and 5) activates nuclear factor-κB (NF-κB), a key transcription factor mediator of inflammatory responses (Lam et al., 2001).
We and others have proposed that enhanced and prolonged activation of astrocytes plays a detrimental role in brain pathology including stroke and AD, and that astroctye-derived S100B is a candidate molecule for this effect, supporting by the following evidence: 1) increased S100B production by activated astrocytes is associated with the occurrence of delayed infarct expansion after cerebral ischemia (Matsui et al., 2002; Mori et al., 2004); 2) overexpression of human S100B acts to exacerbate brain damage and periinfarct reactive gliosis after cerebral ischemia (Mori et al., 2008); 3) pharmacological inhibition of astrocytic S100B synthesis by arundic acid (ONO-2506, Ono Pharmaceutical Co. Ltd., Osaka, Japan) leads to significant amelioration of both astrocytic activation and delayed infarct expansion after ischemia (Asano et al., 2005; Mori et al., 2005; Tateishi et al., 2002); and 4) AD-like pathology (i.e. cerebral amyloidosis and gliosis) in the aged Tg2576 mouse model is attenuated by pharmacological blockade of S100B biosynthesis by arundic acid (Mori et al., 2006). In agreement with these lines of evidence, S100B transgenic and knock-out mice showed worsening and attenuation, respectively, of ischemic brain damage (Wainwright et al., 2004). Collectively, these findings suggest that stroke and AD may share a common pathogenic mechanism – maladaptive astrocytic activation – where S100B acts as a perpetrator of the noxious cytokine cycle leading to neurotoxicity (Mrak and Griffin, 2001).
To dissect the relationship between S100B and AD-like pathology, we took a genetic approach by crossing transgenic mice expressing human S100B (TghuS100B mice) with the Tg2576 mouse model of AD. We then examined AD-like pathology in aged animals, including brain parenchymal and cerebral vascular β-amyloid deposits, amyloid-β (Aβ) levels, β-amyloid deposit-associated gliosis (astrocytosis and microgliosis), and proinflammatory cytokines.
MATERIALS AND METHODS
Mice
We purchased Tg2576 mice (C57BL/6 × SJL background) from Taconic (Germantown, NY). This transgenic mouse model of AD overproduces human Aβ1-40 and Aβ1-42 and develops progressive β-amyloid deposits and learning and memory impairment beginning at 9-10 months of age (Hsiao et al., 1996; Kawarabayashi et al., 2001; Lesné et al., 2006). We obtained TghuS100B mice (carrying approximately 10 copies of the human S100B gene under endogenous regulatory control) from The Jackson Laboratory (Bar harbor, ME) on an outbred CD-1 genetic background (Charles River Laboratories, Wilmington, MA). These mice overexpress S100B in cortical astrocytes by 400% to 600% over CD-1 controls (Friend et al., 1992). We crossed heterozygous TghuS100B mice with heterozygous Tg2576 mice to yield four genotypes of littermates (Tg2576, Tg2576-huS100B, TghuS100B, and wild-type), and characterized offspring by polymerase chain reaction (PCR)-based genotyping for mutant APP and human S100B constructs. We strictly used littermates obtained from this cross-breeding strategy for all of our analyses. Thus, all mice used in the present study are comparable in terms of genetic background (C57BL/6 × SJL × CD-1 background). Animals were housed and maintained in a specific pathogen-free barrier facility under a 12 h/12 h light-dark cycle, with ad libitum access to food and water. All experiments were performed in accordance with the guidelines of the Animal Use Ethics Committee of the Saitama Medical University and NIH guidelines (DHHS publication No. [NIH] 85-23, revised 1985).
Tissue Preparation
We used Tg2576 mice [a total of 72 mice: n = 10 (5 males and 5 females) at 9 months of age; n = 14 each (7 males and 7 females) at 13, 15, and 19 months of age] and Tg2576-huS100B mice [a total of 84 mice: n = 12 (6 males and 6 females) at 9 months of age; n = 16 each (8 males and 8 females) at 13, 15, and 19 months of age] in this study. Furthermore, in a separate set of experiments, we utilized 7-month-old Tg2576, Tg2576-huS100B, TghuS100B, and wild-type littermates [a total of 32 mice: n = 8 each (4 males and 4 females)] to examine cerebral gliosis and proinflammatory cytokine mRNA expression at an age prior to appearance of β-amyloid deposits. In addition, we used TghuS100B [a total of 20 mice: n = 10 each (5 males and 5 females) at 9 and 19 months of age] and wild-type littermates [a total of 20 mice: n = 10 each (5 males and 5 females) at 9 and 19 months of age] for proinflammatory cytokine mRNA expression. At 7, 9, 13, 15, and 19 months of age, animals were anesthetized with sodium pentobarbital (50 mg/kg) and euthanized by transcardial perfusion with ice-cold physiological saline containing heparin (10 U/mL). We rapidly isolated and quartered brains (sagittally at the level of the longitudinal fissure of the cerebrum, and then coronally at the level of the anterior commissure) using a mouse brain slicer (Muromachi Kikai, Tokyo, Japan). Right anterior cerebral quarters were weighed and snap-frozen at −80°C for full-length holo-amyloid precursor protein (APP), β-site APP cleaving enzyme 1 (BACE1), and β-C terminal fragment (β-CTF: phospho-C99 and C99) Western blots as well as β-secretase activity analyses. Right posterior cerebral quarters were weighed and snap-frozen at −80°C for Tris-buffered saline (TBS) - and guanidine-soluble Aβ as well as soluble APPβ (sAPPβ) sandwich enzyme-linked immunosorbance assay (ELISA) analyses. Left anterior cerebral quarters were weighed and immersed in RNA stabilization solution (RNAlater®, Applied Biosystems, Foster City, CA) and then snap-froze them at −80°C for proinflammatory cytokine mRNA analyses. Left posterior cerebral quarters were immersion fixed in 4% paraformaldehyde in 0.1 M phosphate buffer at 4°C overnight, and routinely processed in paraffin for immunohistochemical analyses.
Immunohistochemistry
For paraffin sectioning, we sectioned five coronal sections (per set) with a 100 μm interval and a thickness of 5 μm for each brain region [for cingulate cortex (CC) bregma − 0.10 mm to − 0.82 mm; for hippocampus (H) and entorhinal cortex (EC), bregma −2.92 mm to − 3.64 mm] (Franklin and Paxinos, 2001). We prepared four sets of five sections in each separate region for analyses of S100, glial fibrillary acidic protein (GFAP, as a marker of astrocytosis), and ionized calcium-binding adapter molecule 1 (Iba1, a microgliosis marker) burdens as well as Aβ deposits/β-amyloid plaques (for plaque number and maximum diameter morphometry). Immunohistochemical staining was conducted according to the manufacturer’s protocol using a Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA) coupled with the diaminobenzidine reaction, except that the biotinylated secondary antibody step was omitted for Aβ immunohistochemical staining. The following primary antibodies were used: biotinylated human Aβ monoclonal antibody (4G8; 1:100, Covance Research Products, Emeryville, CA), GFAP and S100 polyclonal antibodies (1:500 and undiluted, respectively, DAKO, Carpinteria, CA), and Iba1 polyclonal antibody (1:1000, Wako, Osaka, Japan). The S100 antibody used for immunohistochemical staining is polyclonal and does not discriminate among different S100 isoforms, hence we adapted the designation of ‘S100′ to express the corresponding results. Normal rabbit or normal mouse serum (isotype control) or phosphate buffer saline (0.1 M, pH 7.4) was used instead of primary antibody or ABC reagent as negative controls.
Image Analysis
Quantitative image analysis was done based on a previously validated method (Tan et al., 2002; Mori et al., 2006; Town et al., 2008). Images were acquired as digitized tagged-image format files to retain maximum resolution using an Olympus BX60 microscope with an attached digital camera system (DP-70, Olympus, Tokyo, Japan), and digital images were routed into a Windows PC for quantitative analyses using SimplePCI software (Compix, Inc. Imaging Systems, Cranberry Township, PA). We captured images of five 5 μm sections through each anatomic region of interest (CC, EC, and H) based on anatomical criteria defined by Franklin and Paxinos (2001), and we obtained a threshold optical density that discriminated staining from background. Each anatomic region of interest was manually edited to eliminate artifacts. For Aβ, S100/GFAP (astrocytosis), and Iba1 (microgliosis) burden analyses, data are reported as the percentage of labeled area captured (positive pixels) divided by the full area captured (total pixels). Selection bias was controlled for by analyzing each region of interest in its entirety. We refer to “plaque associated” astrogiosis or microgliosis based on our observations presented here in Tg2576 and Tg2576-huS100B mice and previous reports (Benzing et al., 1999; Stalder et al., 1999) demonstrating that reactive glia in Tg2576 and APP23 AD model mice are most often found in close association with β-amyloid plaques.
For β-amyloid plaque morphometric analyses (number and maximum diameter), diameters (based on maximum length) of β-amyloid deposits/plaques were measured, and numbers of β-amyloid plaques falling into three mutually exclusive diameter categories (< 25 μm, 25-50 μm, or > 50 μm) were calculated. We present data as mean plaque number per mouse in each region examined. For cerebral amyloid angiopathy (CAA) morphometric analysis, we counted the number of Aβ antibody (4G8)-stained cerebral vessels in each anatomic region of interest, and we present those data as mean CAA deposit number per mouse.
ELISA
Aβ1-40 and Aβ1-42 species were detected by a two-step extraction protocol, similar to previously published methods (Horikoshi et al., 2004; Johnson-Wood et al., 1997; Town et al., 2008). Briefly, we homogenized brains in 0.05% TBS solution containing protease inhibitor cocktail (Sigma, St. Louis, MO) for protein isolation and centrifuged homogenates at 13,000 × g for 30 min at 4°C, removed the supernatant (TBS-soluble fraction), treated the remaining pellet with 5 M guanidine HCl, and solubilized by occasional mixing on ice for 30 min (guanidine HCl-soluble fraction). Aβ1-40 and Aβ1-42 species were separately quantified in individual samples in duplicate using ELISA kits in accordance with the manufacturer’s instructions (Catalog # 27718 for Aβ1-40 and # 27712 for Aβ1-42; IBL, Gunma, Japan). We used dilution factors of 1:5 and 1:50,000 for TBS- and guanidine HCl-soluble brain homogenates, respectively, and all samples fell within the linear range of standard curves. For sAPPβ analysis, TBS-soluble supernatants were used at a 1:20 dilution. We quantified levels of sAPPβ in individual samples in duplicate using ELISA kits in accordance with the manufacturer’s instructions (Catalog # 27723; IBL). All samples fell within the linear range of the standard curve. We report all ELISA values as ng/mg or pg/mg of Aβ1-x/wet g or sAPPβ/wet g of brain.
Quantitative real-time PCR
We quantified tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, and mouse S100B mRNA levels. Total RNA was extracted using RNeasy mini kit (Qiagen, Valencia, CA), and first strand cDNA synthesis was carried out using the QuantiTect reverse transcription kit (Qiagen) in accordance with the manufacturer’s instructions. We diluted cDNA 1:1 in H2O, and carried out quantitative real-time PCR (QPCR) reactions for all genes of interest using cDNA-specific TaqMan primer/probe sets (TaqMan Gene Expression Assays, Applied Biosystems) on an ABI 7700 Real-Time PCR instrument (Applied Biosystems). In each 20 μl of TaqMan reaction, we mixed 2 μl of cDNA with 1 μL of TaqMan Gene Expression Assay, 10 μL of TaqMan Fast Universal PCR Master Mix (Applied Biosystems), and 7 μL of H2O. Thermocycling conditions consisted of: 95°C for 15 s, followed by 40 cycles of 95°C for 1 s and 60°C for 20 s. TaqMan probe/primer sets were as follows: [mouse TNF-α (Catalog # Mm00443258_m1), mouse IL-1β (# Mm00434228_m1), mouse IL-6 (# Mm00446190_m1), mouse S100B (# Mm00485897_m1), and mouse β-actin (# Mm00607939_s1; used as an internal reference control) (Applied Biosystems)]. Samples that were not subjected to reverse transcription were run in parallel as negative controls to rule out genomic DNA contamination as a template for PCR products (data not shown). A “no template control” was also included for each primer set as a further negative control (data not shown). The cycle threshold number (CT) was determined for each PCR. This number was used as an indicator of the relative amount of initial target RNA in each sample. Results are expressed as relative expression levels of target mRNA in Tg2576 and Tg2576-huS100B mice normalized to wild-type littermates according to the comparative CT method (Monney et al., 2002).
Western Blot and β-Secretase Activity Assay
For Western blot analyses of full-length holo APP, β-CTF (phospo-C99 and C99), and BACE1, supernatants extracted in 0.05% TBS solution containing protease inhibitor cocktail followed by TNE buffer (10 mM Tris-HCl, 1% NP-40, 1 mM EDTA, and 150 mM NaCl,) were used. We electrophoretically separated aliquots of brain homogenates containing 10 μg of total protein using 10% or 15% Tris-glycine gels based on molecular weights of the target molecules. We then transferred electrophoresed proteins to polyvinylidene difluoride membranes (Bio-Rad, Richmond, CA), washed in TBS containing 0.05% Tween-20, and blocked for 1 h at ambient temperature in Block Ace (Dainippon Sumitomo Pharma, Osaka, Japan). After blocking, we hybridized membranes for 1 h at ambient temperature with the primary antibodies: N-terminal APP antibody (10D1; 1:100, IBL), C-terminal BACE1 antibody (1:100, IBL), or N-terminal Aβ antibody (82E1; 1:100, IBL). We then washed membranes three times for 30 min each in TBS containing 0.05% Tween-20 and incubated them for 1 h at ambient temperature with HRP-linked anti-mouse IgG antibody (1:2000, IBL) or HRP-linked anti–rabbit IgG (1:4000, IBL) antibodies, depending on the primary antibody. After washing as above for 30 min, we developed blots using ECL Plus Western Blotting Detection Reagents (GE Healthcare Bio-Sciences, Piscataway, NJ). For β-secretase activity analysis in brain homogenates, we used available kits based on secretase-specific peptides conjugated to fluorogenic reporter molecules (R & D Systems, Minneapolis, MN).
Statistical Analysis
Data are presented as the mean ± 1 SEM. All data were found to be normally distributed. In instances of single mean comparisons, Levene’s test for equality of variances followed by t-test for independent samples or Mann-Whitney U test was performed. In instances of multiple mean comparisons, one-way ANOVA was used, followed by post hoc comparison of the means using Bonferroni’s or Dunnett’s T3 methods (where appropriateness was determined using Levene’s test for equality of the variance). P values of less than 0.05 were considered to be significant. All analyses were performed using the Statistical Package for the Social Sciences, release 17.0 (SPSS, Chicago, IL).
RESULTS
The huS100B transgene accelerates cerebral amyloidosis
We hypothesized that expression of the huS100B transgene might enhance inflammatory responses and thereby worsen AD-like pathology. To test this, we bred Tg huS100B mice to the Tg2576 mouse model of AD and began by examining cerebral amyloidosis at 9, 13, 15, and 19 months of age, during which time Tg2576 mice progress to advanced-stage cerebral amyloidosis (Hsiao et al., 1996). We evaluated Aβ/β-amyloid pathology in Tg2576 and Tg2576-huS100B mice by three strategies: Aβ antibody immunoreactivity (conventional Aβ “burden”) analysis, β-amyloid plaque morphometric analysis, and Aβ1-40 or Aβ1-42 sandwich ELISA analysis. Tg2576 mice had typical age-dependent Aβ burden, which was significantly exacerbated in cortical areas (EC and CC) and H in Tg2576-huS100B mice at 9, 13, 15, and 19 months of age (Fig. 1A, ** P < 0.01, *** P < 0.001). We stratified by gender and increased Aβ burden was significant in both male and female mice (data not shown). To assess whether enhanced Aβ burden in Tg2576-huS100B mice was specific to a particular β-amyloid plaque size subset, we performed morphometric analysis of Aβ antibody (4G8)-stained β-amyloid plaques in Tg2576 and Tg2576-huS100B mice. Based on previously described methods (Mori et al., 2006; Tan et al., 2002; Town et al., 2008), we assigned plaques to one of three mutually exclusive categories according to maximum diameter: small (< 25 μm), medium (between 25 and 50 μm) or large (> 50 μm). We performed plaque morphometry at 9, 13, 15, and 19 months of age, and found that all three-subsets of plaques were significantly increased across all three-brain regions examined in Tg2576-huS100B mice at 9, 13, 15, and 19 months of age (Fig. 1B,C, ** P < 0.01, *** P < 0.001). We stratified by gender and this effect was significant in both male and female mice at 15 and 19 months of age (data not shown). It is noteworthy that Tg2576-huS100B mice showed the greatest increase in the medium- and large-sized β-amyloid plaque subsets across all three-brain regions. At 9 months of age, Tg2576 mice had mainly dot-like β-amyloid deposits (< 5 μm in maximum diameter) and no large-sized β-amyloid plaques in brain areas examined. In addition, β-amyloid plaques in Tg2576 littermates were smaller than 100 μm in maximum diameter in all three-brain regions examined, even at 19 months of age. By contrast, Tg2576-huS100B mice had a number of large-sized β-amyloid plaques (> 50 μm in maximum diameter) even at 9 and 13 months of age, and we detected a few gigantic β-amyloid plaques (>150 μm) in the three-brain regions that we examined at 15 and 19 months of age (Fig. 1D).
Fig. 1.


The huS100B transgene accelerates cerebral parencymal β-amyloid deposits in Tg2576 mice. Data were obtained from Tg2576 [n = 10 (5 males and 5 females) at 9 months of age; n = 14 each (7 males and 7 females) at 13, 15, and 19 months of age] and Tg2576-huS100B mice [n = 12 (6 males and 6 females) at 9 months of age; n = 16 each (8 males and 8 females) at 13, 15, and 19 months of age]. (A) Quantitative image analysis for Aβ burden is shown in cingulate cortex (CC, top), hippocampus (H, middle), and entorhinal cortex (EC, bottom). All statistical comparisons are within brain region and between Tg2576 and Tg2576-huS100B mouse groups at 9, 13, 15, and 19 months of age. (B,C) Morphometric analysis of cerebral parencymal β-amyloid plaques is shown in Tg2576 and Tg2576-huS100B mice at 15 and 19 months of age. Brain sections were stained with 4G8 antibody, and positive deposits were counted based on maximum diameter and assigned to: small (< 25 μm; top), medium (between 25 and 50 μm; middle), or large (> 50 μm; bottom). Mean plaque subset number per mouse is shown on the y-axis, and brain region is represented on the x-axis. All statistical comparisons are within brain region and between Tg2576 and Tg2576-huS100B mice at 15 and 19 months of age. (D) Representative photomicrographs of 4G8 immunohistochemistry are shown for cerebral β-amyloid deposits in Tg2576 and Tg2576-huS100B mice at 15 and 19 months of age in each brain region indicated on the left. Scale bar denotes 50 μm. ** P < 0.01, *** P < 0.001 vs. Tg2576 mice.
In addition to brain parenchymal deposition of β-amyloid as senile plaques, 83% of AD patients present with β-amyloid deposits in cerebral vessels, known as cerebral amyloid angiopathy (CAA) (Ellis et al., 1996). Tg2576 mice also develop vascular β-amyloid deposits with age (Robbins et al., 2006). Having shown enhanced brain parenchymal β-amyloid plaques in Tg2576-huS100B mice, we wondered if overexpression of huS100B might also impact cerebral vascular β-amyloid deposits. In both mouse groups, cerebral vascular β-amyloid deposits were predominantly detected at the brain surface containing the pial arteries as well as their penetrating arteries in each anatomic region of interest. We scored Aβ antibody (4G8)-stained cerebral vascular deposits in Tg2576 and Tg2576-huS100B mice and found a striking increase in these deposits in Tg2576-huS100B mouse brains that reached statistical significance in all three-brain regions examined at 15 and 19 months of age (Fig. 2A,B, * P < 0.05, ** P < 0.01). In agreement with histological observations, biochemical analysis of Aβ species in brain homogenates revealed significant increases in both Aβ1-40 and Aβ1-42 levels in the TBS-soluble fraction at 15 and 19 months of age and in the TBS-insoluble (but guanidine-HCl-extractable) fraction at 15, and 19 months of age (Fig. 3A,B, * P < 0.05, ** P < 0.01, *** P < 0.001). Collectively, these data show that cerebral amyloidosis, including bain parenchymal and cerebral vascular β-amyloid deposits and Aβ levels, is exacerbated in Tg2576 mice bearing the huS100B transgene.
Fig. 2.
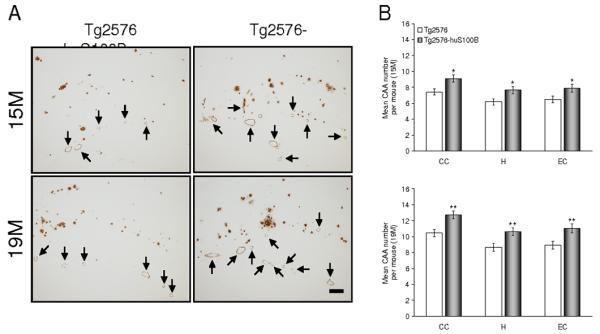
Cerebral vascular β-amyloid deposits are accelerated in Tg2576 mice bearing the huS100B transgene. Data were obtained from Tg2576 [n = 14 each (7 males and 7 females) at 15 and 19 months of age] and Tg2576-huS100B mice [n = 16 each (8 males and 8 females) at 15 and 19 months of age]. (A) Representative photomicrographs of 4G8 immunohistochemistry were taken from hippocampi from Tg2576 and Tg2576-huS100B mice, and show cerebral vascular β-amyloid deposits as indicated (arrows) and cerebral parencymal β-amyloid plaques at 15 and 19 months of age. Scale bar denotes 200 μm. (B) Severity of cerebral amyloid angiopathy (mean CAA deposit number per mouse) is shown on the y-axis with brain region indicated on the x-axis (CC, cingulate cortex; H, hippocampus; EC, entorhinal cortex). All statistical comparisons are within brain region and between Tg2576 and Tg2576-huS100B mice at 15 (upper) and 19 (lower) months of age. * P < 0.05, ** P < 0.01 vs. Tg2576 mice.
Fig. 3.
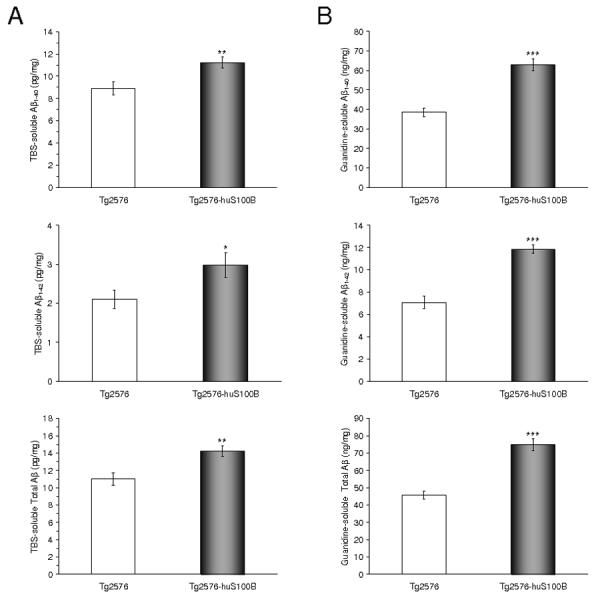
The huS100B transgene increases brain Aβ levels in Tg2576 mice. Data were obtained from Tg2576 [n = 14 (7 males and 7 females)] and Tg2576-huS100B mice [n = 16 (8 males and 8 females) at 15 months of age]. (A,B) The Tris-buffered saline (TBS)-soluble fraction (A) and TBS-insoluble (but 5M guanidine HCl-extractable) fraction (B) from two-step extracted brain homogenates were assayed by ELISA for human Aβ1-40 (top), Aβ1-42 (middle), and total Aβ levels (estimated by summing Aβ1-40 and Aβ1-42). * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Tg2576 mice.
Enhanced amyloidogenic APP metabolism by the huS100B transgene
To examine if accelerated cerebral amyloidosis in Tg2576-huS100B mice could be due to increased expression of transgene-derived APP, we probed brain homogenates from Tg2576 and Tg2576-huS100B mice using an N-terminal APP antibody (which recognizes human transgene-derived APP only), and did not detect a difference between groups (Fig. 4A). To address steady-state APP metabolism, we probed brain homogenates with an N-terminal Aβ antibody that recognizes amino acids 1-16 of human Aβ, and detects the amyloidogenic C-terminal APP fragment (C99, β-CTF). Densitometry confirmed that APP metabolism to phospho-C99 and C99 was significantly enhanced in Tg2576-huS100B mice (Fig. 4A,B, ** P < 0.01, *** P < 0.001). To further determine if the amyloidogenic pathway was preferentially activated in bigenic mice, we probed brain homogenates with a C-terminal BACE1 antibody. Densitometry confirmed that BACE1 expression was significantly increased in Tg2576-huS100B mice (Fig. 4C, ** P < 0.01). Consistent with this result, β-secretase activity was significantly increased in brain homogenates from Tg2576-huS100B vs. Tg2576 mice (Fig. 4D, ** P < 0.01, *** P < 0.001). Further, we measured sAPPβ levels by ELISA in the TBS-soluble brain homogenate fraction from Tg2576 and Tg2576-huS100B mice, and found a significant increase in sAPPβ levels in Tg2576-huS100B mice vs. Tg2576 mice (Fig. 4E, * P < 0.05). These data support the notion that the huS100B transgene accelerates cerebral amyloidosis by promoting amyloidogenic APP metabolism as a consequence of increased β-secretase activity.
Fig. 4.

The huS100B transgene promotes amyloidogenic APP processing in Tg2576 mice. Data were obtained from Tg2576 [n = 14 (7 males and 7 females)] and Tg2576-huS100B mice [n = 16 (8 males and 8 females) at 15 months of age]. (A) Western blots using an N-terminal APP antibody (mAb 10D1, full-length holo-APP and soluble APP species are shown), a C-terminal β-site APP cleaving enzyme 1 antibody (pAb, BACE1), and an-N-terminal Aβ antibody [mAb 82E1, APP-C-terminal fragments generated by amyloidogenic APP cleavage (phospho-C99: P-β-CTF and C99: β-CTF) are shown]. Actin is shown as a loading control for each blot. (B) Densitometry analyses for the ratio of P-C99 or C99 to actin are shown. (C) Densitometry analysis for the ratio of BACE1 to actin is shown. (D) β-secretase activity analysis is shown. (E) The Tris-buffered saline (TBS)-soluble fraction from two-step extracted brain homogenates was also assayed by ELISA for soluble-APPβ. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Tg2576 mice.
The huS100B transgene exacerbates Aβ-associated gliosis and production of proinflammatory cytokines
Reactive gliosis (astrocytosis and/or microgliosis) is often found in close proximity to β-amyloid plaques and increases with age (Benzing et al., 1999; Lim et al., 2000; Mori et al., 2006; Stalder et al., 1999; Town et al., 2008). We next examined β-amyloid plaque-associated gliosis and S100(B) production in Tg2576 and Tg2576-huS100B mice by GFAP/S100 antibody or Iba1 antibody immunoreactivity (conventional astrocytosis or microgliosis “burden” analyses, respectively). Tg2576 mice exhibited enhanced β-amyloid plaque-associated reactive astrocytosis and microgliosis with age, as evidenced by increased expression of GFAP/S100 or Iba1 in glial somata and processes, respectively. S100 immunohistochemistry revealed faint staining of activated astrocytic processes, disclosing delicate structures. Numerous minute S100-positive granules, which were probably within the astrocytic processes, were dispersed between neurons. In addition, S100 and GFAP expression was strongly detected in dystrophic neurites in association with β-amyloid deposits as previously reported (Mori et al., 2006; Mrak et al., 1996). As compared to Tg2576 mice, Tg2576-huS100B mice displayed enhanced β-amyloid plaque-associated reactive astrocytosis with age. It is noteworthy that Tg2576-huS100B mice had hyperplasia and hypertrophy of activated astrocytes (> 50 μm in length), which were strongly GFAP/S100 immunoreactive in all three-brain regions examined at 15 and 19 months of age (Fig. 5,6A). Furthermore, Tg2576-huS100B mice demonstrated increased β-amyloid plaque-associated Iba1 reactive microglia with age.
Fig. 5.

The huS100B transgene further enhances astrocytic S100 expression in Tg2576 mice. Data were obtained from Tg2576 [n = 10 (5 males and 5 females) at 9 months of age; n = 14 each (7 males and 7 females) at 13, 15, and 19 months of age] and Tg2576-huS100B mice [n = 12 (6 males and 6 females) at 9 months of age; n = 16 each (8 males and 8 females) at 13, 15, and 19 months of age]. (A) Representative photomicrographs of S100 immunohistochemistry show β-amyloid plaque-associated astrocytosis in Tg2576 and Tg2576-huS100B mice at 19 months of age in each brain region indicated on the left. Scale bar denotes 50 μm. (B) Quantitative image analysis for S100 burden is shown in cingulate cortex (CC, top), hippocampus (H, middle), and entorhinal cortex (EC, bottom). All statistical comparisons are within brain region and between Tg2576 and Tg2576-huS100B mice at 9, 13, 15, and 19 months of age. ** P < 0.01, *** P < 0.001 vs. Tg2576 mice.
Fig. 6.

The huS100B transgene exacerbates astrocytosis in Tg2576 mice. Data were obtained from Tg2576 [n = 10 (5 males and 5 females) at 9 months of age; n = 14 each (7 males and 7 females) at 13, 15, and 19 months of age] and Tg2576-huS100B mice [n = 12 (6 males and 6 females) at 9 months of age; n = 16 each (8 males and 8 females) at 13, 15, and 19 months of age]. (A) Representative photomicrographs of GFAP immunohistochemistry for β-amyloid plaque-associated astrocytosis are shown in Tg2576 and Tg2576-huS100B mice at 19 months of age in each brain region indicated on the left. Scale bar denotes 50 μm. (B) Quantitative image analysis for glial fibrillary acidic protein (GFAP) burden is shown in cingulate cortex (CC, top), hippocampus (H, middle), and entorhinal cortex (EC, bottom). All statistical comparisons are within brain region and between Tg2576 and Tg2576-huS100B mice at 9, 13, 15, and 19 months of age. *** P < 0.001 vs. Tg2576 mice.
Interestingly, phagocytic microglia have been detected in the aged APP23 AD mouse model in small numbers (Stalder et al., 2005) and in increased abundance in aged Tg2576 mice bearing a CD11c promoter-driven dominant-negative transforming growth factor-β receptor type II transgene (Town et al., 2008) as well as in the ischemic core after cerebral ischemia in the rat brain (Ito et al., 2001). However, microglia in both groups of mice reported here had ramified thin and long cell processes and did not appear to be phagocytic based on morphological criteria (amoeboid structure, puffy cytoplasm, few or no processes resembling brain-infiltrating macrophages) (Streit, 2005). Further, we did not observe co-localization of Iba1 with 4G8 double-positive microglia in any of the sections examined (data not shown) (Fig. 7A). Quantitative microscopy analysis for astrocytosis (using GFAP and S100 antibodies) and microgliosis (using Iba1 antibody) revealed significantly increased burdens in CC, EC, and H regions at 9, 13, 15, and 19 months of age in Tg2576-huS100B mice (Fig. 5-7B, *** P < 0.001). We stratified by gender and increased β-amyloid plaque-associated reactive astrocytosis and microgliosis were significantly increased in both male and female bigenic mice (data not shown). In addition, to determine if inflammatory glial response mediated by the huS100B transgene might precede appearance of β-amyloid deposits, we examined brains of all four groups of mice (Tg2576, Tg2576-huS100B, TghuS100B, and wild-type littermates) at 7 months of age. Tg2576-huS100B mice showed significantly enhanced gliosis (astrocytosis and microgliosis) in CC, EC, and H regions (Fig. 8A,B * P < 0.05, ** P < 0.01, *** P < 0.001 vs. all three other groups of mice). Interestingly, Tg2576 and TghuS100B mice showed a slight increase in astrogliosis as compared to wild-type littermates at this age (Fig. 8A), but this did not reach statistical significance.
Fig. 7.

The huS100B transgene increases Iba1 positive reactive microglia in Tg2576 mice. Data were obtained from Tg2576 [n = 10 (5 males and 5 females) at 9 months of age; n = 14 each (7 males and 7 females) at 13, 15, and 19 months of age] and Tg2576-huS100B mice [n = 12 (6 males and 6 females) at 9 months of age; n = 16 each (8 males and 8 females) at 13, 15, and 19 months of age]. (A) Representative photomicrographs of Iba1 immunohistochemistry for β-amyloid plaque-associated microgliosis are shown in Tg2576 and Tg2576-huS100B mice at 19 months of age in each brain region indicated on the left. Scale bar denotes 50 μm. (B) Quantitative image analysis for Iba1 burden is shown in cingulate cortex (CC, top), hippocampus (H, middle), and entorhinal cortex (EC, bottom). All statistical comparisons are within brain region and between Tg2576 and Tg2576-huS100B mice at 9, 13, 15, and 19 months of age. *** P < 0.001 vs. Tg2576 mice.
Fig. 8.
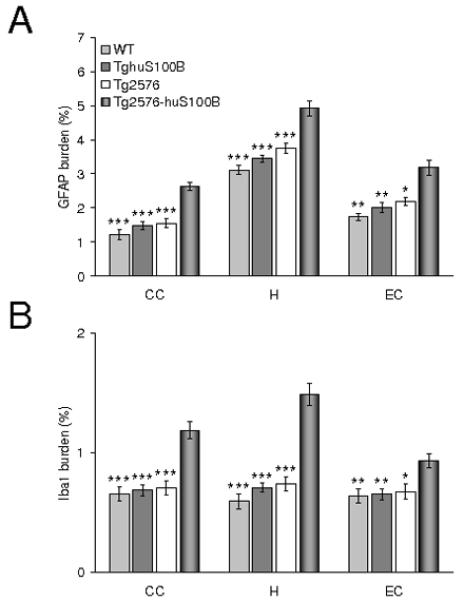
The huS100B transgene enhances cerebral gliosis in Tg2576 mice prior to β-amyloid deposition. Data were obtained from Tg2576, Tg2576-huS100B, TghuS100B, and wild-type (WT) mice [n = 8 each (4 males and 4 females) at 7 months of age]. Quantitative image analyses for GFAP (A) and Iba1 (B) burdens are shown in cingulate cortex (CC), hippocampus (H), and entorhinal cortex (EC). All statistical comparisons are within brain region and among Tg2576-huS100B, Tg2576, TghuS100B, and WT mice. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Tg2576-huS100B mice.
We reasoned that, given the well-established role of S100B in promoting the proinflammatory ‘cytokine cycle’ (Griffin et al., 1998b), forced expression of the huS100B transgene may promote inflammatory responses in the brain. We performed QPCR analyses for TNF-α, IL-1β, and IL-6, as well as mouse S100B mRNA levels in brain homogenates from Tg2576 and Tg2576-huS100B mice in addition to wild-type Tg2576 littermates and TghuS100B mice. We found that bigenic mice demonstrated increased mRNA levels of TNF-α, IL-1β, IL-6, and mouse S100B compared to Tg2576 and TghuS100B mice at 9 and 19 months of age (Fig. 9A,B, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Tg2576-huS100B mice). To determine whether proinflammatory cytokines including TNF-α, IL-1β, IL-6, and mouse S100B might be altered in singly and bigenic transgenic mice at 7 months of age, we examined cytokine mRNAs and were not able to detect any consistent increases in these cytokines as compared to wild-type mice (data not shown). The results from singly transgenic mice are in accord with our cytokine data in 9-month-old mice (Fig. 9A), where TghuS100B or Tg2576 mice did not significantly differ from wild-type animals (all means hovered around 1-fold over wild-type controls). Thus, we do not detect dysregulation of inflammatory response pathways in TghuS100B or Tg2576 mice until 9 months of age. When taken together, these data suggest that the huS100B transgene exacerbated the proinflammatory ‘cytokine cycle’ in Tg2576-huS100B mouse brains as early as 9 months of age, prior to the appearance of frank β-amyloid deposits.
Fig. 9.
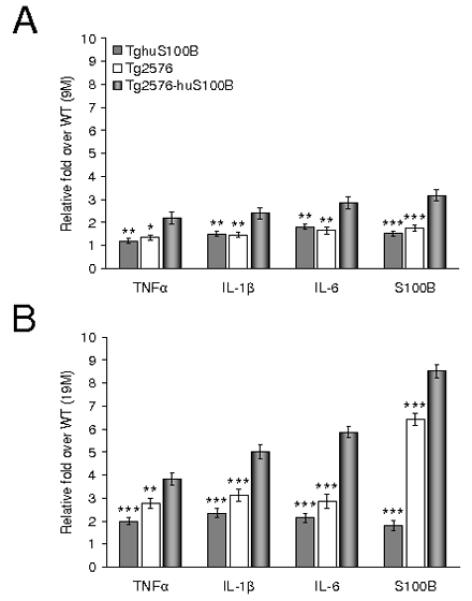
The huS100B transgene up-regulates brain cytokine mRNA expression. Quantitative real-time polymerase chain reaction results are shown at 9 (A) and 19 (B) months of age. Data were obtained from Tg2576 [n = 10 (5 males and 5 females) at 9 months of age; n = 14 each (7 males and 7 females) at 19 months of age], Tg2576-huS100B [n = 12 (6 males and 6 females) at 9 months of age; n = 16 each (8 males and 8 females) at 19 months of age], TghuS100B [n = 10 (5 males and 5 females) at 9 and 19 months of age], and wild-type littermates [n = 10 (5 males and 5 females) at 9 and 19 months of age]. Data were expressed as relative fold over wild-type (WT) mice. All statistical comparisons are among Tg2576-huS100B, Tg2576, and TghuS100B mice. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Tg2576-huS100B mice.
DISCUSSION
We sought to elucidate a putative relationship between S100B and AD-like pathology. To do this, we took a genetic approach to overproduce S100B by crossing TghuS100B mice with the Tg2576 mouse model of AD, and we then examined AD-like pathology. We found that the huS100B transgene acts to exacerbate cerebral amyloidosis, including brain parenchymal and cerebral vascular β-amyloid deposits in Tg2576-huS100B mouse brains. In brain areas examined at 9 months of age, we observed that Tg2576 mice had mainly dot-like β-amyloid deposits (< 5 μm in maximum diameter) but not larger-sized β-amyloid plaques. By contrast, Tg2576-huS100B mice had a number of large-sized β-amyloid plaques (> 50 μm in maximum diameter) that were already present at 9 months of age. In addition, as we did not observe any β-amyloid deposits in Tg2576 and Tg2576-huS100B mice at 7 months of age, the main difference between bigenic and singly-transgenic Tg2576 mice is a higher plaque burden in the former, and not that amyloid deposition starts earlier in Tg2576-huS100B mice. Our findings can thus be interpreted as accelerated AD-like pathology by up to 4 months of age based on increased Aβ burden (but not earlier β-amyloid deposition) in bigenic TghuS100B-Tg2576 mice vs. Tg2576 mice. We observed both histopathological evidence of increased β-amyloid deposits and biochemical evidence of elevated soluble and insoluble Aβ levels as well as of the amyloidogenic β-CTF (C99) and sAPPβ products in bigenic mice. These accelerated amyloidogenic events co-occurred with elevated BACE1 expression and β-secretase activity. Furthermore, these effects were associated with augmented reactive gliosis (astrocytosis and microgliosis), high levels of endogenous mouse S100 expression, and increased levels of proinflammatory cytokines.
The role of inflammatory responses and glial activation in the AD pathological process is complex, and studies have shown both beneficial and deleterious effects of glial activation on AD-like pathology in mouse models depending on the stimulus (Bard et al., 2000; Lim et al., 2000, 2001a, 2001b; Mackenzie and Munoz, 1998; Mori et al., 2006; Schenk et al., 1999; Town et al., 2008). These results have led to the conclusion that there are various forms of glial activation in the context of AD, and not all forms of glial activation are damaging – some are likely even beneficial (Town et al., 2005; Town, 2009). Our data reported here suggest that brain proinflammatory events associated with S100B-dependent glial activation in close proximity to β-amyloid plaques may lead to exacerbation of AD-like pathology, supporting the notion that chronic and prolonged activation of glia is detrimental in the brain (Matsui et al., 2002; Mori et al., 2004, 2008). Our results lend further support to the hypothesis that the astrocyte-derived protein, S100B, exacerbates the proinflammatory cytokine cycle, AD-like pathology, and associated brain injury (Mori et al., 2005, 2006; Tateishi et al., 2002).
Despite low-level, chronic activation of innate immunity and inflammatory response in the AD brain, glial cells ultimately fail to clear β-amyloid deposits. Astrocytes and microglia are the main innate immune response effector cells in the central nervous system. Imbalances between protective and destructive functions of these cells might impact neurotoxicity/synaptotoxicity in the context of neurodegenerative disease. In the AD brain, reactive astrocytes and microglia co-exist in both temporal and spatial proximity to β-amyloid plaques, and it is thought that diffuse β-amyloid deposits attract and activate IL-1-secreting microglia, which in turn activate astrocytes and promote astrocyte-derived S100B synthesis. This self-propagating ‘cytokine cycle’ promotes further release of proinflammatory cytokines and acute phase reactants by both activated microglia and astrocytes, leading to production of oxyradicals and nitric oxide (which are toxic at supraphysiologic levels). These proinflammatory substances in turn promote further enhancement of inflammation and likely bystander neuronal injury in the AD brain (Akiyama et al., 2000; Griffin et al., 1998b).
In view of the interaction between astrocytes and microglia, it is possible that increased astrocyte S100B levels not only promote a proinflammatory response by astrocytes but also exacerbate microgliosis. Several lines of evidence presented here support this: 1) exacerbated Iba1 microgliosis burden concomitant with increased S100- and GFAP-reactive astrocytes in Tg2576-huS100B mice, 2) significantly increased proinflammatory IL-1β mRNA expression and 3) significantly increased proinflammatory IL-6 mRNA expression, and the latter two correlated with up-regulated endogenous murine S100B mRNA in Tg2576-huS100B brains. While our analysis for proinflammatory mediators does not provide information on the cellular source of these products, it is noteworthy that microglia are the primary central nervous system producers of IL-1β and IL-6 (Akiyama et al., 2000), and our immunohistochemical analyses show that increased S100B in bigenic mice is most likely reactive astrocyte-derived.
An important lingering question regarding AD pathology is the relationship between gliosis and β-amyloid plaques. Studies in AD patients and in mouse models of AD clearly demonstrate tight temporal and spatial association between reactive glia and β-amyloid plaques, often interpreted as cerebral amyloidosis driving gliosis (Benzing et al., 1999; Lim et al., 2000; Mori et al., 2006; Stalder et al., 1999; Town et al., 2008). Consistent with this notion, clearance of β-amyloid plaques by Aβ immunotherapy is associated with reduced astrogliosis (Bard et al., 2000; Schenk et al., 1999). However, there is evidence for the converse. For example, non-steroidal anti-inflammatory drugs (NSIADs) reduce numbers of reactive glia (Mackenzie and Munoz, 1998), and numerous epidemiologic studies have shown an inverse risk relationship between NSIAD use and AD (Szekely et al., 2004, 2007). Further, treatment of AD mouse models with the NSIADs ibuprofen or curcumin results in mitigation of gliosis and as much as 50% reduced β-amyloid plaque load (Lim et al., 2000, 2001a, 2001b). Interestingly in our study, we found increased astrocytosis and microgliosis at 9 months of age in Tg2576-huS100B mice, prior to frank β-amyloid plaque deposition in Tg2576 mice (Hsiao et al., 1996). Our findings are consistent with the interpretation that S100B-induced gliosis exacerbates cerebral amyloidosis, and suggest that this form of gliosis is deleterious in the context of AD-like pathology.
Microglia/macrophages resembling Aβ phagocytes have been detected in the aged APP23 AD mouse brain (Stalder et al., 2005), in brains of aged Tg2576 mice bearing a CD11c promoter-driven dominant-negative transforming growth factor-β receptor type II transgene (Town et al., 2008), and in the ischemic core after cerebral ischemia in the rat brain (Ito et al., 2001). Despite careful examination of Iba1-stained brain sections from Tg2576 and Tg2576-huS100B mice, we did not observe phagocytic microglia/macrophages based on morphologic criteria (Streit, 2005). Further, we did not observe Iba1 and 4G8 double-positive microglia in any section examined. Taken together with enhanced brain proinflammatory cytokine levels in bigenic mice, the increased reactive microgliosis that we observed in Tg2576-huS100B mouse brains is consistent with a proinflammatory, anti-phagocytic phenotype (Town et al., 2005). It is possible that increased gliosis might simply be the result of enhanced β-amyloid plaque load in bigenic mice, given that glial activation is proportional to β-amyloid plaque load (Benzing et al., 1999; Lim et al., 2000; Stalder et al., 1999). However, our finding that gliosis precedes increased β-amyloid plaque deposition in Tg2576-huS100B mice instead suggests that S100B-induced inflammatory responses drive accelerated β-amyloid load in these animals. This conclusion is given strength by our findings that Tg2576-huS100B mice showed significantly higher GFAP and Iba1 burdens as compared to Tg2576 mice both during initiation of cerebral amyloidosis (9 months of age) and at an age prior to β-amyloid deposition (7 months of age). However, it should be noted that we also measured proinflammatory cytokines including TNF-α, IL-1β, IL-6, and mouse S100B mRNAs at 7 months of age, and were not able to detect any consistent differences in these cytokines between the four groups of mice (Tg2576, Tg2576-huS100B, TghuS100B, and wild-type littermates; data not shown). Thus, while the huS100B and Tg2576 transgenes are able to synergize on increasing glial surface activation markers prior to formation of β-amyloid plaques (i.e., at 7 months of age), they do not seem to bring about this effect on expression of proinflammatory cytokines until 9 months of age.
Glial cells associated with synapses integrate neuronal inputs and can release transmitters that modulate synaptic activity (Haydon 2001), and astrocyte-derived S100B promotes neurite extension and plays an important role in synaptogenesis and synapse remodeling in the developing and the mature brain under physiological conditions (Donato, 1999). However, in pathological states such as AD, S100B may contribute to neuritic changes associated with β-amyloid plaques. This is suggested by studies in Down’s syndrome showing that astrocytic overexpression of S100B drives dystrophic neurite outgrowth and culminates in the formation of neuritic plaques (Griffin et al., 1998a). Moreover, it has been reported that overexpression of S100B precedes the appearance of neuritic β-amyloid plaques in the APPV717F AD mouse model (Sheng et al., 2000). Thus, S100B may promote conversion of diffuse, non-fibrillar β-amyloid deposits to neuritic β-amyloid plaques, thereby exacerbating the progression of AD pathology (Mrak and Griffin, 2001; Mrak et al., 1996).
It is noteworthy that, while β-amyloid plaque morphometric analysis demonstrated significant increase in all three-plaque size subsets (< 25 μm, 25-50 μm, and > 50 μm) in each of the brain regions examined in Tg2576-huS100B mice, we consistently noted the greatest exacerbation when considering medium- and large-sized plaque subsets. Further, the huS100B transgene produced a number of “gigantic” β-amyloid plaques (>150 μm) in all three-brain regions examined at 15 and 19 months of age in bigenic mice. These data could be interpreted as consistent with a role for S100B in the maturation of β-amyloid deposits. In addition, we demonstrated that the huS100B transgene promotes cerebral vascular β-amyloid deposits. While Aβ synthesis has classically been regarded to predominantly take place at neuronal synapses, it has recently been reported that reactive astrocytes surrounding β-amyloid plaques express β-secretase in AD patient brains and in Tg2576 mice (Hartlage-Rübsamen et al., 2003; Yamamoto et al., 2007). Thus, it is possible that the GFAP promoter-driven huS100B transgene directly impacted astrocytic amyloidogenic APP metabolism. This would explain both the exacerbation of parenchymal and vascular Aβ deposits in bigenic mice. However, further study is warranted to investigate this hypothesis.
One hypothesis is that increasing S100B levels in the brain may directly accelerate amyloidogenicity by promoting cleavage of APP to Aβ. We examined the amyloidogenic β-CTF (C99) and observed enhancement of phospho-C99 and C99 expression in Tg2576-huS100B mice relative to Tg2576 mice. Moreover, we found increased levels of sAPPβ and BACE1, and elevated activity of β-secretase in brain homogenates from Tg2576-huS100B mice. Thus, in addition to its ability to promote brain inflammatory responses, it seems likely that S100B has a previously unappreciated role in directly promoting amyloidogenic APP processing. In conclusion, our data show a pathological relationship between S100B-induced reactive gliosis and cerebral amyloidosis, and offer insight into the etiologic contribution of this form of chronic glial activation to AD-like pathology.
ACKNOWLEDGEMENTS
This work was supported in part by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, Japan (18500279) (T.M.). T. T. is supported by a National Institute of Health/National Institute on Aging “Pathway to Independence” award (4 R00 AG029726-02 and 5 R00 AG029726-03). T. T. is the inaugural holder of the Ben Winters Endowed Chair in Regenerative Medicine.
Grant sponsor: the Japan Society for the Promotion of Science; Grant number: 18500279; Grant sponsor: National Institute of Health/National Institute on Aging; Grant numbers: 4 R00 AG029726-02 and 5 R00 AG029726-03.
REFERENCES
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T, Mori T, Shimoda T, Shinagawa R, Satoh S, Yada N, Katsumata S, Matsuda S, Kagamiishi Y, Tateishi N. Arundic acid (ONO-2506) ameliorates delayed ischemic brain damage by preventing astrocytic overproduction of S100B. Curr Drug Targets CNS Neurol Disord. 2005;4:127–142. doi: 10.2174/1568007053544084. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APPSW transgenic mice. Neurobiol Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim Biophys Acta. 1999;1450:191–231. doi: 10.1016/s0167-4889(99)00058-0. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. From The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Friend WC, Clapoff S, Landry C, Becker LE, O’Hanlon D, Allore RJ, Brown IR, Marks A, Roder J, Dunn RJ. Cell-specific expression of high levels of human S100β in transgenic mouse brain is dependent on gene dosage. J Neurosci. 1992;12:4337–4346. doi: 10.1523/JNEUROSCI.12-11-04337.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, McKenzie JE, Royston MC, Gentleman SM, Brumback RA, Cork LC, Del Bigio MR, Roberts GW, Mrak RE. Life-long overexpression of S100β in Down’s syndrome: implications for Alzheimer pathogenesis. Neurobiol Aging. 1998a;19:401–405. doi: 10.1016/s0197-4580(98)00074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998b;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlage-Rübsamen M, Zeitschel U, Apelt J, Gärtner U, Franke H, Stahl T, Günther A, Schliebs R, Penkowa M, Bigl V, Roßner S. Astrocytic expression of the Alzheimer’s disease β-secretase (BACE1) is stimulus-dependent. Glia. 2003;41:169–179. doi: 10.1002/glia.10178. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Horikoshi Y, Sakaguchi G, Becker AG, Gray AJ, Duff K, Aisen PS, Yamaguchi H, Maeda M, Kinoshita N, Matsuoka Y. Development of Aβ terminal end-specific antibodies and sensitive ELISA for Aβ variant. Biochem Biophys Res Commun. 2004;319:733–737. doi: 10.1016/j.bbrc.2004.05.051. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hu J, Ferreira A, Van Eldik LJ. S100β induces neuronal cell death through nitric oxide release from astrocytes. J Neurochem. 1997;69:2294–2301. doi: 10.1046/j.1471-4159.1997.69062294.x. [DOI] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AG, Koppal T, Akama KT, Guo L, Craft JM, Samy B, Schavocky JP, Watterson DM, Van Eldik LJ. Mechanism of glial activation by S100B: involvement of the transcription factor NFkB. Neurobiol Aging. 2001;22:765–772. doi: 10.1016/s0197-4580(01)00233-0. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001a;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001b;22:983–991. doi: 10.1016/s0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology. 1998;50:986–990. doi: 10.1212/wnl.50.4.986. [DOI] [PubMed] [Google Scholar]
- Matsui T, Mori T, Tateishi N, Kagamiishi Y, Satoh S, Katsube N, Morikawa E, Morimoto T, Ikuta F, Asano T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part I: enhanced astrocytic synthesis of S-100β in the periinfarct area precedes delayed infarct expansion. J Cereb Blood Flow Metab. 2002;22:711–722. doi: 10.1097/00004647-200206000-00010. [DOI] [PubMed] [Google Scholar]
- Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- Mori T, Tan J, Arendash GW, Koyama N, Nojima Y, Town T. Overexpression of human S100B exacerbates brain damage and periinfarct gliosis after permanent focal ischemia. Stroke. 2008;39:2114–2121. doi: 10.1161/STROKEAHA.107.503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Town T, Kobayashi M, Tan J, Fujita SC, Asano T. Augmented delayed infarct expansion and reactive astrocytosis after permanent focal ischemia in apolipoprotein E4 knock-in mice. J Cereb Blood Flow Metab. 2004;24:646–656. doi: 10.1097/01.WCB.0000120787.53851.A4. [DOI] [PubMed] [Google Scholar]
- Mori T, Town T, Tan J, Tateishi N, Asano T. Modulation of astrocytic activation by arundic acid (ONO-2506) mitigates detrimental effects of the apolipoprotein E4 isoform after permanent focal ischemia in apolipoprotein E knock-in mice. J Cereb Blood Flow Metab. 2005;25:748–762. doi: 10.1038/sj.jcbfm.9600063. [DOI] [PubMed] [Google Scholar]
- Mori T, Town T, Tan J, Yada N, Horikoshi Y, Yamamoto J, Shimoda T, Kamanaka Y, Tateishi N, Asano T. Arundic Acid ameliorates cerebral amyloidosis and gliosis in Alzheimer transgenic mice. J Pharmacol Exp Ther. 2006;318:571–578. doi: 10.1124/jpet.106.105171. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 2001;22:915–922. doi: 10.1016/s0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Sheng JG, Griffin WS. Correlation of astrocytic S100β expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:273–27. doi: 10.1097/00005072-199603000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281–286. doi: 10.1002/glia.20205. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, Rebeck GW, Hyman BT, Greenberg SM, Frosch MP, Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;26:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Bales KR, Cordell B, Paul SM, Jones RA, Woodward S, Zhou XQ, McGinness JM, Griffin WS. Overexpression of the neuritotrophic cytokine S100β precedes the appearance of neuritic β-amyloid plaques in APPV717F mice. J Neurochem. 2000;74:295–301. doi: 10.1046/j.1471-4159.2000.0740295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglial cells. In: Kettermann H, Ransom BR, editors. Neuroglia. Oxford Press; New York: 2005. pp. 60–71. [Google Scholar]
- Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology. 2004;23:159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- Szekely CA, Town T, Zandi PP. NSAIDs for the chemoprevention of Alzheimer’s disease. Subcell Biochem. 2007;42:229–248. doi: 10.1007/1-4020-5688-5_11. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Tateishi N, Mori T, Kagamiishi Y, Satoh S, Katsube N, Morikawa E, Morimoto T, Matsui T, Asano T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: suppression of astrocytic activation by a novel agent (R)-(-)-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J Cereb Blood Flow Metab. 2002;22:723–734. doi: 10.1097/00004647-200206000-00011. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-β-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T. Alternative Aβ immunotherapy approaches for Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8:114–127. doi: 10.2174/187152709787847306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright MS, Craft JM, Griffin WS, Marks A, Pineda J, Padgett KR, Van Eldik LJ. Increased susceptibility of S100B transgenic mice to perinatal hypoxia-ischemia. Ann Neurol. 2004;56:61–67. doi: 10.1002/ana.20142. [DOI] [PubMed] [Google Scholar]