

Abstract
Background and purpose:
Hyperlipidaemia interferes with cardioprotective mechanisms, but the cause of this phenomenon is largely unknown, although hyperlipidaemia impairs the cardioprotective NO–cGMP system. However, it is not known if natriuretic peptide–cGMP–protein kinase G (PKG) signalling is affected by hyperlipidaemia. Therefore, we investigated the cardioprotective efficacy of cGMP-elevating agents in hearts from normal and hyperlipidaemic rats.
Experimental approach:
Male Wistar rats were rendered hyperlipidaemic by feeding with 2% cholesterol-enriched chow for 12 weeks. Hearts isolated from normal and hyperlipidaemic rats were perfused (Langendorff mode) and subjected to 30 min occlusion of the left main coronary artery, followed by 120 min reperfusion. 8-Br-cGMP (CG, 10 nM), B-type natriuretic peptide-32 (BNP, 10 nM), S-nitroso-N-acetyl-penicillamine (SNAP, 1 µM) were perfused from 10 min prior to coronary occlusion until the 15th min of reperfusion. Infarct size (% of ischaemic risk zone) was determined by triphenyltetrazolium staining.
Key results:
Treatment with CG, SNAP or BNP decreased infarct size significantly in normal hearts from its control value of 41.6 ± 2.9% to 15.5 ± 2.4%, 23.3 ± 3.0% and 25.3 ± 4.6%, respectively (P < 0.05). Protection by BNP was abolished by co-perfusion of PKG inhibitors KT5823 (600 nM) or Rp-8pCPT-PET-cGMPs (1 µM), confirming its PKG dependence. In hearts from hyperlipidaemic rats, CG, SNAP or BNP failed to decrease infarct size. Hyperlipidaemia did not alter basal myocardial PKG content, but decreased its activity as assessed by phosphorylation of cardiac troponin I.
Conclusions and implications:
This is the first demonstration that defects in the cardioprotective cGMP–PKG system could be a critical biochemical anomaly in hyperlipidaemia.
Keywords: protein kinase G, BNP, cGMP, hyperlipidaemia, ischaemia, troponin I, rat
Introduction
Guanosine-3′,5′-cyclic monophosphate (cGMP) is synthesized by soluble (sGC) and particulate guanylyl cyclases. Particulate guanylyl cyclases are known to be natriuretic peptide receptors (NPRs). In cardiac myocytes, the major activator of the sGC is nitric oxide (NO), whereas the activity of NPRs is regulated by the natriuretic peptides, atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP) and C-type natriuretic peptide (CNP). Elevation of intracellular cGMP concentration results in the activation of several distal cGMP-dependent mechanisms, including the activation of cGMP-dependent protein kinase (PKG). PKG is a serine–threonine phosphokinase that can phosphorylate several key proteins involved in the regulation of heart function, such as phospholamban or troponin I (Raeymaekers et al., 1988; Mery et al., 1993; Yuasa et al., 1999; Munzel et al., 2003). Elevation of intracellular cGMP concentration maintains not only physiological functions of the heart, but numerous studies indicate that cGMP plays a key role in cytoprotection against ischaemia/reperfusion injury (see Burley et al., 2007). For example, increased intracellular cGMP following exogenous NO induced cardioprotection and reduced infarct size in rats (Qin et al., 2004; Xu et al., 2004; Cuong et al., 2006). We reported previously that BNP was protective against ischaemia/reperfusion injury in rat heart, resulting in decreased infarct size which correlated with elevations in tissue cGMP concentration (D'Souza et al., 2003). In addition, we demonstrated (Burley and Baxter, 2007) demonstrated that BNP limits infarct size when given specifically at reperfusion. Although cardioprotection associated with cGMP elevation is not definitively confirmed to be the result of PKG activation, there is limited evidence suggesting that PKG inhibitors can prevent the protective effects of interventions that increase cGMP concentration (see Burley et al., 2007).
The myocardial NO–cGMP pathway seems to be impaired in hyperlipidaemia. We have previously shown a reduced NO production in hearts of hyperlipidaemic rats (Giricz et al., 2003; Onody et al., 2003). Schwemmer et al. (2000) found decreased cGMP levels in hearts of hypercholesterolaemic guinea pigs. In a recent study, Penumathsa et al. (2007) reported that in hypercholesterolaemic rats, phosphorylation of endothelial NO synthase and protein kinase B was decreased compared to controls, which may result in a decreased NO production, increased apoptosis and the loss of cardioprotective interventions. However, the exact mechanisms of NO–cGMP pathway disruption in hyperlipidaemia are still unknown. We have previously shown that animals rendered hyperlipidaemic by cholesterol feeding exhibit increased superoxide production compared to control animals (Onody et al., 2003), which is known to decrease the bioavailability of NO (see Ferdinandy and Schulz, 2001; 2003;). It has also been demonstrated that cardioprotective mechanisms such as pre- and post-conditioning in which the NO–cGMP pathway plays an important role are lost in hyperlipidaemia (Szilvassy et al., 1994; Ferdinandy and Schulz, 2003; Giricz et al., 2006; Iliodromitis et al., 2006). The underlying mechanisms by which hyperlipidaemia prevents the protective effect of pre- and post-conditioning are not known. The impairment might occur through down-regulation of NO synthesis, disruption of sGC activity or decreased PKG expression/activation (Ueda et al., 1999; Tang et al., 2005). Despite the well-recognized impairment of the NO–cGMP pathway in hyperlipidaemia, it is not known if the alternative NPR–cGMP system, activated by natriuretic peptides, remains effective.
Therefore, in the present study, we have investigated the effects of hyperlipidaemia on cardioprotective response to exogenous NO and BNP, and whether disruption of the NO–cGMP–PKG pathway plays a role in the loss of cardioprotection against ischaemia/reperfusion injury in hyperlipidaemia, by using different cGMP-elevating agents such as the NO-donor S-nitroso-N-acetyl-penicillamine (SNAP), the stable cGMP analogue 8-Br-cGMP and BNP.
Methods
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996), and were approved by the local animal ethics committee of the University of Szeged.
Induction of hyperlipidaemia
Eighteen-week-old male Wistar rats were fed control or 2% cholesterol-enriched diet for 12 weeks. The body weights of the animals at the time of experimentation after the diet period were 420–500 g, with no significant difference between control and cholesterol-fed groups. Wistar rats were chosen for the study because they show moderate increase in serum cholesterol level, after a high-cholesterol diet, without developing substantial atherosclerosis (Giricz et al., 2006). The 12 week cholesterol-enriched diet increased serum cholesterol from 1.41 ± 0.05 to 1.87 ± 0.1 mM (n= 8 in each group, P < 0.05), and serum triglyceride from 0.47 ± 0.07 to 1.23 ± 0.18 (n= 8–12, P < 0.05) mM, similar to our previous studies (Puskas et al., 2004; Giricz et al., 2006).
Heart perfusion
At the end of the diet period, the rats were anaesthetized with diethyl ether, and heparinized (500 IU·kg−1 i.p.). The hearts were then isolated and perfused in Langendorff mode at constant pressure (80 cm H2O) with Krebs–Henseleit (KH) buffer of the following composition (mM): NaCl, 118.4; KCl, 4.1; CaCl2, 2.5; NaHCO3, 25; KH2PO4, 1.17; MgCl2, 1.46; and glucose, 11.1, then gassed with 95% O2 and 5% CO2, pH 7.4 at 37°C. A fluid-filled latex balloon connected to a pressure transducer was inserted into the left ventricle via the left atrium, and inflated to give a baseline end diastolic pressure of 5–10 mm Hg. Coronary flow rate was determined by timed collection of the coronary effluent. After 15 min stabilization, the hearts were subjected to 30 min regional ischaemia, induced by occlusion of the left main coronary artery using a silk suture positioned around the vessel close to its origin. The suture was then released to permit reperfusion for 120 min (see Figure 1).
Figure 1.
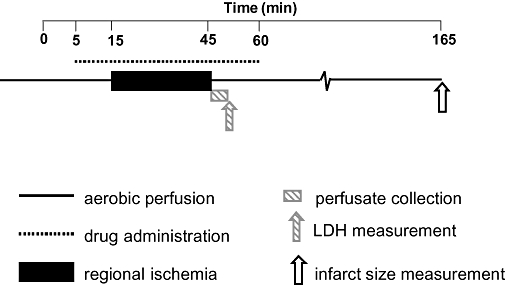
Heart perfusion protocol. Hearts of control or hyperlipidaemic animals were perfused in Langendorff mode for an initial 5 min with Krebs–Henseleit buffer. Between the 5th and 60th min of perfusion, 10 nM 8-Br-cGMP, 1 mM S-nitroso-N-acetyl-penicillamine or 10 nM B-type natriuretic peptide (BNP) was added to the perfusing solution. Additionally, a group of hearts from control chow-fed rats received 10 nM BNP and 600 nM KT, 10 nM BNP and 1 µM PET, 600 nM KT or 1 µM PET, at similar times. From the 15th to the 45th min of the perfusion, no-flow ischaemia was induced by occlusion of the left coronary artery. In the first 5 min of the reperfusion, coronary effluent was collected for lactate dehydrogenase measurement. After 120 min reperfusion, the hearts were stained for infarct size measurement.
Treatment protocols
The experimental protocol is illustrated in Figure 1. Control hearts from normal and hyperlipidaemic animals underwent ischaemia–reperfusion, but received no treatment. To compare the cardioprotective effects of cGMP-elevating agents, hearts from normal and hyperlipidaemic animals were randomized to perfusion with either the cGMP analogue 8-Br-cGMP (10 nM), the NO donor SNAP (1 mM) or recombinant rat B-type natriuretic peptide-32 (BNP-32; 10 nM). Agents were added to the perfusion buffer from 10 min prior to occlusion of the left coronary artery until 15 min of reperfusion. To examine the dependency of BNP-induced cardioprotection on PKG activation, additional hearts from normal rats received treatment with or without BNP-32 (10 nM) in combination with the PKG inhibitors KT-5823 (600 nM; Tocris, Bristol, UK) or Rp-8-pCPT-PET-cGMPS (1 µM; BioLog, Bremen, Germany). Perfusion with KT or PET only was performed in separate experiments together with a new batch of corresponding control hearts. As the infarct size of the new control hearts was different from the previous control batch, we normalized the infarct size data of the KT and PET groups to the previous set of data.
A group of hearts from normal or hyperlipidaemic animals received no ischaemia or pharmacological treatment, and ventricles were freeze-clamped after 60 min of aerobic perfusion in liquid nitrogen for Western blot studies.
Determination of lactate dehydrogenase (LDH) release and infarct size
LDH release was measured in coronary perfusate collected for 5 min at the onset of reperfusion using an LDH-P kit (Diagnosticum, Budapest, Hungary). LDH release was expressed as mU·min−1·(g wet weight)−1 of the ischaemic risk zone.
After 120 min reperfusion, the left coronary artery was re-occluded, and 2 mL of 0.1% Evans blue dissolved in KH buffer was slowly injected for 1 min into the aorta to stain the non-ischaemic myocardium. The Evans blue-stained hearts were frozen (−20°C), cut into approximately 3 mm thick slices, incubated in 1% 2,3,5-triphenyl-tetrazolium chloride (TTC) dissolved in Krebs buffer for 10 min, then in 3% formaldehyde for 15 min. Slices were then scanned between glass plates. TTC-stained viable, unstained infarcted and blue non-ischaemic areas of images were quantified by planimetry (ImageJ, National Institutes of Health, Bethesda, MA, USA). Infarct size was expressed as the percentage of infarcted tissue within the ischaemic risk zone (infarct size as % risk zone).
Myocardial expression of PKG
To characterize changes in expression and activation of PKG as a possible consequence of hyperlipidaemia, we measured the basal expression and the conformational distribution of PKG in ventricular myocardium, and the phosphorylation of a PKG substrate, the cardiac protein, troponin I. Protein levels were determined in ventricular tissue of isolated hearts after 60 min of aerobic perfusion. Samples were homogenized by ultrasonic homogenizer. Protein concentrations were measured by the bicinchoninic acid assay with BSA as a standard. Samples were boiled in Laemmli buffer at 95°C for 5 min. These were then analysed by SDS–electrophoresis, and blotted onto nitrocellulose membranes. Loading of equal amount of protein was checked by Ponceau staining of the membranes. The membranes were then blocked overnight at 4°C in Tris-buffered saline containing 0.1% Tween-20 and 5% skimmed milk powder. For determination of the ratio of dimeric and monomeric forms of PKG, samples were prepared without boiling with the addition of 2-mercaptoethanol. The membranes were incubated either with anti-PKG-Iα/β (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho-troponin I (Cardiac, Ser23/24) antibody or anti-troponin I (both from Cell Signaling Technology, Danvers, MA, USA) antibody and HRP-conjugated rabbit anti-mouse or goat anti-rabbit secondary antibody (DakoCytomation, Glostrup, Denmark). The membranes were then developed with an enhanced chemiluminescence kit (ECL Plus, Amersham Biosciences, Uppsala, Sweden), exposed to X-ray film and scanned. Band density was calculated by integrating the area (in pixels × intensity, expressed in arbitrary units). For troponin I, data were represented as a ratio of the band densities of the phosphorylated/non-phosphorylated proteins.
Statistical analysis
Results are expressed as mean ± SEM, and analysed by SigmaStat software (Jandel Scientific, Costa Madre, CA, USA). Differences between mean values were analysed by one-way analysis of variance followed by Newman–Keuls post hoc or by unpaired Student's t-test, and were considered statistically significant when P < 0.05.
Materials
Unless otherwise stated, all chemicals were purchased from Sigma (St Louis, MO, USA). Nomenclature of enzymes and receptors follows (Alexander et al., 2008).
Results
Technical exclusions
A total number of 90 animals were included in the final analysis of the infarct study. Hearts with coronary flow rate more than 30% higher than the mean coronary flow rate of the corresponding group at the 10th min of stabilization period (four hearts), or with coronary flow rate reduction by less than 30% during ischaemia (six hearts) were excluded from data analysis.
Infarct size and LDH release in isolated hearts
The major anatomical determinant of infarct size in the rat heart, which lacks any substantial pre-formed collateral circulation, is the size of the ischaemic risk zone. Ischaemic risk zones were between 40 and 50% in all groups (data not shown). Infarct size, normalized to risk zone, is shown in Figure 2. Following 30 min occlusion of the left coronary artery and 120 min reperfusion, TTC-determined infarct size was not significantly different in normal and hyperlipidaemic hearts (P= 0.143). In normal hearts, significant infarct size limitation was observed with 8-Br-cGMP, SNAP or BNP-32 (Figure 2A). The protective effect of BNP-32 effect was prevented by co-treatment with the PKG inhibitors KT or PET (Figure 2C), while KT and PET alone did not affect infarct size. In contrast, in hearts from hyperlipidaemic rats, there was no statistically significant effect of 8-Br-cGMP, SNAP or BNP on infarct size (Figure 2E).
Figure 2.
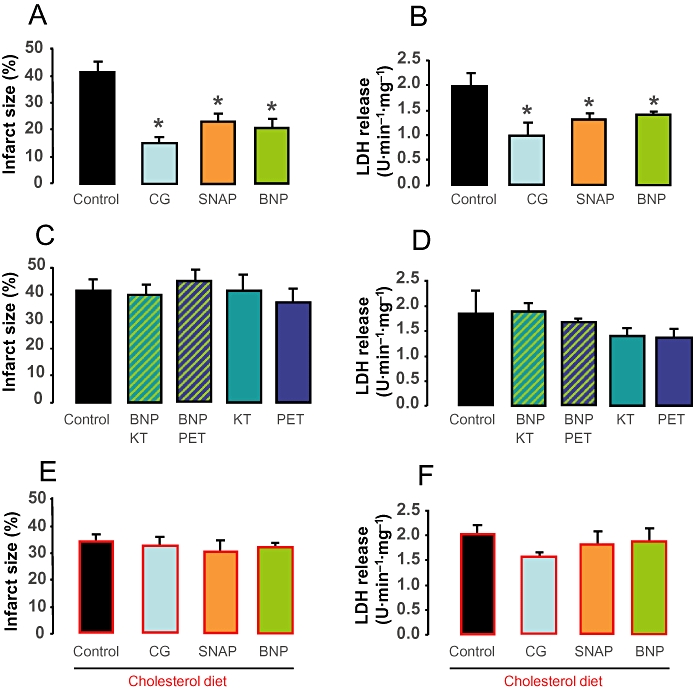
In A, C, E, the infarct size (expressed as % of risk zone) and in B, D, F, the corresponding values of lactate dehydrogenase (LDH) in coronary perfusate are shown. In hearts from control animals, treatment with 10 nM 8-Br-cGMP, 1 mM S-nitroso-N-acetyl-penicillamine (SNAP) or 10 nM B-type natriuretic peptide (BNP) significantly decreased infarct size (A) and LDH release (B; n= 5–9, *P < 0.05 vs. control). In the presence of 600 nM KT or 1 µM PET, the cardioprotective effect of BNP was abolished as assessed by infarct size (C) or LDH release (D). In hearts from cholesterol-fed hyperlipidaemic animals, neither 10 nM 8-Br-cGMP, 1 mM SNAP nor 10 nM BNP decreased infarct size (E) or LDH release (F) significantly.
LDH release into the coronary perfusate during early reperfusion was used as an additional measure of irreversible ischaemia/reperfusion injury. During the first 5 min of reperfusion, a marked LDH release from normal and hyperlipidaemic hearts was observed, indicative of wash-out of the enzyme from irreversibly injured cells. 8-Br-cGMP, SNAP or BNP-32 significantly attenuated LDH release in hearts of normal animals (Figure 2B), and co-administration with BNP-32 of KT or PET abolished the effect of BNP on LDH release (Figure 2D). In contrast to their protective effects in normal hearts, 8-Br-cGMP, SNAP or BNP-32 failed to attenuate LDH release in hyperlipidaemic hearts (Figure 2F). Thus, the pattern of LDH release in early reperfusion mirrors the infarct size data.
Coronary flow and heart rate
Basal coronary flow rate was similar in normal and hyperlipidaemic hearts (Table 1). Treatment with SNAP, but not 8Br-cGMP or BNP, caused a significant increase in coronary flow rate in both normal and hyperlipidaemic hearts, indicative of a vasodilator response. During occlusion of the left coronary artery, coronary flow rate decreased to a similar extent (35–50% reduction compared to pre-ischaemic value) in all groups. Spontaneous heart rate (Table 2) was similar in normal and hyperlipidaemic hearts before coronary occlusion and at the end of ischaemia. None of the drug treatments showed any effect on heart rate.
Table 1.
Coronary flow of hearts from normal chow-fed animals (A) and cholesterol-fed animals (B)
| A.time (min) |
Coronary Flow (mL × min−1 × gwet weight) |
|||||
|---|---|---|---|---|---|---|
| Control(n = 8) | CG(n = 8) | BNP(n = 8) | SNAP(n = 8) | BNP + KT(n = 8) | BNP + PET(n = 6) | |
| 5 | 9.76 ± 0.81 | 11.47 ± 1.16 | 9.95 ± 1.28 | 9.48 ± 0.46 | 8.44 ± 0.72 | 8.7 ± 1.13 |
| 15 | 9.36 ± 0.81 | 10.3 ± 1.14 | 9.32 ± 1.06 | 12.06 ± 0.44# | 9.79 ± 0.67 | 7.36 ± 0.54 |
| 45 | 6.31 ± 0.45* | 5.87 ± 0.72* | 4.91 ± 0.29* | 5.81 ± 0.21* | 5.4 ± 0.4* | 4.53 ± 0.18* |
| 60 | 10.64 ± 0.94 | 10.13 ± 0.8 | 10.58 ± 0.99 | 10.87 ± 0.52 | 9.5 ± 0.63 | 8.21 ± 0.33 |
| 165 | 8.58 ± 1.04 | 7.93 ± 0.85 | 6.04 ± 0.77 | 6.72 ± 0.5 | 6.04 ± 0.46 | 6.27 ± 0.45 |
| B.time (min) |
Coronary Flow (mL × min−1 × gwet weight) |
|||||
| Chol(n = 8) | Chol + CG(n = 8) | Chol + BNP(n = 8) | Chol + SNAP(n = 8) | |||
| 5 | 8.42 ± 0.83 | 7.49 ± 0.55 | 8.42 ± 0.65 | 9.55 ± 0.69 | ||
| 15 | 7.31 ± 0.66 | 7.71 ± 0.64 | 7.92 ± 0.51 | 12.38 ± 0.8# | ||
| 45 | 4.88 ± 0.36* | 4.34 ± 0.31* | 4.8 ± 0.31* | 8.23 ± 0.81* | ||
| 60 | 9.43 ± 0.36 | 8.72 ± 0.3 | 9.18 ± 0.31 | 11.13 ± 0.88 | ||
| 165 | 6.29 ± 0.36 | 4.42 ± 0.63 | 6.4 ± 0.69 | 6.39 ± 0.32 | ||
P < 0.05 versus corresponding 15 min (pre-ischaemic) value.
P < 0.05 versus corresponding 5 min (pre-drug) value.
Table 2.
Heart rate of hearts from normal chow-fed animals (A) and cholesterol-fed animals (B)
| A. Time (min) |
Heart rate (beats·min−1) |
|||||
|---|---|---|---|---|---|---|
| Control(n=8) | CG(n=8) | BNP(n=8) | SNAP(n=8) | BNP+KT(n=8) | BNP+PET(n=6) | |
| 15 | 259 ± 20 | 321 ± 29 | 313 ± 20 | 296 ± 11 | 286 ± 17 | 287 ± 23 |
| 45 | 282 ± 38 | 310 ± 16 | 304 ± 8 | 305 ± 8 | 275 ± 24 | 305 ± 14 |
| B. Time (min) |
Heart rate (beats·min−1) |
|||||
| Chol(n=8) | Chol+CG(n=8) | Chol+BNP(n=8) | Chol+SNAP(n=8) | |||
| 15 | 267 ± 16 | 256 ± 14 | 275 ± 23 | 258 ± 3 | ||
| 45 | 253 ± 32 | 250 ± 23 | 263 ± 28 | 288 ± 14 | ||
Expression and activation of PKG
We examined the effect of hyperlipidaemia on basal expression and its activity of PKG by the help of the phosphorylation of a major myocardial substrate of PKG, the protein troponin I. In addition, we investigated the distribution of the monomeric and dimeric form of PKG by non-reducing Western blots. We observed no difference in basal expression of PKG in ventricular myocardium of hyperlipidaemic rats compared with normal rats (Figure 3A). Although total cardiac expression of PKG was not affected by hyperlipidaemia, in hearts of hyperlipidaemic rats, the dimmer : monomer ratio of PKG was significantly elevated compared to normal hearts (n= 5 per group, Figure 3B). In contrast, phosphorylation of cardiac troponin I was significantly attenuated in ventricular myocardium of hyperlipidaemic rats compared with normal rats (Figure 3C).
Figure 3.
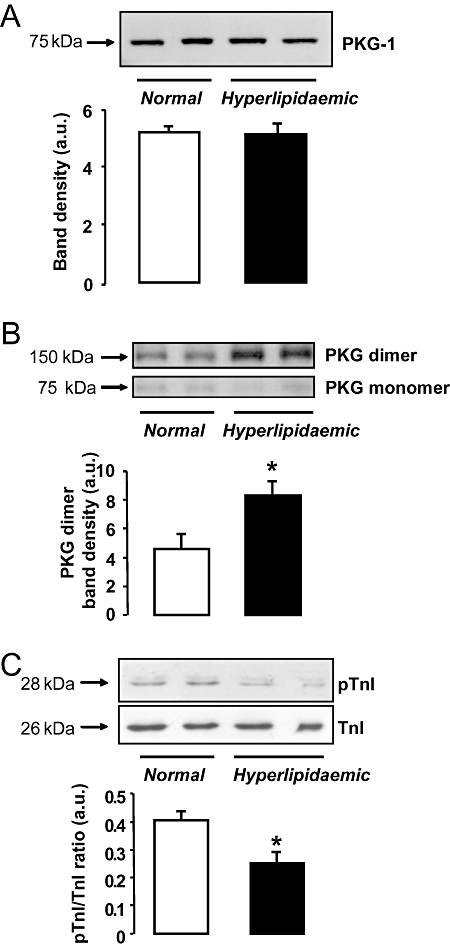
Immunoblot analysis of left ventricular myocardium from normal and cholesterol-fed hyperlipidaemic rats. Basal protein expression of protein kinase G (PKG) was not influenced by hyperlipidaemia (A); however, the ratio of dimeric : monomeric forms of PKG was elevated in hearts of hyperlipidaemic animals after 60 min of aerobic perfusion (B; *P < 0.05, Student's t-test). Phosphorylation of troponin I (TnI) was determined as an indicator of PKG activity. Hyperlipidaemia was associated with significantly reduced phosphorylation of troponin I, as compared to controls (C; *P < 0.05, Student's t-test). All panels show representative Western blots and optical densitometry data (n= 5 per group).
Discussion
The principal findings of the present studies are as follows. In hearts from normal rats, cGMP-elevating agents administered during ischaemia and reperfusion resulted in cardioprotection. The protective effect of cGMP was evident when either a cGMP analogue, an NO donor or a natriuretic peptide (BNP-32) was perfused, suggesting that both sGC- and NPR-derived cGMP is protective. In normal hearts, the protective effect of BNP-32 was abolished by two chemically distinct inhibitors of PKG activation, KT5823 or pCPT-PET-cGMPS, suggesting that PKG activation is a critical mediator of natriuretic peptide/cGMP-induced cytoprotection in ischaemia–reperfusion. In hyperlipidaemic hearts, the NO donor, BNP-32, and exogenous cGMP failed to induce cardioprotection, suggesting that the defect in cytoprotective signalling in the hyperlipidaemic myocardium may reside downstream of cGMP elevation. Hyperlipidaemia also reduced the basal activity of PKG, but not its protein expression, indicating that compromised PKG activation may contribute to the loss of cytoprotective interventions in hyperlipidaemia. This is the first demonstration that impairment of the cGMP–PKG signalling could be a critical biochemical anomaly in hyperlipidaemia, leading to the loss of cardioprotection normally provided by cGMP-elevating agents.
Considered together, the infarct size measurements in this study, supported by measurements of early LDH release, suggest that pharmacological activation of PKG significantly limited irreversible ischaemia/reperfusion injury in normal rat myocardium, but that this protective mechanism cannot be recruited in myocardium from chronically hyperlipidaemic animals. The lack of any substantial coronary vasodilator response to BNP in this model is consistent with our previous observations (D'Souza et al., 2003; Burley and Baxter, 2007).
Cytoprotective cGMP/PKG signalling
There is a growing appreciation of the role of cGMP signalling pathways in the mediation of protection against myocardial ischaemia–reperfusion injury. Until relatively recently, a major focus of research has been the involvement of endogenous NO as a modulator of sGC activity and cGMP generation. This research has been stimulated to a large extent by the recognition that endogenous NO is a pivotal mediator of the endogenous cardioprotective signalling pathways activated by ischaemic preconditioning and ischaemic post-conditioning (see Cohen et al., 2006). However, more recently, attention has turned to investigation of the role of natriuretic peptide/NPR signalling pathways in the mediation of cardioprotection. The actions of the peptides (ANP, BNP and CNP) are brought about by binding to membrane-bound receptors (NPR-1 and NPR-2), leading to activation of a guanylyl cyclase domain and elevation of intracellular cGMP. NPRs are distinct from the NO-activated sGC. Experimental evidence suggests that exogenous ANP and BNP are cardioprotective in a variety of ischaemia–reperfusion models. For example, ANP is anti-arrhythmic in coronary artery occlusion models (Rastegar et al., 2000), and both ANP and BNP limit infarct size when administered prior to coronary occlusion (Takagi et al., 2000; D'Souza et al., 2003), and specifically when administered at the onset of reperfusion (Yang et al., 2006; Burley and Baxter, 2007). These protective effects are clearly related to elevation of cGMP concentration, but the distal effectors of the survival signalling cascade are unclear. We have proposed that PKG is a salvage kinase at reperfusion (Burley et al., 2007), and demonstrated that pharmacological inhibition of PKG abolishes the infarct-limiting effect of post-conditioning (Burley et al., unpubl. data). Evidence from such studies suggests that therapeutic activation of the natriuretic peptide/cGMP pathway may have potential to salvage myocardium from the injurious consequences of reperfusion. However, most of the work undertaken to date has studied responses in the hearts of juvenile and/or naive animals, without any modifying co-morbidities.
Modification of cardioprotective signalling in hyperlipidaemia
There is a growing awareness that co-morbid conditions, particularly those that are also major risk factors for the development of ischaemic heart disease such as diabetes mellitus, hypertension and hyperlipidaemia, may modify cytoprotective responses in acute ischaemia–reperfusion (Ferdinandy et al., 2007). For example, in hearts from obese Zucker diabetic fatty and lean Goto–Kakizaki type 2 diabetic rats, ischaemic preconditioning did not afford protection against reperfusion injury (Kristiansen et al., 2004). In hyperlipidaemia, cardioprotection induced by ischaemic preconditioning and post-conditioning is lost (Giricz et al., 2006; Iliodromitis et al., 2006), and it has also been reported that the infarct size-limiting effect of ischaemic preconditioning is lost in hyperlipidaemic rabbits (Juhasz et al., 2004). Consistent with these observations, in the present study we have observed that neither exogenous NO, cGMP nor BNP protects the myocardium of hyperlipidaemic rats against ischaemia–reperfusion injury, as illustrated by the lack of decrease in infarct size and myocardial LDH efflux. As the majority of the effects of cGMP are exerted through PKG (see Burley et al., 2007; Ferdinandy et al., 2007), we investigated its myocardial expression and oxidative status. Here, we found that hyperlipidaemia did not modulate the basal expression of PKG, but the ratio of its oxidized dimeric form was more abundant in hearts of hyperlipidaemic animals. Although the underlying reason for the increased oxidative dimerization in hyperlipidaemia is still unknown, we have reported previously an increased superoxide production in hearts of hyperlipidaemic rats (Onody et al., 2003) that may be responsible for this phenomenon. In addition, increased vascular superoxide and decreased phosphorylation of a PKG target were found in hyperlipidaemic Watanabe rabbits (Oelze et al., 2000). Moreover, in hyperlipidaemic Watanabe rabbits treated with a direct NO donor (glyceryl trinitrate), there was increased vascular production of peroxynitrite in parallel with a substantially decreased PKG activity (Warnholtz et al., 2002). These results suggest that in hyperlipidaemia, the increased oxidative stress may contribute to the decreased PKG activity, and that the use of NO donors such as glyceryl trinitrate or SNAP in hyperlipidaemic animals can further increase oxidative stress, resulting in a decreased PKG activity. In accordance with this suggestion, Burgoyne et al. (2007) reported that hydrogen peroxide increases the formation of PKG dimers in vitro and in isolated hearts. The somewhat controversial findings that dimerization activated PKG (Burgoyne et al., 2007), and that hyperlipidaemia increased PKG dimerization, but compromised the activity of PKG suggest that in hyperlipidaemia, the oxidative state of the PKG is not a major determinant of its downstream effects. This hypothesis is further supported by the finding that treatments elevating intracellular cGMP concentration do not induce PKG-dependent cardioprotection in hyperlipidaemia. It has been previously shown that targeted disruption of PKG signalling leads to the loss of cGMP-dependent cardioprotection (Das et al., 2008), but this phenomenon has not been studied in connection with hyperlipidaemia. In this study, we assessed the activity of PKG indirectly by investigating the phosphorylation of troponin I, which was decreased in hyperlipidaemia. The functional relevance of decreased phosphorylation of troponin I in the attenuation of PKG-induced cardioprotection observed in hyperlipidaemia is not clear. However, it has been shown that PKG-mediated phosphorylation of troponin I could result in a reduction of responsiveness of myofilament Ca2+ in rat ventricular myocytes (Layland et al., 2002) that might be responsible for some of the cardioprotection in ischaemia/reperfusion injury (Stamm and del Nido, 2004).
The major limitation of the present study is the lack of direct evidence for decreased activity of PKG and of the effect of the treatment protocols on myocardial cGMP concentration. Unfortunately, specific substrates of PKG that permit unambiguous estimation of the kinase's activity in intact tissues are not known. As we have indicated previously (Burley et al., 2007), global estimates of cGMP concentration in tissue extracts may not reflect localization and biological activity of the cyclic nucleotide. Therefore, it is not possible to conclude firmly at this stage that hyperlipidaemia-induced attenuation of PKG activation accounts for the loss of the cardioprotective effects of the applied treatments. Clearly, further studies investigating alterations in PKG activity, guanylyl cyclase and phosphodiesterase activity and cGMP content and localization are required to provide comprehensive insights into the detrimental effects of chronic hyperlipidaemia on the cGMP–PKG system.
In conclusion, these data reveal that hyperlipidaemia is associated with loss of cardioprotective responses through interruption of a major cardioprotective signalling mechanism in the myocardium. In contrast to the normal myocardium, the hyperlipidaemic myocardium is clearly insensitive to the protective effects of NO and natriuretic peptide. Although our present study has not identified the nature of the cardioprotective signalling defect in hyperlipidaemia, we speculate that this could lie downstream of PKG activation, as we found evidence of down-regulation of basal PKG activity and troponin I phosphorylation. However, detailed exploration of the effects of hyperlipidaemia on PKG activation and its putative downstream substrates associated with cardioprotection is warranted, especially in view of the increasing recognition of this pathway as a pharmacological target in ischaemia–reperfusion injury and the prevalence of hyperlipidaemia in human patients at risk of ischaemia/reperfusion injury.
Acknowledgments
This work was supported by Wellcome Trust Collaborative Initiative project grant (GR074567MA), and grants from the National Research and Technology Development Office (Asboth-2005, Jedlik Med-Food), the Hungarian Ministry of Health (ETT 597/2006). DSB was supported by a PhD Studentship awarded by Heart Research UK.
Glossary
Abbreviations:
- BNP/BNP-32
B-type natriuretic peptide(-32)
- 8-Br-cGMP
8-bromo- cyclic guanosine 3,5 monophosphate
- KT
KT5823
- LDH
lactate dehydrogenase
- NPR
natriuretic peptide receptor
- PET
Rp-8-pCPT-PET-CGMPS
- PKG
cGMP-dependent protein kinase, protein kinase G
- sGC
soluble guanylyl cyclase
- SNAP
S-nitroso-N-acetyl-penicillamine
- TTC
2,3,5-triphenyl-tetrazolium chloride
Conflicts of interest
The authors declare no conflicts of interest.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317(5843):1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- Burley DS, Baxter GF. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res Cardiol. 2007;102(6):529–541. doi: 10.1007/s00395-007-0672-1. [DOI] [PubMed] [Google Scholar]
- Burley DS, Ferdinandy P, Baxter GF. Cyclic GMP and protein kinase-G in myocardial ischaemia–reperfusion: opportunities and obstacles for survival signaling. Br J Pharmacol. 2007;152(6):855–869. doi: 10.1038/sj.bjp.0707409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Nitric oxide is a preconditioning mimetic and cardioprotectant and is the basis of many available infarct-sparing strategies. Cardiovasc Res. 2006;70(2):231–239. doi: 10.1016/j.cardiores.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Cuong DV, Kim N, Youm JB, Joo H, Warda M, Lee JW, et al. Nitric oxide–cGMP–protein kinase G signaling pathway induces anoxic preconditioning through activation of ATP-sensitive K+ channels in rat hearts. Am J Physiol Heart Circ Physiol. 2006;290(5):H1808–H1817. doi: 10.1152/ajpheart.00772.2005. [DOI] [PubMed] [Google Scholar]
- Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283(43):29572–29585. doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza SP, Yellon DM, Martin C, Schulz R, Heusch G, Onody A, et al. B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am J Physiol Heart Circ Physiol. 2003;284(5):H1592–H1600. doi: 10.1152/ajpheart.00902.2002. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Peroxynitrite: toxic or protective in the heart? Circ Res. 2001;88(2):E12–E13.. doi: 10.1161/01.res.88.2.e12. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia–reperfusion injury and preconditioning. Br J Pharmacol. 2003;138(4):532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59(4):418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Csonka C, Onody A, Csont T, Ferdinandy P. Role of cholesterol-enriched diet and the mevalonate pathway in cardiac nitric oxide synthesis. Basic Res Cardiol. 2003;98(5):304–310. doi: 10.1007/s00395-003-0412-0. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Lalu MM, Csonka C, Bencsik P, Schulz R, Ferdinandy P. Hyperlipidemia attenuates the infarct size-limiting effect of ischemic preconditioning: role of matrix metalloproteinase-2 inhibition. J Pharmacol Exp Ther. 2006;316(1):154–161. doi: 10.1124/jpet.105.091140. [DOI] [PubMed] [Google Scholar]
- Iliodromitis EK, Zoga A, Vrettou A, Andreadou I, Paraskevaidis IA, Kaklamanis L, et al. The effectiveness of postconditioning and preconditioning on infarct size in hypercholesterolemic and normal anesthetized rabbits. Atherosclerosis. 2006;188(2):356–362. doi: 10.1016/j.atherosclerosis.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Juhasz B, Der P, Turoczi T, Bacskay I, Varga E, Tosaki A. Preconditioning in intact and previously diseased myocardium: laboratory or clinical dilemma? Antioxid Redox Signal. 2004;6(2):325–333. doi: 10.1089/152308604322899396. [DOI] [PubMed] [Google Scholar]
- Kristiansen SB, Lofgren B, Stottrup NB, Khatir D, Nielsen-Kudsk JE, Nielsen TT, et al. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia. 2004;47(10):1716–1721. doi: 10.1007/s00125-004-1514-4. [DOI] [PubMed] [Google Scholar]
- Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540(2):457–467. doi: 10.1113/jphysiol.2001.014126. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mery PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J Biol Chem. 1993;268(35):26286–26295. [PubMed] [Google Scholar]
- Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3′,5′-cyclic monophosphate-dependent protein kinase [corrected. Circulation. 2003;108(18):2172–2183. doi: 10.1161/01.CIR.0000094403.78467.C3. [DOI] [PubMed] [Google Scholar]
- Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A, et al. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res. 2000;87(11):999–1005. doi: 10.1161/01.res.87.11.999. [DOI] [PubMed] [Google Scholar]
- Onody A, Csonka C, Giricz Z, Ferdinandy P. Hyperlipidemia induced by a cholesterol-rich diet leads to enhanced peroxynitrite formation in rat hearts. Cardiovasc Res. 2003;58(3):663–670. doi: 10.1016/s0008-6363(03)00330-4. [DOI] [PubMed] [Google Scholar]
- Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B, Zhan L, Pant R, et al. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J Mol Cell Cardiol. 2007;42(3):508–516. doi: 10.1016/j.yjmcc.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskas LG, Nagy ZB, Giricz Z, Onody A, Csonka C, Kitajka K, et al. Cholesterol diet-induced hyperlipidemia influences gene expression pattern of rat hearts: a DNA microarray study. FEBS Lett. 2004;562(1–3):99–104. doi: 10.1016/S0014-5793(04)00189-9. [DOI] [PubMed] [Google Scholar]
- Qin Q, Yang XM, Cui L, Critz SD, Cohen MV, Browner NC, et al. Exogenous NO triggers preconditioning via a cGMP- and mitoKATP-dependent mechanism. Am J Physiol Heart Circ Physiol. 2004;287(2):H712–H718. doi: 10.1152/ajpheart.00954.2003. [DOI] [PubMed] [Google Scholar]
- Raeymaekers L, Hofmann F, Casteels R. Cyclic GMP-dependent protein kinase phosphorylates phospholamban in isolated sarcoplasmic reticulum from cardiac and smooth muscle. Biochem J. 1988;252(1):269–273. doi: 10.1042/bj2520269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastegar MA, Vegh A, Papp JG, Parratt JR. Atrial natriuretic peptide reduces the severe consequences of coronary artery occlusion in anaesthetized dogs. Cardiovasc Drugs Ther. 2000;14(5):471–479. doi: 10.1023/a:1007828804553. [DOI] [PubMed] [Google Scholar]
- Schwemmer M, Sommer O, Koeckerbauer R, Bassenge E. Cardiovascular dysfunction in hypercholesterolemia associated with enhanced formation of AT1-receptor and of eicosanoids. J Cardiovasc Pharmacol Ther. 2000;5(1):59–68. doi: 10.1177/107424840000500108. [DOI] [PubMed] [Google Scholar]
- Stamm C, del Nido PJ. Protein kinase C and myocardial calcium handling during ischemia and reperfusion: lessons learned using Rhod-2 spectrofluorometry. Thorac Cardiovasc Surg. 2004;52(3):127–134. doi: 10.1055/s-2004-817978. [DOI] [PubMed] [Google Scholar]
- Szilvassy Z, Ferdinandy P, Bor P, Jakab I, Lonovics J, Koltai M. Ventricular overdrive pacing-induced anti-ischemic effect: a conscious rabbit model of preconditioning. Am J Physiol. 1994;266(5):H2033–H2041. doi: 10.1152/ajpheart.1994.266.5.H2033. Pt 2. [DOI] [PubMed] [Google Scholar]
- Takagi G, Kiuchi K, Endo T, Yamamoto T, Sato N, Nejima J, et al. Alpha-human atrial natriuretic peptide, carperitide, reduces infarct size but not arrhythmias after coronary occlusion/reperfusion in dogs. J Cardiovasc Pharmacol. 2000;36(1):22–30. doi: 10.1097/00005344-200007000-00003. [DOI] [PubMed] [Google Scholar]
- Tang XL, Takano H, Xuan YT, Sato H, Kodani E, Dawn B, et al. Hypercholesterolemia abrogates late preconditioning via a tetrahydrobiopterin-dependent mechanism in conscious rabbits. Circulation. 2005;112(14):2149–2156. doi: 10.1161/CIRCULATIONAHA.105.566190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Kitakaze M, Komamura K, Minamino T, Asanuma H, Sato H, et al. Pravastatin restored the infarct size-limiting effect of ischemic preconditioning blunted by hypercholesterolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol. 1999;34(7):2120–2125. doi: 10.1016/s0735-1097(99)00440-4. [DOI] [PubMed] [Google Scholar]
- Warnholtz A, Mollnau H, Heitzer T, Kontush A, Moller-Bertram T, Lavall D, et al. Adverse effects of nitroglycerin treatment on endothelial function, vascular nitrotyrosine levels and cGMP-dependent protein kinase activity in hyperlipidemic Watanabe rabbits. J Am Coll Cardiol. 2002;40(7):1356–1363. doi: 10.1016/s0735-1097(02)02133-2. [DOI] [PubMed] [Google Scholar]
- Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286(4):H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- Yang XM, Philipp S, Downey JM, Cohen MV. Atrial natriuretic peptide administered just prior to reperfusion limits infarction in rabbit hearts. Basic Res Cardiol. 2006;101(4):311–318. doi: 10.1007/s00395-006-0587-2. [DOI] [PubMed] [Google Scholar]
- Yuasa K, Michibata H, Omori K, Yanaka N. A novel interaction of cGMP-dependent protein kinase I with troponin T. J Biol Chem. 1999;274(52):37429–37434. doi: 10.1074/jbc.274.52.37429. [DOI] [PubMed] [Google Scholar]