

Abstract
Background and purpose:
The present study investigated whether the pathophysiological changes induced by burn and smoke inhalation are modulated by parenteral administration of Na2S, a H2S donor.
Experimental approach:
The study used a total of 16 chronically instrumented, adult female sheep. Na2S was administered 1 h post injury, as a bolus injection at a dose of 0.5 mg·kg−1 and subsequently, as a continuous infusion at a rate of 0.2 mg·kg−1·h−1 for 24 h. Cardiopulmonary variables (mean arterial and pulmonary arterial blood pressure, cardiac output, ventricular stroke work index, vascular resistance) and arterial and mixed venous blood gases were measured. Lung wet-to-dry ratio and myeloperoxidase content and protein oxidation and nitration were also measured. In addition, lung inducible nitric oxide synthase expression and cytochrome c were measured in lung homogenates via Western blotting and enzyme-linked immunosorbent assay (elisa) respectively.
Key results:
The H2S donor decreased mortality during the 96 h experimental period, improved pulmonary gas exchange and lowered further increase in inspiratory pressure and fluid accumulation associated with burn- and smoke-induced acute lung injury. Further, the H2S donor treatment reduced the presence of protein oxidation and 3-nitrotyrosine formation following burn and smoke inhalation injury.
Conclusions and implications:
Parenteral administration of the H2S donor ameliorated the pulmonary pathophysiological changes associated with burn- and smoke-induced acute lung injury. Based on the effect of H2S observed in this clinically relevant model of disease, we propose that treatment with H2S or its donors may represent a potential therapeutic strategy in managing patients with acute lung injury.
Keywords: acute lung injury, protein oxidation, hydrogen sulphide, smoke inhalation, sheep
Introduction
Acute lung injury develops in patients suffering from major trauma caused by cutaneous burn and smoke inhalation injury. Current definitions require the exclusion of left atrial hypertension and heart failure during clinical assessment (Brown, 1998;Abraham et al., 2000). The primary physiological abnormalities are severe arterial hypoxaemia as well as marked increase in ventilation and pulmonary hypertension. Most of these patients require assisted ventilation with positive pressure.
The ovine model of lung injury induced by inhalation of cotton smoke, alone, or in combination with burn injury, has been used extensively by several laboratories to investigate the mechanisms and experimental therapy of acute lung injury (Traber et al., 1986; Demling et al., 1994; Lalonde et al., 1994; Hales et al., 1997; Fukuda et al., 1999; Efimova et al., 2000; Riedel et al., 2006; Wang et al., 2006; Hamahata et al., 2008). It has been established that combined burn and smoke inhalation injury impairs hypoxic pulmonary vasoconstriction (HPV), the vasoconstrictive response to hypoxia, thereby mismatching ventilation with perfusion (Westphal et al., 2006). Gas exchange is affected by increases in the dispersion of both alveolar ventilation and cardiac output because bronchial and vascular functions are altered by injury-related factors, such as the effects of inflammatory mediators on airway and vascular smooth muscle tone. Because oxygen exchange is determined by alveolar ventilation, areas of high alveolar ventilation/blood flow (VA/Q) ratio and dead space in acute lung injury increase the ventilation required to keep the arterial partial pressure of oxygen (PaO2) level constant. Therefore, the work to expand the lungs to maintain the PaO2 level must also increase as lung compliance decreases. Additionally, hypoxaemia produced by alveolar oedema impairs gas exchange by creating areas of low VA/Q ratio and shunt. Elevated levels of nitric oxide (NO) and elevated reactive nitrogen and oxygen species production are one of the features of this model, which is associated with increased pulmonary shunting (Westphal et al., 2006; Westphal et al., 2008): pharmacological inhibition of NO overproduction, or therapy with antioxidants exerts protective effects in this model (Ahn et al., 1990; Bone et al., 2002; Enkhbaatar et al., 2006; Westphal et al., 2008). Activation of pro-inflammatory pathways is another important feature of this model (Hales et al., 1997; Kimura et al., 1998; Shimoda et al., 2003; Cox et al., 2009).
The current study used sodium sulphide (Na2S) to study the potential effect of attenuated reactive nitrogen species formation on survival and pulmonary function outcomes, including PaO2/FiO2 (fraction of inspired oxygen) ratio, pulmonary shunt fraction and inspiratory peak and pause airway pressures. H2S is an endogenous biological mediator formed from L-cysteine by two pyridoxal-5′-phosphate-dependent enzymes: cystathionine β-synthase and cystathionine γ-lyase. The organ- and cell-specific expression of the cystathionine β-synthase and cystathionine γ-lyase enzymes, and thus production of endogenous H2S includes the liver, kidneys and vascular and non-vascular smooth muscle (Kamoun 2004;Lowicka and Beltowski, 2007; Szabo, 2007; Li and Moore, 2008). Sulphide production ranges from 1 to 10 pmol·s−1·(mg protein)−1 and has been shown to possess cytotoxic effects at higher micromolar concentration in in vitro studies (see Szabo, 2007). Sulphide formulations have previously been demonstrated to have pharmacological effects as vasodilators (Zhao et al., 2001; Koenitzer et al., 2007; Kiss et al., 2008; Webb et al., 2008) peroxynitrite scavengers (Whiteman et al., 2004), cytoprotective agents (Johansen et al., 2006; Jha et al., 2008; Tang et al., 2008) and as inhibitors of leukocyte adhesion and leukocyte-mediated inflammation (Zanardo et al., 2006; Wallace et al., 2007; Andruski et al., 2008; Sivarajah et al., 2009). H2S attenuates translocation of the p65 subunit of nuclear factor-kappa B (NF-κB) from the cytosol to the nucleus (Sivarajah et al., 2009) and may therefore decrease transcription of NF-κB-dependent proteins such as inducible NO synthase (iNOS).
Recently, we have demonstrated that H2S therapy has beneficial effects in a murine model of smoke and burn injury as evidenced by improved survival and reduced pulmonary oxidation (Esechie et al., 2008). In order to extend these findings to a clinically relevant model, in the present study we have examined the effect of Na2S treatment in a clinically relevant ovine model of burn- and smoke inhalation-induced acute lung injury. We have followed various physiological parameters of haemodynamics and pulmonary function. In addition, as an indirect measure of peroxynitrite formation, we have quantified protein 3-nitrotyrosine formation in lung homogenates at 96 h post injury.
Methods
Animal model
The following procedures were approved by the Animal Care and Use Committee of the University of Texas Medical Branch and were in compliance with the guidelines for the care and use of laboratory animals of the National Institutes of Health and the American Physiological Society. The ovine model of burn- and smoke-induced acute lung injury has been described previously (Westphal et al., 2006). Briefly, adult merino sheep, weighing between 33 ± 3 kg, were surgically instrumented for chronic study with a femoral artery catheter, a thermodilution catheter and a left atrial catheter. The sheep were given 5–7 days to recover from the surgical procedure with free access to food and water.
Injury and resuscitation protocol
The study protocol is shown in Figure 1, and the injury protocol has been previously described in detail (Westphal et al., 2006; Enkhbaatar et al., 2008;Westphal et al., 2008). Briefly, the animals were anaesthetized using halothane and a tracheostomy performed. The animal's wool was shaved, and a 20% total body surface area third-degree flame burn was made on each flank. This procedure produces a third-degree burn, with destroys the nerve endings in the skin and is considered a painless injury. We administered buprenorphine (0.03 mg, i.v.) during the burn and every 12 h subsequently, to provide analgesia for the edges of the burn, which may be second-degree burns.
Figure 1.
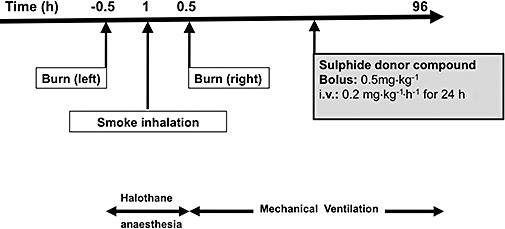
Experimental protocol. Administration of the H2S donor was started 1 h after smoke inhalation injury. Fluid resuscitation using Ringer lactate was started immediately after the injury.
Inhalation injury was induced using a modified bee smoker that was filled with 50 g of burning cotton towelling and connected to the tracheostomy tube via a modified endotracheal tube containing an indwelling thermistor from a Swan-Ganz catheter. During the insufflation procedure, the temperature of the smoke was monitored carefully not to exceed 40°C. The sheep was insufflated with a total of 48 breaths of cotton smoke. Na2S was administered 1 h post injury, as a bolus injection at a dose of 0.5 mg·kg−1 and subsequently, as a continuous infusion at a rate of 0.2 mg·kg−1·h−1 for 24 h. Control animals received equivalent volume of saline vehicle. The dose of Na2S was selected based on preliminary range-finding studies (data not shown) that were based on previous studies utilizing hydrogen sulphide donors in large animals (Simon et al., 2008; Sodha et al., 2008) as well as pharmacokinetic considerations based on the short half-life of parenterally infused H2S donors (Szabo, 2007; Insko et al., 2009).
All sheep were mechanically ventilated with a Servo Ventilator (Model 900C Siemens; Elema, Sweden) throughout the experimental period. Ventilation was performed with a positive end-expiratory pressure of 5 cm H2O and a tidal volume of 15 mL·kg−1. The respiratory rate set to maintain normocapnia and adjusted according to blood gas analysis. All animals were fluid-resuscitated 1 h after injury, initially with an infusion rate of 4 mL·(% burned surface area)−1·kg−1 for the first 24 h and 2 mL·(% burned surface area)−1·kg−1·day−1 for the next 48 h. Urine output recorded every 6 h. During this experimental period, the animals were allowed free access to food, but not to water, to allow accurate determination of fluid balance.
Monitoring and euthanasia
Animals were subject to monitoring 24 h a day and received the full-time attention of veterinary staff. At the end of the 96 h experimental period, the animals were given 20 mg (1 mL, s.c.) of xylazine and then anaesthetized with 5 mg·kg−1 of ketamine intravenously. Once they were deeply anaesthetized, a lethal injection of saturated KCl (0.5 mL·kg−1) was administered. During the study, animals that exhibited distress (as judged by a set of pre-determined criteria) were killed in the same manner.
Cardiopulmonary variables measured
Measured physiological parameters were not considered valid until the animals were fully awake and standing. Mean arterial, pulmonary arterial, left atrial and central venous pressures (in mmHg) were measured with pressure transducers (model P X 3 X 3, Edwards Lifesciences, Irvine, CA, USA). Cardiac output was measured with a cardiac output computer (Model COM-1; Edwards Lifesciences, Irvine, CA, USA). Cardiac index, ventricular stroke work index, vascular resistance index and shunt fraction were calculated using standard equations. Arterial and mixed venous blood samples were measured with a blood gas analyzer (model IL Synthesis 15, Instrumentation Laboratory, Lexington, MA, USA). Oxyhaemoglobin saturation and carboxyhaemoglobin concentration were analysed with a CO-oximeter (Model IL482; Instrumentation Laboratory, Lexington, MA, USA). Haematocrit (Hct) was measured in heparinized microhaematocrit capillary tubes (Fisher, Inc., Pittsburgh, PA, USA).
Lung tissue preparation
Tissue was sampled from the animals that survived the 96 h experimental period. Briefly, animals were killed, and lung tissue was sampled from the lower right lobe, immediately frozen in liquid nitrogen and stored at −80°C. Tissue preparation was performed using a modified protocol (Mineta et al., 2002). Lung tissue was weighed, suspended in 1 mL cold lysis buffer on ice [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Triton X-100]; 10 µL stock protease inhibitor cocktail (Sigma Aldrich, Saint Louis, MO, USA) was added to produce a final concentration of 4-(2-aminoethyl) benzenesulphonyl fluoride hydrochloride (AEBSF) (1 mM), aprotinin (800 nM), E-64 (15 µM), leupeptin (20 µM), bestatin (50 µM) and pepstatin A (10 µM). Additionally, 10 µL of stock phosphatase inhibitor (Pierce, Rockford, IL, USA) was added to produce a final concentration of sodium fluoride (50 mM), sodium orthovanadate (1 mM), sodium pyrophosphate (10 mM) and β-glycerophosphate (10 mM), and 1 µL of 1 M dithiothreitol (DTT) (all from Sigma Aldrich, Saint Louis, MO, USA) was added to the lysis buffer mixture and homogenized with a LabGen 125 (Cole Parmer, Vernon Hills, IL, USA). The lysate was centrifuged at 10 000×g for 25 min at 4°C. The supernatant (cytoplasmic extract) was collected for total protein measurement.
The pellet containing the nuclei was resuspended in 250 µL cold nuclear extraction buffer containing 0.1 M DTT (Sigma Aldrich, Saint Louis, MO, USA) and 1.5 µL of the stock protease inhibitor cocktail (Sigma Aldrich, Saint Louis, MO, USA). Samples were agitated vigorously for 15 s every 10 min for 30 min. The mixture is again centrifuged at ∼20 000×g for 5 min. The supernatant containing the nuclear proteins was collected, and total protein was determined using the Bradford method (Bradford, 1976).
Preparation of mitochondrial protein extracts
The mitochondrial fraction was isolated using a modified protocol (Cox and Emili, 2008). Lung tissue samples were homogenized, and the lysate was centrifuged at 800×g for 15 min. The pellet was discarded and the supernatant further centrifuged at 6000×g for 15 min. The supernatant was discarded, and the pellet was resuspended in 10 volumes of a hypotonic lysis buffer containing 10 mM HEPES (pH 7.9), 1 mM DTT and 1 mM phenylmethylsulphonyl fluoride (PMSF) for extraction of soluble mitochondrial proteins. This lysate was again centrifuged at 11 000×g for 20 min.
Western blotting protocol
Eighty micrograms of the protein extract or 20 µg of nuclear extract was boiled in 2× Laemmli sample buffer, separated on a 4–20% SDS-polyacrylamide gradient gel by electrophoresis and transferred to an Immobilon–P PVDF (polyvinylidene fluoride) membrane. Membranes were blocked in blocking solution [Tris-buffered saline-Tween 20: 10 mM Tris-HCl (pH 7.4), 154 mM NaCl, 0.05% Tween 20 (v/v)] containing 5% non-fat dry milk powder (w/v) for 1 h at room temperature. The membranes were immunoblotted at 4°C overnight with primary antibodies diluted in Tris-buffered saline-Tween 20 [10 mM Tris-HCl (pH 7.4), 154 mM NaCl, 0.05% Tween 20 (v/v)] containing 1% non-fat dry milk powder (w/v). The following antibodies were used: rabbit anti-poly (ADP-ribose) (PAR; catalogue No. 4336-BPC-100) purchased from Trevigen (Gaithersburg, MD, USA) was diluted to 1:1000; biotinylated mouse anti-nitrotyrosine (Catalog No. 10006966) purchased from Cayman Chemicals (Ann Arbor, MI, USA) was diluted to 1:1500; and rabbit anti-NOS-2 (Catalog No. sc-650) diluted to 1:200 and goat anti-β-actin (Catalog No. sc-1616) purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA) was diluted to 1:500. The primary antibody was detected using the following horseradish peroxidase-conjugated secondary antibodies: goat anti-rabbit (Catalog No. HAF008) diluted to 1:4000 or donkey anti-goat (Catalog No. HAF109) antibody diluted to 1:3000 or streptavidin-horseradish peroxidase (Catalog No. DY998) diluted at 1:200 purchased from R&D Systems (Minneapolis, MN, USA). The membranes were subjected to chemiluminescence using the SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA) according to the manufacturer's instructions, and intensity of immunoreactivity was measured using NIH ImageJ software (http://rsb.info.nih.gov/nih-image/).
Enzyme-linked immunosorbent assay (elisa) for lung cytochrome c
The assay was performed according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). Briefly, 100 µL of standards or lung mitochondrial extracts were added to each well pre-coated with a monoclonal antibody specific for cytochrome c and incubated for 2 h at room temperature (20–25°C). The plates were aspirated and washed three times with 400 µL of wash buffer. Two hundred microlitres of the cytochrome c conjugate antibody was added to each well and incubated for 2 h. The microplate was washed, and 200 µL of substrate solution was added to each well. The microplate was protected from light during this step and incubated for 30 min at room temperature. Fifty microlitres of a stop solution was added to each well, and the intensity of the colour was measured at 450 nm with a wavelength correction at 540 nm using a spectophotometer (MRX, Dynatech Laboratories Inc., Chantilly, VA, USA).
Lung wet-to-dry weight ratio
The blood wet-to-dry ratio was calculated to determine the bloodless lung wet-to-dry ratio of the lung. The right lung was then removed, and the lower one-half of the right lower lobe was used for the determination of wet-to-dry weight ratio (Pearce et al., 1965).
Measurement of lung myeloperoxidase content
Myeloperoxidase (MPO), an indicator of polymorphonuclear leukocyte accumulation, was determined directly in whole lung homogenates (Elrod et al., 2007). Frozen lung tissue (50 mg) was weighed and homogenized in 0.5 mL of 0.5% (w/v) hexa-decyl-trimethyl-ammonium bromide (HTAB, Sigma, St. Louis, MO, USA) in 50 mM phosphate buffer (pH 6). An additional 0.5 mL of the HTAB buffer was added to yield a final concentration of 50 mg tissue per millilitre of buffer. The sample was suspended by vortex mixing and then centrifuged for 20 min at 19 000×g at 4°C. Twenty microlitres of supernatant from each sample was added to 200 µL of a 1.0% hydrogen peroxide solution (H2O2) in 50 µM phosphate buffer (pH 6.0) containing 4.175 mg·mL−1 O-dianisidine hydrochloride (ODN). Each sample was assayed for MPO activity in duplicate wells in a 96-well plate. Using a plate reader (MRX, Dynatech Laboratories Inc., Chantilly, VA, USA) absorbance was measured at 450 nm immediately and at 60 s intervals for a total of 10 readings. One unit of MPO activity was defined as the degradation of 1 µmol H2O2·min−1 at 25°C, which gives a change in absorbance (ΔA) of 0.0113/min.
Data analysis
Summary statistics of data are expressed as mean ± SEM. Comparisons were made using two-way analysis of variance with a Tukey-Kramer post hoc procedure. A P-value ≤ 0.05 was considered to be statistically significant.
Results
Mortality study and cardiopulmonary responses
Figure 1 describes the study protocol followed in this study. The number of animals used in the survival analysis was eight for both the control and the Na2S-treated group. The animals in the Na2S-treated group had 100% survival at the end of the experimental period compared with the untreated control animals that showed 75% mortality (Figure 2; P < 0.005). Table 1 describes the haemodynamic changes observed during the 96 h experimental time period. There was no significant difference in cardiac index, mean arterial pressure and mean pulmonary artery pressure between the control group and the Na2S-treated group. Left atrial pressure was reduced in the Na2S-treated group at 30 and 36 h post injury. There was a significant difference in central venous pressure at 30 and 72 h in the Na2S-treated animals while pulmonary vascular resistance index was attenuated at 18 h and at 30–60 h post injury. Sulphide treatment also attenuated the calculated left venticular stroke work index at 18 and 30 h post injury. Although there was a tendency towards haemo-concentration (Hct) in the control animals, a significantly higher Hct value was recorded at 30 and 36 h in the control animals when compared with the sulphide-treated animals. Likewise, haemoglobin was significantly lowered at 30 h post injury in the treated animals.
Figure 2.
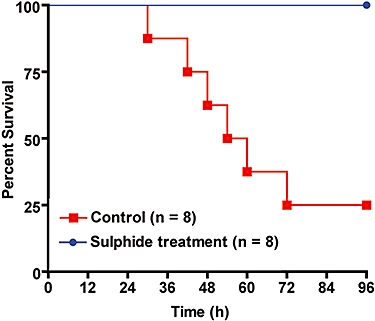
Kaplan-Meier curve showing survival over time in the vehicle control group and the Na2S treatment group. There were only two animals in the control group from 72 to 96 h.
Table 1.
Cardiopulmonary hemodynamics
| Parameter |
Times post injury (h) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 12 | 18 | 24 | 30 | 36 | 48 | 60 | 72 | 84 | 96 | |
| CI LI (min−1·m−2) | |||||||||||
| Vehicle | 5.8 ± 0.2 | 5.5 ± 0.3 | 5.3 ± 0.4 | 5.9 ± 0.4 | 6.1 ± 0.3 | 6.7 ± 0.5 | 7.6 ± 0.6 | 8.1 ± 1.0 | 7.4 ± 0.9 | 7.1 ± 1.7 | 8.2 ± 2.4 |
| Sulphide Treatment | 6.9 ± 0.2 | 7.1 ± 0.8 | 6.8 ± 0.5 | 6.5 ± 0.2 | 6.7 ± 0.3 | 6.4 ± 0.3 | 7.0 ± 0.6 | 7.1 ± 0.6 | 7.0 ± 0.6 | 7.7 ± 0.6 | 7.5 ± 0.7 |
| MAP (mmHg) | |||||||||||
| Vehicle | 99 ± 2 | 108 ± 3 | 104 ± 4 | 104 ± 3 | 103 ± 3 | 101 ± 3 | 105 ± 2 | 105 ± 5 | 105 ± 8 | 93 ± 3 | 88 ± 3 |
| Sulphide Treatment | 94 ± 2 | 105 ± 4 | 108 ± 4 | 104 ± 3 | 104 ± 3 | 105 ± 3 | 104 ± 3 | 104 ± 4 | 102 ± 3 | 101 ± 4 | 100 ± 5 |
| MPAP (mmHg) | |||||||||||
| Vehicle | 20.9 ± 0.7 | 25.9 ± 1.6 | 28.9 ± 1.4 | 28.9 ± 1.2 | 29.9 ± 1.4 | 29.4 ± 1.4 | 29.2 ± 1.3 | 26.3 ± 2.8 | 25.0 ± 3.0 | 23.0 ± 1.0 | 24.0 ± 2.0 |
| Sulphide Treatment | 19.9 ± 0.5 | 27.4 ± 1.3 | 27.4 ± 1.5 | 28.0 ± 1.3 | 26.8 ± 1.3 | 26.6 ± 1.6 | 28.1 ± 1.0 | 26.0 ± 1.2 | 26.6 ± 1.1 | 26.9 ± 1.3 | 27.3 ± 1.4 |
| LAP (mmHg) | |||||||||||
| Vehicle | 9.5 ± 0.7 | 9.6 ± 0.8 | 10.3 ± 1.1 | 12.0 ± 0.8 | 13.3 ± 1.1 | 13.1 ± 1.1 | 15.1 ± 1.5 | 14.0 ± 1.7 | 14.0 ± 1.0 | 13.0 ± 2.5 | 15.0 ± 3.5 |
| Sulphide Treatment | 8.0 ± 0.6 | 11.1 ± 1.4 | 11.4 ± 1.3 | 11.6 ± 0.8 | 8.0 ± 0.8* | 11.0 ± 0.6* | 12.4 ± 1.2 | 11.3 ± 1.3 | 11.0 ± 1.0 | 12.3 ± 1.2 | 9.9 ± 1.0 |
| CVP (mmHg) | |||||||||||
| Vehicle | 6.6 ± 0.9 | 9.0 ± 1.2 | 11.4 ± 2.6 | 10.1 ± 0.7 | 13.4 ± 1.6 | 11.9 ± 1.4 | 16.0 ± 5.0 | 11.3 ± 5.0 | 12.0 ± 2.1 | 13.0 ± 4.0 | 13.5 ± 2.5 |
| Sulphide Treatment | 5.6 ± 0.6 | 10.9 ± 0.7 | 9.5 ± 0.8 | 9.9 ± 0.6 | 11.1 ± 1.1* | 10.0 ± 0.8 | 9.4 ± 1.9 | 8.4 ± 1.3 | 6.3 ± 1.0* | 9.0 ± 1.0 | 9.0 ± 1.0 |
| PVRI (dyne·s−1·cm−5·m−2) | |||||||||||
| Vehicle | 150 ± 7 | 184 ± 28 | 227 ± 30 | 187 ± 20 | 183 ± 21 | 162 ± 13 | 112 ± 14 | 77 ± 7 | 136 ± 31 | 98 ± 35 | 92 ± 47 |
| Sulphide Treatment | 120 ± 7 | 146 ± 21 | 127 ± 16* | 151 ± 16 | 127 ± 11* | 135 ± 19* | 149 ± 15* | 137 ± 17* | 156 ± 16 | 135 ± 13 | 144 ± 19 |
| LVSWI (g·m−1·m−2) | |||||||||||
| Vehicle | 74 ± 4 | 61 ± 3 | 51 ± 4 | 56 ± 5 | 53 ± 2 | 57 ± 4 | 68 ± 4 | 86 ± 17 | 82 ± 17 | 67 ± 16 | 72 ± 20 |
| Sulphide Treatment | 83 ± 4 | 69 ± 7 | 71 ± 7* | 66 ± 6 | 69 ± 7* | 67 ± 6 | 69 ± 7 | 77 ± 8 | 65 ± 5 | 70 ± 4 | 60 ± 4 |
| Hct (%PCV) | |||||||||||
| Vehicle | 23.0 ± 0.8 | 25.0 ± 2.0 | 25.3 ± 2.1 | 27.3 ± 2.0 | 29.0 ± 1.5 | 27.3 ± 1.5 | 25.0 ± 1.0 | 22.0 ± 1.2 | 24.3 ± 0.9 | 22.0 ± 3.0 | 24.0 ± 3.5 |
| Sulphide Treatment | 21.8 ± 1.4 | 23.1 ± 1.6 | 23.8 ± 1.6 | 23.4 ± 1.1 | 24.5 ± 1.2* | 23.1 ± 1.5* | 24.0 ± 1.8 | 22.0 ± 1.8 | 22.0 ± 1.6 | 22.3 ± 2.1 | 23.0 ± 2.3 |
| Hb (g·mL−1) | |||||||||||
| Vehicle | 0.08 ± 0.004 | 0.09 ± 0.006 | 0.09 ± 0.007 | 0.09 ± 0.006 | 0.1 ± 0.005 | 0.09 ± 0.005 | 0.09 ± 0.005 | 0.08 ± 0.003 | 0.08 ± 0.001 | 0.08 ± 0.009 | 0.09 ± 0.01 |
| Sulphide Treatment | 0.08 ± 0.006 | 0.08 ± 0.004 | 0.08 ± 0.005 | 0.08 ± 0.005 | 0.09 ± 0.005* | 0.08 ± 0.006 | 0.08 ± 0.006 | 0.08 ± 0.007 | 0.08 ± 0.007 | 0.08 ± 0.007 | 0.08 ± 0.008 |
| FiO2 (%) | |||||||||||
| Vehicle | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.3 ± 0.1 | 0.5 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.1 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.9 ± 0.1 | 0.7 ± 0.3 |
| Sulphide Treatment | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.4 ± 0.1 | 0.5 ± 0.1* | 0.7 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.1 | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.5 ± 0.1 |
Values are presented as mean ± SEM.
Comparisons were made using two-way analysis of variance with a Tukey-Kramer post hoc procedure. There were only two animals in the control group from 72 to 96 h.
P≤ 0.05 versus control.
CI, cardiac index; CVP, central venous pressure; FiO2, fraction of inspired oxygen; Hb, haemoglobin; Hct, haematocrit; LAP, left atrial pressure; LVSWI, left venticular stroke work index; MAP, mean arterial pressure; MPAP, mean pulmonary arterial pressure; PVRI, pulmonary vascular resistance index.
Pulmonary gas exchange
Pulmonary gas exchange (PaO2/FiO2 and pulmonary shunt fraction) were calculated in all animals and are shown in Figure 3. Panel A and B compares the differences between the two experimental groups with (Figure 3A) and without (Figure 3B) the animals in the control group that survived the 96 h experiment. The combined injury was associated with a deterioration of pulmonary gas exchange, as shown by a reduction in the PaO2/FiO2 ratio (top, Figure 3A) associated with a marked increase in pulmonary shunt fraction (bottom, Figure 3) in vehicle-treated control animals. The PaO2/FiO2 ratio in these animals reached a level below 200 at 24 h post injury and steadily declined in the subsequent 72 h, indicating the presence of severe acute respiratory distress syndrome. Treatment with sulphide prevented the impaired gas exchange caused by burn and smoke inhalation. The PaO2/FiO2 ratio remained above 200 in treated animals for 42 h after the initial trauma (top, Figure 3A). Moreover, we observed an improvement in gas exchange (increase in PaO2/FiO2 above 200) in the treated group beginning at 72–96 h. The increase in pulmonary shunt fraction was markedly decreased in the animals treated with sulphide at 24, 36, 40 and 60 h (bottom, Figure 3A). The PaO2/FiO2 ratio in the non-surviving control animals (top, Figure 3A) remained below 200. Inspiratory peak and pause airway pressures were recorded from the ventilator readout. Both peak (top, Figure 4A) and pause (bottom, Figure 4A) airway pressures were especially pronounced in the control animals compared with the measured airway pressures in the treated animals. This increase in ventilatory pressures was markedly increased in the first 48 h in both groups. However, at 30 and 42 h the peak pressure was significantly higher in the control group compared with the Na2S-treated group.
Figure 3.
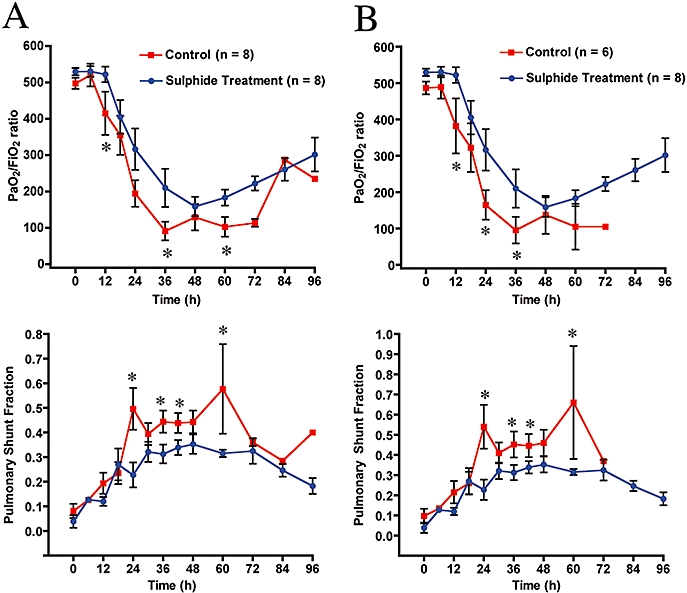
Effect of burn and smoke injury and Na2S treatment on pulmonary gas exchange. (A) Na2S treatment versus all control animals; (B) Na2S treatment versus non-surviving animals during the experimental period. Note that Na2S treatment after combined injury resulted in arterial partial pressure of oxygen/fraction of inspired oxygen (PaO2/FiO2) ratio recovery after 48 h. Comparisons were made using two-way analysis of variance with a Tukey-Kramer post hoc procedure. There were only two animals in the control group from 72 to 96 h. *P≤ 0.05 versus control.
Figure 4.
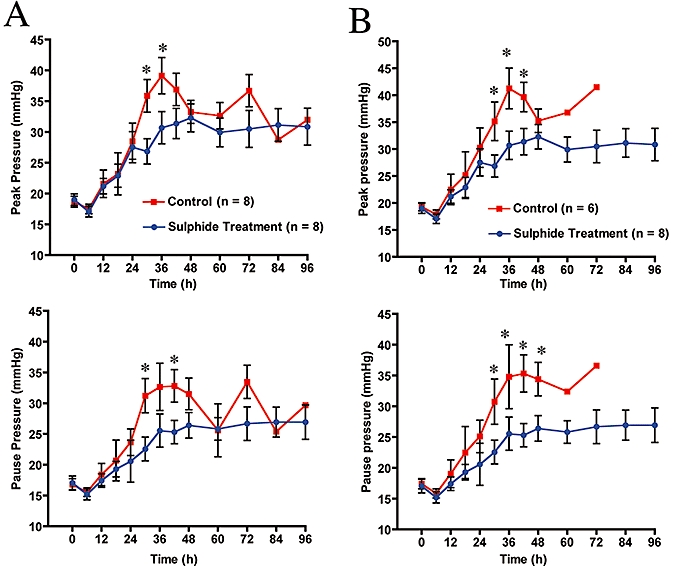
Effect of burn and smoke injury and Na2S treatment on airway pressure. (A) Na2S treatment versus all control animals; (B) Na2S treatment versus non-surviving animals during the experimental period. Comparisons were made using two-way analysis of variance with a Tukey-Kramer post hoc procedure. There were only two animals in the control group from 72 to 96 h. *P≤ 0.05 versus control.
Changes in urine output and fluid accumulation
The effect of sulphide administration on fluid input and urine output are shown in Figure 5A and 5B. At 24 and 36 h post injury, treatment with Na2S improved urine output. Our data show that the sulphide-treated animals had a negative fluid balance beginning at 30 h post injury, and this was significantly different from the control animals at 36 h (middle, Figure 5B). A comparison of the control animals that survived versus the non-surviving animals at the 96 h experimental study period showed that sulphide reduced fluid accumulation (bottom, Figure 5C vs. bottom, Figure 5B).
Figure 5.
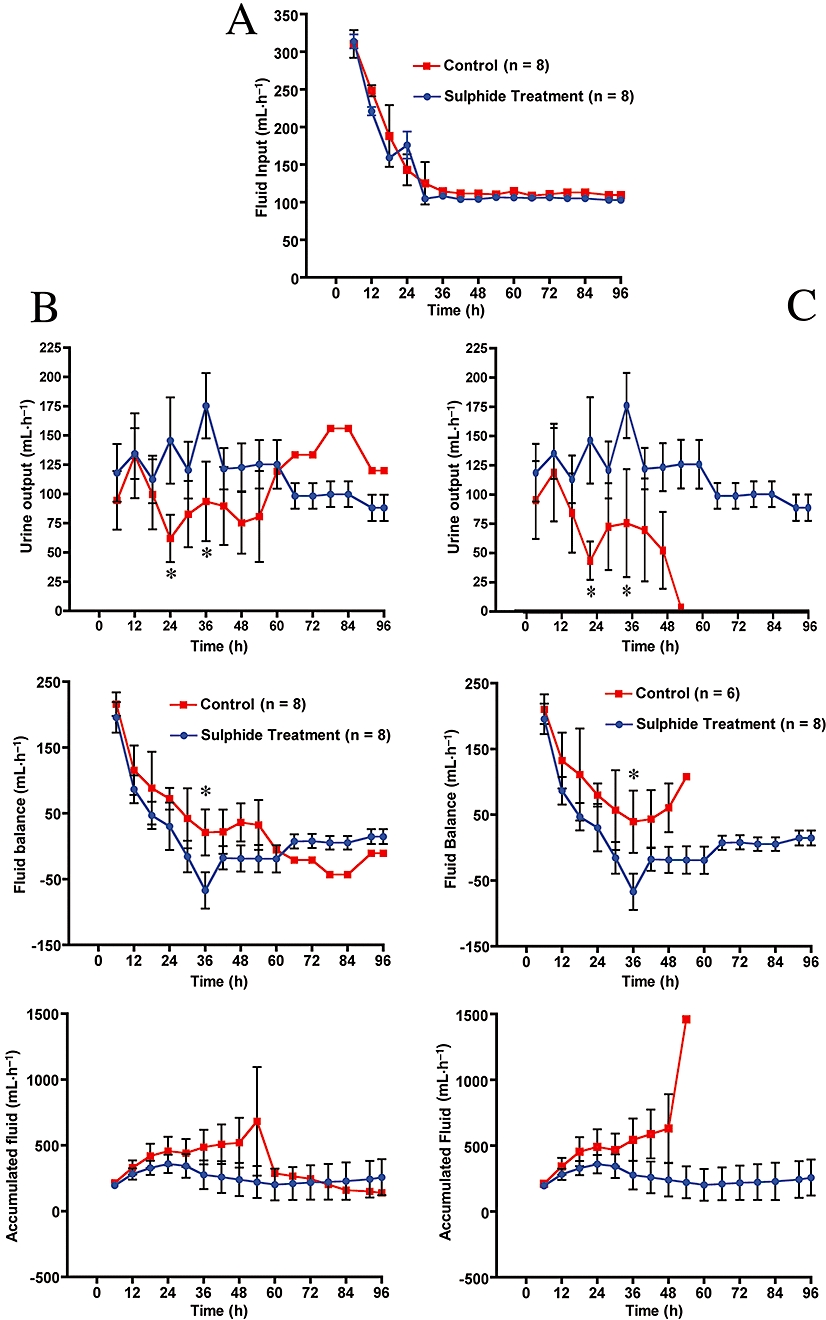
Effect of burn and smoke injury and Na2S treatment on fluid intake, urine output, fluid balance and accumulated fluid. (A) Na2S did not affect fluid input; (B) and (C) Na2S treatment versus surviving and non-surviving animals. n= 2 between T= 72 and 96 h. *P≤ 0.05 versus control.
Protein nitration and oxidation in lung homogenate
In order to determine the mechanism(s) through which Na2S exerted its effects, lung tissue lysates from control and treated animals were analysed. The control group included samples collected from two animals in a parallel 96 h control study that underwent identical experimental conditions. As shown in Figure 6, sulphide treatment significantly reduced iNOS protein expression in the lung homogenates, as compared with vehicle-treated animals (P < 0.05). Moreover, the degree of peroxynitrite formation in the lung tissue (as measured by 3-nitrotyrosine formation) was elevated in the injured control animals.
Figure 6.
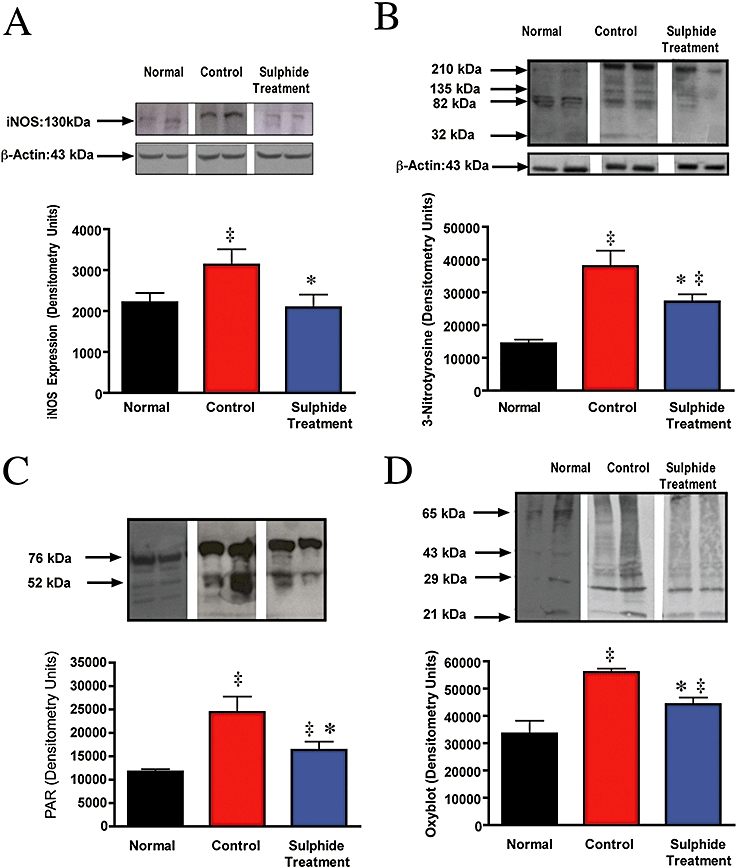
inducible nitric oxide synthase (iNOS), 3-nitrotyrosine, poly (ADP ribose) polymer (PAR) and protein oxidation formation in sheep lung lysate after combined burn and smoke injury and Na2S treatment. Representative Western blots of lung homogenate using (A) rabbit anti-iNOS, (B) mouse anti-3-nitrotyrosine, (C) rabbit anti-PAR antibody and (D) rabbit anti-dinitrophenol (DNP) after protein derivatization with DNP-hydrazone. Histograms represent the integrated band intensities obtained from quantitative results of samples from four separate sheep in each group. Arrows indicate protein molecular weight. n= 4 per group. ‡P≤ 0.05 versus normal; *P≤ 0.05 versus control.
elisa for lung cytochrome c
As shown in Figure 7, mitochondrial cytochrome c levels were higher in the animals subjected to Na2S treatment than in the vehicle-treated animals subjected to burn and smoke inhalation injury (P < 0.01). There were no significant differences in cytochrome c levels in the cytoplasmic fraction in the control group compared with the Na2S group. Treatment with sulphide markedly reduced peroxynitrite formation in the lung tissue (P < 0.05). Protein oxidation (as measured by protein carbonyl formation) was also significantly attenuated in the control animals by sulphide treatment (Figure 6D).
Figure 7.
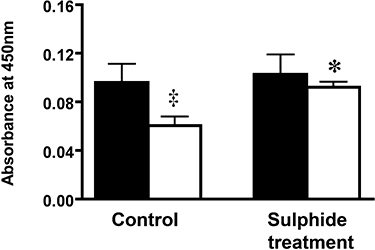
Changes in lung cytochrome c measurement in lung tissue after burn and smoke inhalation injury and Na2S treatment. Solid bars represent cytoplasmic fraction (n= 4), and the open bars represent mitochondrial fraction (n= 4). Lung tissue was homogenized, subjected to centrifugation, and the supernatant containing the cytoplamic fraction was withheld for cytochrome c measurement. The mitochondrial fraction was isolated following the protocol described in the Methods section. This experiment was repeated at least four times, and the data are presented as mean ± SEM. n= 4 per group. ‡P≤ 0.05 versus cytoplasmic cytochrome c; *P≤ 0.05 versus control.
Lung wet-to-dry ratios and lung MPO content
Treatment with Na2S exhibited a trend towards decreased lung MPO content, a marker of neutrophil infiltration (4.0 ± 0.3 for control, 2.8 ± 0.5 for treatment). The difference between groups was not statistically significant (P= 0.059). Lung wet-to-dry weight ratio in the sulphide-treated group (6.7 ± 0.6) was not significantly different from values measured in the control animals (8.9 ± 0.5).
Discussion
We have previously described the beneficial effect of H2S donors by negatively regulating the pro-inflammatory response associated with burn- and smoke-induced acute lung injury in a murine model as evidenced by improvement of survival, reduction and oxidative stress and increases in the production of the anti-inflammatory cytokine IL-10 (Esechie et al., 2008). In this study, we have investigated whether in a clinically relevant large animal model of acute lung injury, the hydrogen sulphide donor affects the measured outcomes associated with this trauma. We hypothesized that administration of intravenous Na2S, a H2S donor compound, after burn and smoke inhalation trauma may attenuate the declining pulmonary function by reducing nitrosative and oxidative stress caused by up-regulation of iNOS. We found that the H2S donor compound significantly reduced animal mortality compared with the vehicle-treated control animals. By 42 h post injury the total number of sheep in the control group was reduced by 50%. Furthermore, H2S treatment was found to not only decrease protein nitration (decreased 3-nitrotyrosine formation) and oxidation (decreased protein oxidation) in lung tissue after injury, but also attenuated iNOS protein expression.
There are several mechanisms that may underlie the improvement in survival seen with H2S donor administration. First, the animals that expired early in the control group had severely depressed oxygenation status, compared with that in the control animals which survived for 96 h. The sulphide-treated animals maintained a better oxygenation status as shown by the change in PaO2/FiO2 ratio (Figure 3). Although the PaO2/FiO2 ratio fell significantly from baseline in both groups, the rate of decline was more pronounced in the vehicle-treated group suggesting a much more immediately compromised gas exchange. There is a strong correlation between mean bronchiolar obstruction and PaO2/FiO2 ratio in sheep (Cox et al., 2003). These authors addressed the idea of gravitational migration of obstructive cast material at 72 h in the absence of therapeutic intervention. They observed that the bronchioles were maximally obstructed at 72 h post injury while terminal bronchioles were obstructed at 48 h post injury. It is well known that the obstructive material consists of sloughed airway epithelia cells, mucous and inflammatory cells. However goblet cells only exist in the cartilaginous bronchi. The mucous from goblet cells was found as far down the airway as the alveoli, supporting the downward movement of airway secretions and debris.
Moderate increases in the inspiratory airway pressure were observed in the injured untreated animals in our study compared with the treated animals (Figure 4). This was paralleled by an increase in pulmonary shunt fraction (Figure 2B). Our data suggest that sulphide treatment not only improves the ventilatory outcomes after injury but also blunts HPV associated with this dual trauma. Work from our laboratory has also reported on a loss of HPV after inhalation injury in relation to the vasorelaxation aspects of NO (Westphal et al., 2006; Westphal et al., 2008).
Several reports describe significant changes in H2S plasma levels in various disease states. Hui et al. (2003) proposed that the endogenous concentrations of H2S and NO are markedly increased during inflammatory states such as sepsis and shock in rats, suggesting that both molecules may work independently and/or modulate each other's activity in pathological conditions. However, patients with coronary heart disease had reduced plasma sulphide levels, from ∼25 to 50 µM (Chang et al., 2008). It should be pointed out, however, that recent studies have questioned the methodology and validity of these previous reports (Whitfield et al., 2008). In our study, plasma H2S concentration was not measured. However, administration of a H2S donor, followed by increased survival and improved pulmonary function suggests a beneficial effect in increasing circulating H2S levels after combined burn and smoke inhalation injury.
The pathogenesis of burn and smoke inhalation injury is exacerbated by both local and systemic inflammation. As demonstrated in this study, H2S treatment significantly reduced iNOS protein expression in the lung after injury. This is in agreement with recent work by Oh et al. (2006) who also found that NF-κB was sequestered to the cytosol after H2S treatment thus decreasing κB-dependent transcription. A reduction in iNOS protein in our study suggests that a similar pattern of NF-κB inhibition may be a potential mechanism in attenuating the negative effects of smoke inhalation with associated thermal injury. A reduction in iNOS was paralleled with a decrease in the peroxynitrite response and protein oxidation that is characteristic of acute lung injury. Although neutrophil accumulation in this study was not significantly attenuated (P= 0.059 vs. the control), there was a tendency for lower MPO activity in the lung after injury.
Interestingly, treatment of the sheep with the H2S donor inhibited cytochrome c translocation from the mitochondria into the cytoplasm. Within the inter-mitochondrial space, cytochrome c is actively involved in ATP synthesis through the oxidative phosphorylation pathway. In the mitochondrion, cytochrome c shuttles electrons from the cytochrome c reductase complex to the cytochrome c oxidase complex. This transports excess electrons along the respiratory pathway and generates ATP for energy-dependent processes. However, in response to oxidative or nitrosative stress, cytochrome c can be released from mitochondria into the cytoplasm as one of the initial steps before cell death (Hong et al., 2004). A recent study (Du et al., 2003) demonstrated that peroxynitrite can exert deleterious effects on the mitochondria resulting in: (i) changes in ultrastructural integrity; (ii) a reduction in the mitochondrial membrane potential; and (iii) increased generation of reactive oxygen species. In this study, we detected an increase in peroxynitrite formation and protein oxidation after injury that paralleled our finding of increased cytochrome c translocation. The reduced mitochondrial release of cytochrome c may also reduce cellular apoptosis. In this context it is noteworthy that H2S formulations have previously been demonstrated to reduce apoptosis in various models of myocardial and hepatic injury (Jha et al., 2008; Sodha et al., 2008). We observed a significant reduction in the generation of nuclear PAR polymer after H2S treatment. As activation of the PAR polymerase (PARP) is downstream from oxidative/nitrosative stress-induced DNA damage (Virag and Szabo, 2002; Szabo et al., 2007), the current results showing decreased nitrosative stress are consistent with the reduced PARP activation. Decreased nitrosative stress and diminished PARP activation appears to be a general feature of H2S therapy in diseases associated with oxidative and nitrosative stress, as similar reductions in PAR and nitrotyrosine staining were also previously noted in a porcine model of myocardial ischaemia/reperfusion injury (Sodha et al., 2008). Some of the current findings, however, are in contrast to a recent report (Baskar et al., 2007) that suggests that H2S might act through cytochrome c and Bax protein to promote DNA damage. It is important to note that the effects of H2S are dose-dependent, and at lower concentrations H2S exerts physiological, cytoprotective effects, while at high local concentrations can become pro-oxidant and cytotoxic (Szabo, 2007). It is also important to note that Baskar et al. (2007) conducted their studies in an in vitro setting. Indeed, we have observed in unpublished preliminary studies that sulphide therapy, at doses 10 times higher than the one used in the current study, no longer elicits beneficial effects. The U-shaped dose–response curve for the cytoprotective effect for H2S was also noted by Lefer's group in a murine myocardial infarction study (Elrod et al., 2007).
Several studies examining the effect of smoke alone and burn alone injuries on mitochondrial integrity have consistently reported a lower cytoplasmic cytochrome c level compared with cytochrome c mitochondrial levels in an unstimulated state (Ramage et al., 2006; Zang et al., 2007). These studies, moreover, have shown that after injury with either smoke exposure (Ramage et al., 2006) or thermal injury (Zang et al., 2007), there is an immediate and significant increase in cytoplasmic cytochrome c protein levels within the first 4 h. The latter study (Zang et al., 2007) demonstrated that cytoplasmic cytochrome c levels decrease to baseline levels at 72 h post burn injury. Our data are limited in that they we did not measure cytochrome c in un-injured normal sheep, and we can therefore not determine how combined burn and smoke inhalation injury affect cytochrome c translocation and if the observed changes were restored to normal levels after combined injury and treatment.
In conclusion, we have demonstrated that, after burn- and smoke-induced acute lung injury and acute respiratory distress syndrome, treatment with Na2S (a H2S donor) attenuated iNOS expression, peroxynitrite formation (as measured by 3-nitrotyrosine), protein oxidation (as measured by protein carbonyl formation) and PARP-1 activity in vivo. Due to logistical limitations typical to large animal studies, we did not include additional groups (e.g. with NOS inhibitors; peroxynitrite decomposition catalysts) where the individual contribution of each of these pathways could have been tested. Thus, the direct causal role of these pathways was not directly demonstrated in the current experiments. Nevertheless, based on studies previously completed in the current experimental model, which demonstrate that antioxidant therapy (Bone et al., 2002; Hamahata et al., 2008), NO synthase inhibition (Enkhbaatar et al., 2006; Westphal et al., 2008) and PARP inhibition (Shimoda et al., 2003) are beneficial, we speculate that actions of H2S on some or all of these pathways may have contributed to the beneficial effects of H2S observed in the current model, culminating in an improvement in survival. Nevertheless, we cannot exclude the role of potential additional mechanisms, including indirect effects. For instance, improvements in pulmonary gas exchange, microvascular vasodilatory effects of H2S or local metabolic modulatory effects of H2S may have also contributed to the protection observed. The current results and the results of several other studies (Blackstone and Roth, 2007; Esechie et al., 2008; Jha et al., 2008; Morrison et al., 2008; Simon et al., 2008; Sodha et al., 2008) support the view that H2S therapy may be beneficial in various conditions of critical illness.
Acknowledgments
This work was supported by National Institute for General Medical Sciences Grants GM66312, Grants 8954, 8450, and 8460 from the Shriners of North America and Ikaria Inc. (Seattle, WA, USA).
Glossary
Abbreviations:
- AEBSF
4-(2-aminoethyl) benzenesulphonyl fluoride hydrochloride
- CI
cardiac index
- CVP
central venous pressure
- DNP
dinitrophenol
- DTT
dithiothreitol
- elisa
enzyme-linked immunosorbent assay
- FiO2
fraction of inspired oxygen
- Hb
haemoglobin
- Hct
haematocrit
- HPV
hypoxic pulmonary vasoconstriction
- HTAB
hexa-decyl-trimethyl-ammonium bromide
- iNOS
inducible NO synthase
- LAP
left atrial pressure
- LVSWI
left venticular stroke work index
- MAP
mean arterial pressure
- MPAP
mean pulmonary arterial pressure
- MPO
myeloperoxidase
- NF-κB
nuclear factor-kappa B
- ODN
O-dianisidine hydrochloride
- PaO2
arterial partial pressure of oxygen
- PaO2/FiO2
arterial partial pressure of oxygen/fraction of inspired oxygen
- PAR
poly (ADP-ribose) polymer
- PARP
poly (ADP-ribose) polymerase
- PMSF
phenylmethylsulphonyl fluoride
- PVDF
polyvinylidene fluoride
- PVRI
pulmonary vascular resistance index
- VA/Q
alveolar ventilation/blood flow
Conflict of interest
CS is Chief Scientific Officer of Ikaria Inc. (Seattle, WA), a for-profit organization involved in the development of H2S-based therapies.
References
- Abraham E, Matthay MA, Dinarello CA, Vincent JL, Cohen J, Opal SM, et al. Consensus conference definitions for sepsis, septic shock, acute lung injury, and acute respiratory distress syndrome: time for a reevaluation. Crit Care Med. 2000;28:232–235. doi: 10.1097/00003246-200001000-00039. [DOI] [PubMed] [Google Scholar]
- Ahn SY, Sugi K, Talke P, Theissen JL, Linares HA, Traber LD, et al. Effects of allopurinol on smoke inhalation in the ovine model. J Appl Physiol. 1990;68:228–234. doi: 10.1152/jappl.1990.68.1.228. [DOI] [PubMed] [Google Scholar]
- Andruski B, McCafferty DM, Ignacy T, Millen B, McDougall JJ. Leukocyte trafficking and pain behavioral responses to a hydrogen sulfide donor in acute monoarthritis. Am J Physiol Regul Integr Comp Physiol. 2008;295:R814–R820. doi: 10.1152/ajpregu.90524.2008. [DOI] [PubMed] [Google Scholar]
- Baskar R, Li L, Moore PK. Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. Faseb J. 2007;21:247–255. doi: 10.1096/fj.06-6255com. [DOI] [PubMed] [Google Scholar]
- Blackstone E, Roth MB. Suspended animation-like state protects mice from lethal hypoxia. Shock. 2007;27:370–372. doi: 10.1097/SHK.0b013e31802e27a0. [DOI] [PubMed] [Google Scholar]
- Bone HG, Sakurai H, Schenarts PJ, Traber LD, Traber DL. Effects of manganese superoxide dismutase, when given after inhalation injury has been established. Crit Care Med. 2002;30:856–860. doi: 10.1097/00003246-200204000-00024. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brown SD. ARDS: history, definitions, and physiology. Respir Care Clin N Am. 1998;4:567–582. [PubMed] [Google Scholar]
- Chang L, Geng B, Yu F, Zhao J, Jiang H, Du J, et al. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids. 2008;34:573–585. doi: 10.1007/s00726-007-0011-8. [DOI] [PubMed] [Google Scholar]
- Cox B, Emili A. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc. 2008;1:1872–1878. doi: 10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- Cox RA, Burke AS, Soejima K, Murakami K, Katahira J, Traber LD, et al. Airway obstruction in sheep with burn and smoke inhalation injuries. Am J Respir Cell Mol Biol. 2003;29:295–302. doi: 10.1165/rcmb.4860. [DOI] [PubMed] [Google Scholar]
- Cox RA, Burke AS, Jacob S, Oliveras G, Murakami K, Shimoda K, et al. Activated nuclear factor Kappa B and airway inflammation after smoke inhalation and burn injury in sheep. J Burn Care Res. 2009;30:489–498. doi: 10.1097/BCR.0b013e3181a28e13. [DOI] [PubMed] [Google Scholar]
- Demling R, Lalonde C, Picard L, Blanchard J. Changes in lung and systemic oxidant and antioxidant activity after smoke inhalation. Shock. 1994;1:101–107. doi: 10.1097/00024382-199402000-00004. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Efimova O, Volokhov AB, Iliaifar S, Hales CA. Ligation of the bronchial artery in sheep attenuates early pulmonary changes following exposure to smoke. J Appl Physiol. 2000;88:888–893. doi: 10.1152/jappl.2000.88.3.888. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Kiss L, Olah G, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkhbaatar P, Murakami K, Traber LD, Cox R, Parkinson JF, Westphal M, et al. The inhibition of inducible nitric oxide synthase in ovine sepsis model. Shock. 2006;25:522–527. doi: 10.1097/01.shk.0000209525.50990.28. [DOI] [PubMed] [Google Scholar]
- Enkhbaatar P, Kiss L, Olah G, Cox RA, Nakano Y, Hamahata A, et al. Combined anticoagulants ameliorate acute lung injury in sheep after burn and smoke inhalation. Clin Sci (Lond) 2008;114:321–329. doi: 10.1042/CS20070254. [DOI] [PubMed] [Google Scholar]
- Esechie A, Kiss L, Olah G, Horváth EM, Hawkins H, Szabo C, et al. Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation. Clin Sci (Lond) 2008;115:91–97. doi: 10.1042/CS20080021. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Kim DK, Chin MR, Hales CA, Bonventre JV. Increased group IV cytosolic phospholipase A2 activity in lungs of sheep after smoke inhalation injury. Am J Physiol. 1999;277(3):L533–L542. doi: 10.1152/ajplung.1999.277.3.L533. Pt 1. [DOI] [PubMed] [Google Scholar]
- Hales CA, Elsasser TH, Ocampo P, Efimova O. TNF-alpha in smoke inhalation lung injury. J Appl Physiol. 1997;82:1433–1437. doi: 10.1152/jappl.1997.82.5.1433. [DOI] [PubMed] [Google Scholar]
- Hamahata A, Enkhbaatar P, Kraft ER, Lange M, Leonard SW, Traber MG, et al. Gamma-Tocopherol nebulization by a lipid aerosolization device improves pulmonary function in sheep with burn and smoke inhalation injury. Free Radic Biol Med. 2008;45:425–433. doi: 10.1016/j.freeradbiomed.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Hui Y, Du J, Tang C, Bin G, Jiang H. Changes in arterial hydrogen sulfide (H(2)S) content during septic shock and endotoxin shock in rats. J Infect. 2003;47:155–160. doi: 10.1016/s0163-4453(03)00043-4. [DOI] [PubMed] [Google Scholar]
- Insko MA, Deckwerth TL, Hill P, Toombs CF, Szabó C. Detection of exhaled hydrogen sulfide gas in rats exposed to intravenous sodium sulfide. Br J Pharmacol. 2009;157:944–951. doi: 10.1111/j.1476-5381.2009.00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen D, Ytrehus K, Baxter GF. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury – evidence for a role of K ATP channels. Basic Res Cardiol. 2006;101:53–60. doi: 10.1007/s00395-005-0569-9. [DOI] [PubMed] [Google Scholar]
- Kamoun P. Endogenous production of hydrogen sulfide in mammals. Amino Acids. 2004;26:243–254. doi: 10.1007/s00726-004-0072-x. [DOI] [PubMed] [Google Scholar]
- Kimura R, Traber L, Herndon D, Niehaus G, Flynn J, Traber DL. Ibuprofen reduces the lung lymph flow changes associated with inhalation injury. Circ Shock. 1998;24:183–191. [PubMed] [Google Scholar]
- Kiss L, Deitch EA, Szabo C. Hydrogen sulfide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sci. 2008;83:589–594. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenitzer JR, Isbell TS, Patel HD, Benavides GA, Dickinson DA, Patel RP, et al. Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. Am J Physiol Heart Circ Physiol. 2007;292:H1953–H1960. doi: 10.1152/ajpheart.01193.2006. [DOI] [PubMed] [Google Scholar]
- Lalonde C, Demling R, Brain J, Blanchard J. Smoke inhalation injury in sheep is caused by the particle phase, not the gas phase. J Appl Physiol. 1994;77:15–22. doi: 10.1152/jappl.1994.77.1.15. [DOI] [PubMed] [Google Scholar]
- Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008;29:84–90. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Lowicka E, Beltowski J. Hydrogen sulfide (H2S) – the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- Mineta H, Miura K, Ogino T, Takebayashi S, Misawa K, Ueda Y. Vascular endothelial growth factor (VEGF) expression correlates with p53 and ki-67 expressions in tongue squamous cell carcinoma. Anticancer Res. 2002;22:1039–1044. [PubMed] [Google Scholar]
- Morrison ML, Blackwood JE, Lockett SL, Iwata A, Winn RK, Roth MB. Surviving blood loss using hydrogen sulfide. J Trauma. 2008;65:183–188. doi: 10.1097/TA.0b013e3181507579. [DOI] [PubMed] [Google Scholar]
- Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, et al. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med. 2006;41:106–119. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Pearce ML, Yamashita J, Beazell J. Measurement of pulmonary edema. Circ Res. 1965;16:482–488. doi: 10.1161/01.res.16.5.482. [DOI] [PubMed] [Google Scholar]
- Ramage L, Jones AC, Whelan CJ. Induction of apoptosis with tobacco smoke and related products in A549 lung epithelial cells in vitro. J Inflamm (Lond) 2006;3:3. doi: 10.1186/1476-9255-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel T, Fraser JF, Dunster K, Fitzgibbon J, Schibler A. Effect of smoke inhalation on viscoelastic properties and ventilation distribution in sheep. J Appl Physiol. 2006;101:763–770. doi: 10.1152/japplphysiol.01635.2005. [DOI] [PubMed] [Google Scholar]
- Shimoda K, Murakami K, Enkhbaatar P, Traber LD, Cox RA, Hawkins HK, et al. Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am J Physiol Lung Cell Mol Physiol. 2003;285:L240–L249. doi: 10.1152/ajplung.00319.2002. [DOI] [PubMed] [Google Scholar]
- Simon F, Giudici R, Duy CN, Schelzig H, Oter S, Gröger M, et al. Hemodynamic and metabolic effects of hydrogen sulfide during porcine ischemia/reperfusion injury. Shock. 2008;30:359–364. doi: 10.1097/SHK.0b013e3181674185. [DOI] [PubMed] [Google Scholar]
- Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E, et al. Antiapoptotic and inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009;31:267–274. doi: 10.1097/SHK.0b013e318180ff89. [DOI] [PubMed] [Google Scholar]
- Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, et al. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur J Cardiothorac Surg. 2008;33:906–913. doi: 10.1016/j.ejcts.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Tang XQ, Yang CT, Chen J, Yin WL, Tian SW, Hu B, et al. Effect of hydrogen sulphide on beta-amyloid-induced damage in PC12 cells. Clin Exp Pharmacol Physiol. 2008;35:180–186. doi: 10.1111/j.1440-1681.2007.04799.x. [DOI] [PubMed] [Google Scholar]
- Traber DL, Herndon DN, Stein MD, Traber LD, Flynn JT, Niehaus GD. The pulmonary lesion of smoke inhalation in an ovine model. Circ Shock. 1986;18:311–323. [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- Wang D, Zwischenberger JB, Savage C, Miller L, Deyo DJ, Alpard S, et al. High-frequency percussive ventilation with systemic heparin improves short-term survival in a LD100 sheep model of acute respiratory distress syndrome. J Burn Care Res. 2006;27:463–471. doi: 10.1097/01.BCR.0000226003.18885.E8. [DOI] [PubMed] [Google Scholar]
- Webb GD, Lim LH, Oh VM, Yeo SB, Cheong YP, Ali MY, et al. Contractile and vasorelaxant effects of hydrogen sulfide and its biosynthesis in the human internal mammary artery. J Pharmacol Exp Ther. 2008;324:876–882. doi: 10.1124/jpet.107.133538. [DOI] [PubMed] [Google Scholar]
- Westphal M, Cox RA, Traber LD, Morita N, Enkhbaatar P, Schmalstieg FC, et al. Combined burn and smoke inhalation injury impairs ovine hypoxic pulmonary vasoconstriction. Crit Care Med. 2006;34:1428–1436. doi: 10.1097/01.CCM.0000215828.00289.B9. [DOI] [PubMed] [Google Scholar]
- Westphal M, Enkhbaatar P, Schmalstieg FC, Kulp GA, Traber LD, Morita N, et al. Neuronal nitric oxide synthase inhibition attenuates cardiopulmonary dysfunctions after combined burn and smoke inhalation injury in sheep. Crit Care Med. 2008;36:1196–1204. doi: 10.1097/CCM.0b013e31816a1a0c. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Zang Q, Maass DL, White J, Horton JW. Cardiac mitochondrial damage and loss of ROS defense after burn injury: the beneficial effects of antioxidant therapy. J Appl Physiol. 2007;102:103–112. doi: 10.1152/japplphysiol.00359.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]