

Abstract
Background and purpose:
During development of thoracic aortic aneurysms in a mouse model of Marfan syndrome, upregulation of matrix metalloproteinase (MMP)-2 and -9 was accompanied by compromised aortic constriction and endothelium-dependent relaxation. Losartan has been proposed for the prevention of thoracic aortic aneurysm. We hypothesized that losartan would suppress MMP-2/-9 activation and improve aortic vasomotor function in this model.
Experimental approach:
A well-characterized mouse model of Marfan syndrome (Fbn1C1039G/+) was used. Starting at 6 weeks old, Marfan mice were untreated or given losartan (0.6 g·L−1 in drinking water, n= 30). The littermate Fbn1+/+ mice served as control. Thoracic aortas were studied at 3, 6 and 9 months by histology and by contractility assays in isolated segments in vitro.
Key results:
Losartan improved elastic fibre organization and increased aortic breaking stress. Losartan reduced the activity and protein expression of MMP-2 and MMP-9 at all ages. Aortic constriction in response to membrane depolarization or phenylephrine was increased by losartan at 3 and 9 months by 100–200%. Active force of aortic smooth muscle was also increased at 6 and 9 months. Acetylcholine-induced endothelium-dependent relaxation was improved by 30% after 3 months of losartan treatment, but such improvement disappeared with longer duration of treatment, accompanied by reduced phosphorylation of endothelial nitric oxide (NO) synthaseSer1177, AktThr308 and AktSer473, compared with the control.
Conclusions and implications:
Losartan improved the contractile function of aorta and reduced MMP activation. However, the endothelial NO pathway remained suppressed in the thoracic aorta during losartan treatment, which might limit its long-term benefits in Marfan syndrome.
Keywords: losartan, Marfan syndrome, thoracic aortic aneurysm, long-term treatment, aortic contractile function, endothelial-dependent relaxation
Introduction
Marfan syndrome is an autosomal dominant disorder of connective tissue resulting from mutations in the gene encoding fibrillin-1 (FBN-1). FBN-1 is a principal component of the extracellular microfibrils, which are crucial as organizing scaffolds in the formation and maturation of elastic fibres. Abnormal elastic fibres alter the load-bearing capacity of the aorta and cause micro-dissection and degeneration of the media in the aorta. Consequently, thoracic aortic aneurysm, dissection and rupture account for over 90% of the mortality in Marfan syndrome (Dietz et al., 1991; Judge and Dietz, 2005). Clinical management of thoracic aortic aneurysm in Marfan syndrome aims to decrease the rate of aortic root dilatation and reduce the risk of dissection and rupture. Blockade of β-adrenoceptors with atenolol, propanolol, etc. has been advocated for preventive therapy (Simpson et al., 1968; Shores et al., 1994). However, this treatment does not prevent important clinical end points including aortic regurgitation, surgery, aortic dissection and death, and no clinical benefit is observed in patients with marked dilatation (Yin et al., 1989; Rios et al., 1999; Baumgartner et al., 2006; Gersony et al., 2007). Therefore, there is need for the development of more effective pharmacological strategies for the management of thoracic aortic aneurysms in Marfan syndrome.
Aortic aneurysm is characterized by impairment of vascular cell functions, destruction of extracellular matrix integrity and deterioration of the mechanical properties of the vessel wall. We have demonstrated that the progression of thoracic aortic aneurysm in Marfan syndrome is associated with upregulation of matrix metalloproteinase (MMP)-2 and -9, which is concomitant with extensive degeneration of elastic fibres, alterations in aortic mechanical properties, endothelial dysfunction and reduction of smooth muscle contractility (Chung et al., 2007a,b,c; 2008a,b;). Recent investigations have demonstrated that perturbation of transforming growth factor (TGF)-β/Smad2-signalling contributes to the prominent clinical manifestations in Marfan syndrome. Antagonizing TGF-β through TGF-β neutralizing antibodies or losartan, an angiotensin AT1 receptor antagonist, has been shown to prevent aortic elastic fibre degeneration, aortic root dilatation, mitral valve prolapse, alveolar septation and skeletal muscle dysfunction in a mouse model of Marfan syndrome (Neptune et al., 2003; Judge et al., 2004; Ng et al., 2004; Habashi et al., 2006; Cohn et al., 2007). Therefore, the prolonged administration of losartan as a pharmacological strategy for the management of thoracic aortic aneurysm in patients with Marfan syndrome is considered to be of great potential (Lacro et al., 2007; Brooke et al., 2008; Matt et al., 2008). A multicentre, randomized, clinical trial has been initiated for the comparison of the effectiveness of losartan and atenolol in patients with Marfan syndrome (Lacro et al., 2007). However, we believe that a cautious approach is needed to address important questions: will the losartan effect be consistent and offer significant vascular protection over the long-term? Will losartan modify MMP activation? Will there be unanticipated side effects during the long-term losartan treatment?
The present study was designed to fill the gap in the current knowledge related to the effects of losartan on MMP activation and aortic vascular function in a mouse model of Marfan syndrome. In this study, we demonstrated that although losartan normalized the elastic fibre architecture, suppressed MMP-2 and -9 activation, and enhanced aortic contractility, it did not improve the endothelium-dependent relaxation in the long term. The downregulated endothelial NO signalling pathway could impair vascular health and such findings might limit the long-term benefits of losartan in the management of thoracic aortic aneurysm in Marfan syndrome.
Methods
Experimental animals and tissue preparation
All animal care and experimental procedures were approved by the institutional Animal Ethics Board and were in compliance with the Guide for the Care and Use of Laboratory Animals (Chung et al., 2007a,b,c; 2008a,b;). Heterozygous (Fbn1C1039G/+, the most common class of mutation in Marfan syndrome) mice were bred with wild-type mice (C57BL/6) to generate ‘control’ (Fbn1+/+) and ‘Marfan’ mice, which were housed in the institutional animal facility. To better translate the current findings into human conditions, we provided a rough guide of the developmental stages of this mouse model based on the ‘reproductive stage’. For example, mice become ‘reproductively mature’ by 6–7 weeks of age; female mice reach the menopausal stage at the age of 7–8 months.
In the primary prevention study investigating the vascular effects of losartan, the treatment dosage and timing followed the previous study (Habashi et al., 2006). Starting at 6 weeks of age, Marfan mice were given losartan (0.6 g·L−1 in drinking water, n= 30) or left untreated. The losartan solution was changed once a week. Mice at the age of 3, 6 and 9 months were anaesthetized with a mixture of ketamine hydrochloride (80 mg·kg−1) and xylazine hydrochloride (12 mg·kg−1) i.p. for experimentation. From each mouse, a segment of aortic arch (2.5 mm in length) was fixed for histological analysis. A 1.8-mm segment was used for the functional study (ascending aorta was not used because of its short length). The rest of the thoracic aorta was flash-frozen for sample homogenization and protein extraction.
Movat's staining
Aortic segments were formalin fixed and embedded in paraffin. Because of the limited samples from each mouse, perfusion fixation was not performed. 3 µm cross-sections were prepared and stained with modified Movat's pentachrome (Chung et al., 2007b; 2008a,b;). Image acquisition and processing were performed using a Nikon MicroPhot microscope. Average aortic wall architecture score was assessed by three observers who were unaware of the genotype and treatments. Elastic fibre integrity was assessed in four representative areas on a scale from 1 to 4: 1, extensive fragmentation and degradation; 2, local fragmentation and degradation; 3, mild disorganization without fragmentation; 4: completely intact with wavy organization.
Measurement of isometric force
Aortic segments were mounted isometrically in a small vessel myograph (A/S Danish Myotechnology, Aarhus N, Denmark) for force generation measurement as described previously (Chung et al., 2007a,b,c; 2008a,b;). They were stretched to the resting tension (6.0 mN) for 20 min and challenged twice with 60 mmol·L−1 KCl before experiments were continued.
Mechanical properties of the aorta
The measurement of ‘breaking stress’ was performed as previously described (Chung et al., 2007b; 2008a,b;). Briefly, in a small vessel myograph, the aortic segment was stretched by increasing the distance between the two stainless wires. Initially, two wires were adjusted to Lo, at which the vessel was not stretched. Inside circumference of the aortic segment was measured as twice the distance between two wires, plus the wire circumference, plus two wire radii (2 × 20 µm). The distance between the two wires was then increased by 100 µm, and the new length was noted as ‘L’. The developed force (mN) was divided by the surface area (= inside circumference of the segment × length of the segment) of the aorta segment (mm2) to calculate the wall stress (mN·mm−2). The procedure was repeated until the vessel was unable to maintain its resting tension. The stress at which rupture occurred was reported as ‘breaking stress’.
‘Active force’ is the difference between ‘Total force’ and ‘Passive force’. ‘Passive force’ was measured by repeating the above procedures in a calcium-free Krebs solution prepared by replacing CaCl2 with 320 µmol·L−1 EGTA to eliminate smooth muscle cell contractility. ‘Total force’ was determined by assessing the active contractility at each level of stretch in response to 60 mmol·L−1 KCl.
Gelatinolytic zymography
The gelatinolytic activity was analysed by separating protein on 8% SDS-PAGE gels copolymerized with gelatin (2 mg·mL−1) (Chung et al., 2007b; 2008b;). The gelatinolytic activity was identified as transparent bands against the background of Coomassie blue-stained gelatin.
Western immunoblotting
Because of the limited sample from each mouse, aortic segments were pooled during homogenization in groups with the same strain and age. The procedures of protein homogenization and Western immunoblotting were previously described (Chung et al., 2007a,b,c; 2008a,b;). In brief, protein samples were separated on 6% (for endothelial nitric oxide synthase; eNOS) or 9% (MMP-2, MMP-9), or 13% (for Akt, tissue inhibitor of matrix metalloproteinase [TIMP]-1, TIMP-2 and β-actin) sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and the separated proteins were transferred to polyvinyldifluoride membranes. The membranes were then incubated with the appropriate antibodies as follows: monoclonal antibodies to MMP-2, MMP-9 (1:250), TIMP-1, TIMP-2 (1:400), eNOS (1 µg·mL−1) and β-actin (1:5000); rabbit polyclonal antibody to phospho-eNOSSer1177 (1:1000), Akt (1:1000), phospho-AktThr308 (1:1000) or phospho-AktSer473 (1:1000). Afterwards, membranes were incubated with anti-rabbit or anti-mouse IgG peroxidase-conjugated secondary antibodies (1:2500).
Data analysis
Data are reported as mean ± standard error mean. Statistical analysis and stress-strain exponential curves were prepared using GraphPad Prism software (San Diego, CA, USA). Unpaired Student's t-test and one-way anova were used for comparisons between two and multiple groups respectively. Statistical significance was defined as P value <0.05.
Materials
Ketamine hydrochloride and xylazine hydrochloride (Research Biochemicals International, Natick, MA, USA); phenylephrine, acetylcholine, KCl, chemicals for preparing Krebs solution, monoclonal β-actin antibody, anti-rabbit and mouse IgG peroxidase-conjugated secondary antibodies (Sigma-Aldrich, Oakville, ON, USA); MMP-2, MMP-9, TIMP-1 and TIMP-2 antibodies (Calbiochem, San Diego, CA, USA); Akt, phospho-eNOSSer1177, phospho-AktThr308, phospho-AktSer473 antibodies (Santa Cruz, Santa Cruz, CA, USA); eNOS antibody (BD Biosciences, Mississauga, ON, USA); ECL western blotting detection kit (Amersham Life Sciences, Arlington Heights, IL, USA).
Results
Losartan improves structural integrity of elastic fibres in thoracic aorta from Marfan mice
The structure of elastic fibres in aorta is shown in histological slides with Movat's staining (Figure 1A), and the averaged architecture score of aortic wall is reported in Figure 1B. Compared with control, Marfan aortic elastic fibre exhibited severe fragmentation, thinning and disorganization. Losartan treatment greatly improved the elastic fibre structure and restored its wavy organization which was indistinguishable from that in the control, non-Marfan mice (Figure 1).
Figure 1.
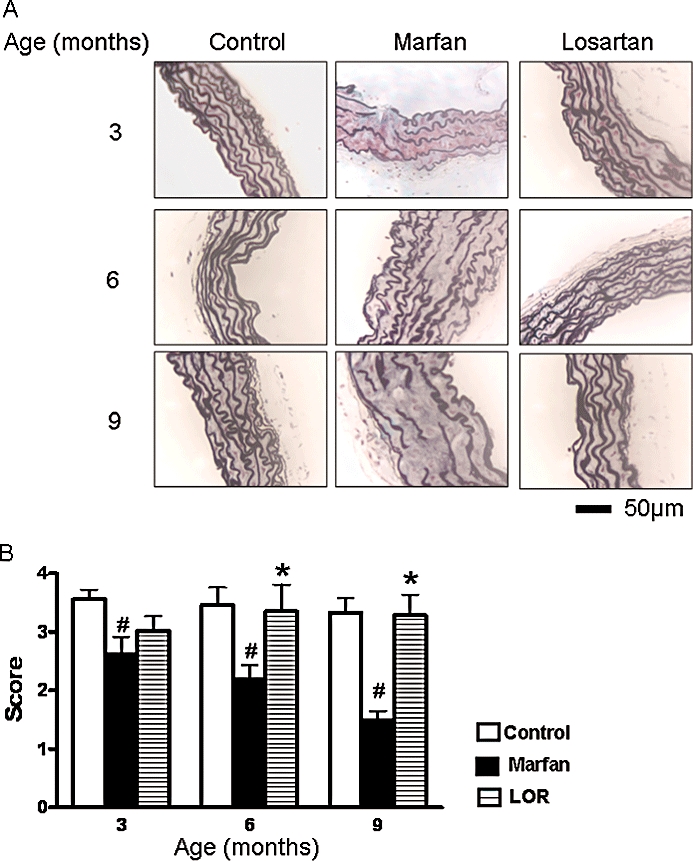
Structural integrity of elastic fibres in aorta. (A) Organization of elastic fibres was examined in Movat's stained, cross-sections of thoracic aorta from control mice, Marfan mice and Marfan mice treated with losartan (LOR), at 3, 6 and 9 months of age. Elastic fibres are stained as black fibrils. (B) Averaged architecture score of aortic wall. Three aortic segments from each mouse of each group were examined and scored as described in the Methods. n= 7–10, #P < 0.05 vs. age-matched control; *P < 0.05 vs. age-matched Marfan.
To investigate the structural stability of the aorta, we measured the breaking stress, which represents the breaking of the association of elastic fibre and smooth muscle, and at which the vessel could no longer maintain a stable resting tension. Marfan syndrome lowered the breaking stress of the thoracic aorta at 6 and 9 months of age by 33% and 22% respectively, but losartan significantly improved the breaking stress, to the values in aortas from control mice (Figure 2).
Figure 2.
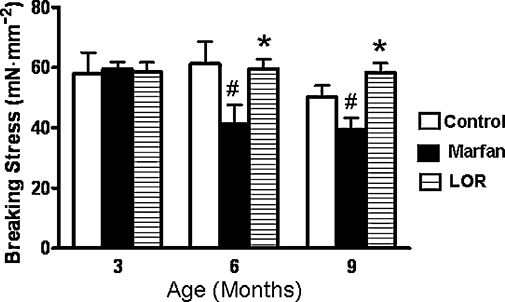
Measurement of breaking stress (mN·mm−2) of thoracic aorta at 3, 6 and 9 months of age during losartan (LOR) treatment of Marfan mice, n= 7–10, #P < 0.05 vs. age-matched control; *P < 0.05 vs. age-matched Marfan mice.
Losartan downregulates MMP-2 and -9
We have demonstrated that the extensive elastic fibre degradation in Marfan syndrome could be associated with elevated activities of MMP-2 and -9 in the thoracic aorta (Chung et al., 2007b; 2008b;). In the present work, losartan markedly reduced the gelatinolytic activity of MMP-9 at 3 and 6 months of age, and prevented the upregulation of MMP-2 activity (Figure 3A).
Figure 3.
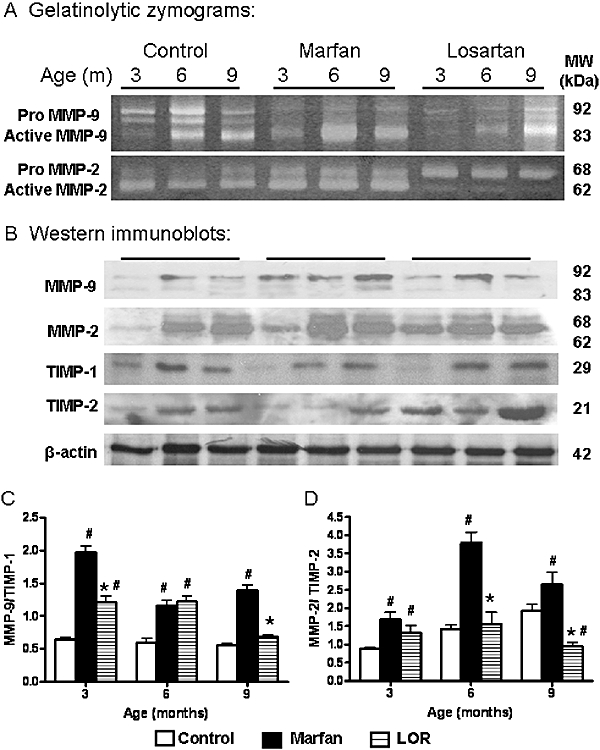
(A) Gelatinolytic zymograms showing activities of MMP-2 and -9 in homogenized aortic samples (8µg) from control, Marfan, and Marfan mice treated with losartan (LOR). (B) Western immunoblots showing the immunoreactivity of MMP-2 and -9, and TIMP-1 and -2 in homogenized aortic samples (30µg). Densitometric analysis showing the ratio of protein expressions of (C) MMP-9/TIMP-1 and (D) MMP-2/TIMP-2. *vs. Marfan, P < 0.05; #vs. age-matched control, P < 0.05, n= 7–10. MMP, matrix metalloproteinase; TIMP, tissue inhibitor of matrix metalloproteinase.
The endogenous protease inhibitor, TIMP-1, has preferential inhibitory capability against MMP-9, while TIMP-2 at high concentration selectively inhibits MMP-2 activation. At 3 and 9 months, the ratio of MMP-9/TIMP-1 protein expression in the losartan group was reduced to 60% and 40%, respectively, of that in the age-matched untreated group (Figure 3C). At 3 months, MMP-2/TIMP-2 protein expression was not altered by losartan. However, at 6 months, losartan effectively suppressed the increased MMP-2/TIMP-2 ratio in the Marfan group, and this ratio was further reduced by 65% compared with the untreated at 9 months (Figure 3D).
Losartan improves aortic smooth muscle contractile function
Aneurysm formation is accompanied by a significant reduction in aortic contractility (Chung et al., 2007a,b,c; 2008a,b;). However, we found that losartan improved the depressed KCl-stimulated contraction in the Marfan aorta by 100–200% (Figure 4A). Similarly, the reduced phenylephrine-contraction in the Marfan aorta at 3 and 9 months was greatly improved by losartan treatment. Losartan also increased the sensitivity to phenylephrine-contraction, denoted as pEC50 values, to 7.0 from 6.7 in the age-matched untreated group at 9 months of age. However, it remained significantly reduced compared with the control (P < 0.05).
Figure 4.
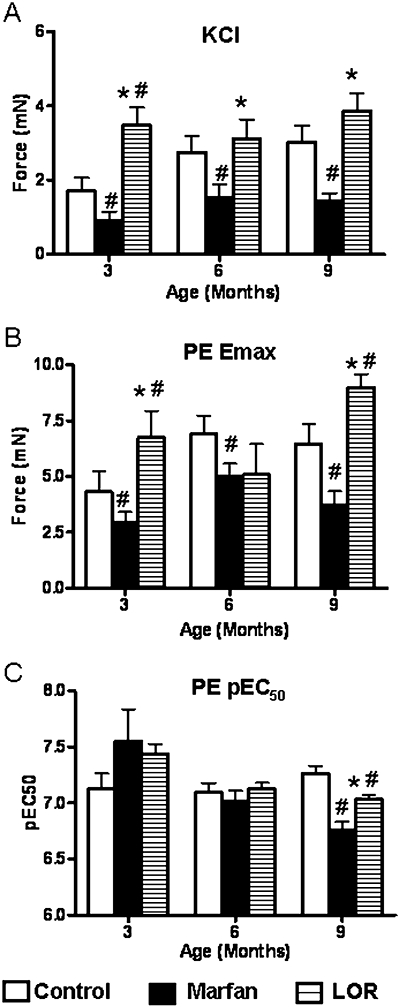
Contractility of thoracic aorta. Maximum force generated in the aortic segments from control, Marfan, and Marfan mice treated with losartan (LOR), from 3 to 9 months, in response to (A) 60 mmol·L−1 potassium chloride (KCl), and (B) 3 µmol·L−1 phenylephrine (PE). (C) pEC50 values of PE. *vs. Marfan, P < 0.05; #vs. age-matched control, P < 0.05, n= 7–10.
To further investigate the contribution of smooth muscle in the development of contractile force, we measured the active force in segments from aortas of Marfan mice during KCl-stimulated contraction. The active force in the untreated Marfan group was significantly less compared with the control at all ages. However, after 3 months of treatment, losartan normalized the active force to the control values (Figure 5A). At 6 and 9 months, the aortic segments from the losartan-treated group of Marfan mice exhibited an active force, comparable to that in the control group, over a strain range <2.0. Over the strain ranges between 2.0 and 3.0, these aortic segments had higher active force compared with the control (Figure 5B,C).
Figure 5.
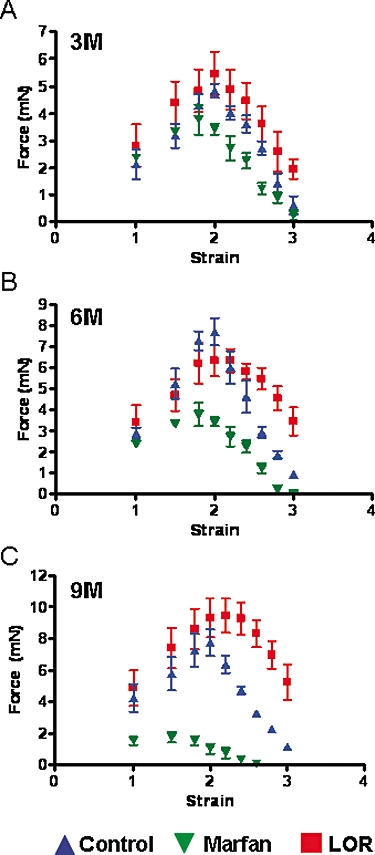
Active force from smooth muscle cells generated by stimulation with 60 mmol·L−1 potassium chloride (KCl) in the thoracic aorta from control, Marfan, and Marfan mice treated with losartan (LOR) at (A) 3, (B) 6 and (C) 9 months of age.
Endothelium-dependent relaxation remains downregulated with losartan treatment
The ACh-stimulated endothelium-dependent relaxation was attenuated in the Marfan aorta compared with the aortas from control mice at all ages (Figure 6A,B). Although losartan treatment of Marfan mice improved the ACh-relaxation (Emax = 73%; pEC50= 8.2) at 3 months of age, such benefits were lost with longer treatment periods. Thus at 9 months, aortic segments from the losartan-treated group exhibited relaxation to ACh similar to that in the untreated Marfan group (Figure 6A–C).
Figure 6.
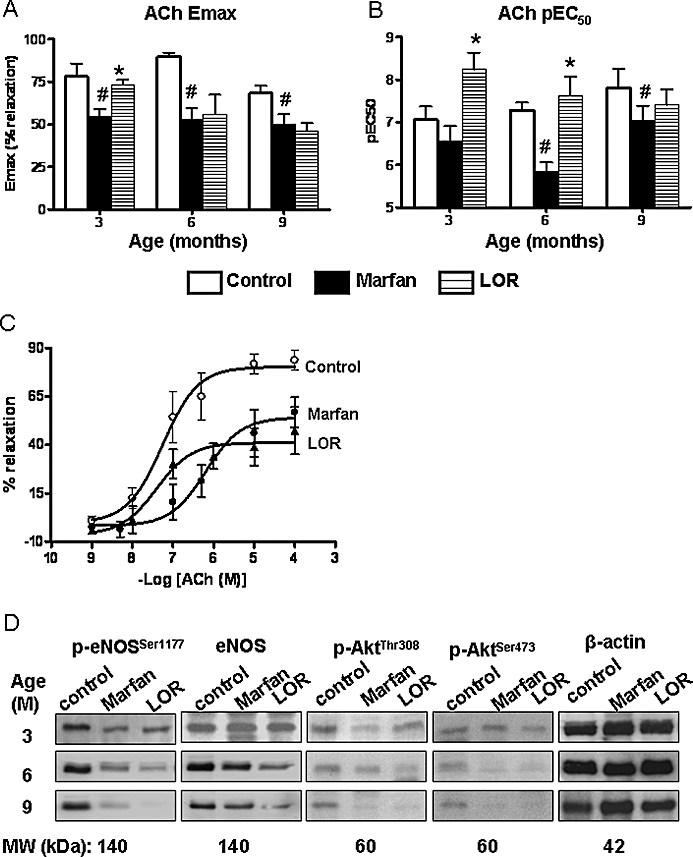
Endothelial-dependent relaxation in the thoracic aorta. (A) Acetylcholine (ACh)-induced maximum relaxation (Emax) and (B) pEC50 values in the thoracic aortas from control, Marfan, and Marfan mice treated with losartan (LOR) from 3 to 9 months. Aortic segments were precontracted with 3 µmol·L−1 phenylephrine before addition of ACh (n= 6–10, *P < 0.05 vs. Marfan; #vs. age-matched control). (C) Concentration-response curves showing the ACh-induced relaxation in the phenylephrine-precontracted aortae at 6 months of age. (D) Western immunoblotting showing protein expression of eNOS, phosphorylated-eNOSSer1177, phosphorylated-AktThr308, and phosphorylated-AktSer473 in homogenized aortic samples (20 µg). Expression of β-actin served as loading control. eNOS, endothelial nitric oxide synthase.
The activity of eNOS is upregulated via phosphorylation at Ser1177 by Akt phosphorylated at Thr308 or Ser473 (Dimmeler et al., 1999). At 3 months, the protein expression of eNOS and phosphorylated eNOS, an indicator of eNOS activation, in aortic tissue from the losartan-treated group of Marfan mice was similar to that in the control and untreated Marfan groups. However, from 6 months onward, expression of phosphorylated-eNOS in aorta was clearly reduced in the losartan-treated group compared with that in the control group. At 9 months, both eNOS and phosphorylated-eNOS in the losartan group were reduced compared with the untreated Marfan group (Figure 6D).
In the losartan-treated group, at the age of 6 months, the phosphorylated AktThr308 began to be downregulated and gradually reduced with longer treatments. Levels of phosphorylated AktSer473 in aortas from the losartan group were similar to those in the untreated Marfan group. The level of phosphorylated Akt was comparable to the level of phosphorylated eNOS (Figure 6D). The basal level of Akt was not different among groups (data not shown).
Discussion
Despite major advances in the medical and surgical management of Marfan syndrome, morbidity persists. Existing medical therapies, that is, β-adrenoceptor blockers and calcium channel blockers, only reduce the haemodynamic stress on the aorta (Simpson et al., 1968; Shores et al., 1994), but do not target the pathogenic basis for Marfan syndrome. Recent publications have demonstrated that deficiency of FBN-1-containing microfibrils results in excessive TGF-β activation, and antagonism of TGF-β by losartan has been shown to prevent aortic root dilatation, mitral valve prolapse, lung disease and skeletal muscle dysfunction in a mouse model of Marfan syndrome (Neptune et al., 2003; Judge et al., 2004; Ng et al., 2004; Habashi et al., 2006; Cohn et al., 2007). The present study is the first to investigate the long-term effects of losartan on the aortic vasculature during the progression of Marfan syndrome in a mouse model. There are three new findings in this study: first, during losartan treatment, activation of MMP-2 and -9 was markedly suppressed, suggesting cross-talk between TGF-β and MMP in the pathogenesis of the disease. Second, losartan effectively enhanced aortic contractility and improved endothelial-dependent relaxation during short-term treatment. Finally,the improvement in endothelial function was lost over the longer term, probably because of the downregulation of Akt/eNOS signalling. The results from this study indicate that although losartan offered short-term aneurysm-suppressing effects, its long-term benefits might be limited because of the lack of improvement in endothelial function.
Although a clinical trial has been initiated for the comparison of effectiveness of losartan and atenolol in patients with Marfan syndrome (Lacro et al., 2007; Brooke et al., 2008; Matt et al., 2008), we believe that a cautious approach is needed in order to address several important questions: will losartan inhibit MMP activation, another crucial mediator in aneurysm formation? Will losartan offer significant vascular protection over the long term?
To address the above questions at the pre-clinical level, this study was designed to elucidate the underlying mechanisms of beneficial effects of losartan. We have previously shown that the disruption of the MMP/TIMP balance could be associated with the excessive elastic fibre degradation in thoracic aortic aneurysm (Chung et al., 2007b; 2008b;). Therefore, the remarkable reduction of MMP/TIMP ratios by losartan suggested that proteolytic activity in the vasculature and vascular remodelling during aortic dilatation could be reduced by losartan treatment. Furthermore, the normalization of the elastic fibre architecture of the thoracic aorta treated by losartan could contribute to the favourably increased the threshold of breaking stress, which could explain the reduced incidence of aortic dissection and rupture (Habashi et al., 2006).
We suggested that the aneurysm-suppressing effect of losartan could be associated with the inhibition of MMPs. Evidence for cross-talk between MMP and TGF-β has been extensively presented. TGF-β over-expression and its downstream signalling (TGF-β receptor/Smad) are associated with MMP upregulation at the transcriptional and translational levels, and MMP-2 and -9 have been identified as latent TGF-β activators (Han et al., 2001; Nataatmadja et al., 2003; Jones et al., 2005; Ikonomidis et al., 2006). Within the aortic wall during aging, TGF-β activation is dependent on the age-associated increase in MMP-2 activity (Wang et al., 2006). Increased TGF-β signalling could cause valve calcification and dysfunction through apoptotic events and increase in MMP-9 (Clark-Greuel et al., 2007). Over-expression of TGF-β and the concomitant upregulation of various MMPs and TIMPs are associated with the increased apoptosis, impaired progenitor cell recruitment and abnormal directional migration, all of which are likely to contribute to aneurysm development in Marfan syndrome (Nataatmadja et al., 2003; Ikonomidis et al., 2006). Dura from homozygotes stained for increased presence of activated TGF-β and MMP-2 and suggested that dural ectasia in Marfan syndrome may be due to TGF-β over-activation and MMP-2-mediated elastolysis and collagen breakdown (Jones et al., 2005).
The significant improvement in the aortic contractility following the losartan treatment could be explained by the normalization of elastic fibre integrity. It has been shown that growing smooth muscle cells on elastin preserves the contractile phenotype, as demonstrated by the presence of contractile myofilaments, and that mice deficient in elastin would develop less contractile force (Karnik et al., 2003). From ultrastructural analysis, elastic laminae connect to adjacent endothelial and smooth muscle cells through an intermediate structure composed of microfibrils (Davis, 1993; 1994;). It has been shown that the RGD sequence of FBN-1 can support cellular adhesion via integrin α5β3 (Pfaff et al., 1996; Sakamoto et al., 1996; D'Arrigo et al., 1998), and these interactions are believed to contribute to the structural integrity of the vessel wall and to coordinate contractile and elastic tensions (Davis, 1993). Additionally, during losartan treatment the reduction of MMP could improve the contractility as increased levels of MMP-2 and -9 are associated with impaired contraction (Chew et al., 2004; Chung et al., 2007b; 2008b;). Nevertheless, TGF-β has been demonstrated to impair vasoconstriction directly. For example, in mouse cerebral artery, high TGF-β inhibited endothelin-1-induced contraction by inducing MAP-phosphatase 1 (Tong and Hamel, 2007). TGF-β could cause vascular dysfunction of early diabetes by inhibiting calcium transients and impairing angiotensin II-induced contraction in vascular smooth muscle cells (Sharma et al., 2003).
Although losartan exerted beneficial effects on aortic contractile function at all ages in the Marfan mice, the improvement in endothelial function was lost over long-term treatment and was concomitant with the downregulation of eNOS/Akt signalling. The short-term improvement in endothelium-dependent relaxation with losartan at 3 months of age could be due to the reduction of MMP-2 which has been shown to be reciprocally related to NO bioavailability (Wang et al., 2005). The increased relaxation response could also be the result of the preservation of elastic fibres, as functions of endothelial cells could be modulated by matrix integrity (Davis, 1994; Cohn et al., 2007). However, such vascular benefits disappeared during the long-term treatment, probably because of the prolonged inhibition of the cardioprotective effects of TGF-β. Low blood levels of TGF-β are associated with the pathogenesis of atherosclerosis, vascular hypertrophy and the severity of vascular disease (Grainger et al., 1995; Schleicher and Sessa, 2008). The protective effect of TGF-β on endothelium has been demonstrated in the epicardial coronary artery and splanchnic artery during occlusion with reperfusion (Karasawa et al., 1991; Kenny et al., 1994). The TGF-β-mediated stimulation of PI3K/Akt and p42/p44 MAPK signalling cascades is essential for endothelial cell survival and formation of capillary-like structures during angiogenesis (Viñals and Pouysségur, 2001). Through the transcriptional activation of the eNOS gene promoter, TGF-β and the downstream nuclear translocation of Smad-2 increase eNOS expression and NO production in endothelial cells (Inoue et al., 1995; Saura et al., 2002). It is widely believed that NO mediates vasorelaxation but it also regulates the balance of vascular smooth muscle cell proliferation and apoptosis, governing important aspects of vessel caliber and remodelling (Schleicher and Sessa, 2008). Therefore, the downregulation of nitric oxide signalling during the losartan treatment might exert negative effects on vascular health and be associated with aberrant vascular remodelling.
The findings from this study may have significant implications in the use of angiotensin receptor blockers for treatment of aortic aneurysm in Marfan syndrome. We believe that data generated from the mouse aorta could be extrapolated to the human aorta. Elastin is the dominant arterial extracellular matrix protein, comprising 50% of the dry weight of the mammalian aorta (Davis, 1993). Elastin, collagen and smooth muscle in the aortic media are oriented in concentric layers called lamellar units, which account for viscoelastic properties that determine the static and dynamic mechanical features. It has been reported that the number of lamellar units in the media of adult mammalian aortas is nearly proportional to aortic diameter regardless of species, animal body weight or medial thickness. Importantly, the average tension per lamellar unit of an aortic media was remarkably constant independent of species (e.g. mouse, pig, sheep, dog, human, cow) despite wide variation in aortic diameter and stress (Wolinsky and Glagov, 1967).
In conclusion, we are the first to show that losartan offered short-term aneurysm-suppressing effects, with reduced MMP activation and improved aortic contractile and endothelial function in a mouse model of Marfan syndrome. However, losartan did not improve endothelial function in the long-term treatment. The compelling results of losartan therapy in the management of thoracic aortic aneurysm in mice with Marfan syndrome indeed prompted a desire to translate these results systematically to humans (Habashi et al., 2006). A multicentre, randomized, clinical trial has been initiated to compare aortic root growth and other short-term cardiovascular outcomes in individuals with Marfan syndrome randomized to either atenolol or losartan for 3 years (Lacro et al., 2007; Brooke et al., 2008; Matt et al., 2008). However, based on the data obtained in the present study, we believe that the long-term use of losartan should be monitored carefully for the potential adverse effects because of the compromised vascular endothelial function. Our study offers better understanding of optimal treatment for Marfan syndrome, which is important to lessen the risks of aortic dilatation, dissection and rupture, and reduce the associated morbidity and mortality.
Acknowledgments
This study was supported by an operating grant from Canadian Institutes of Health Research. A.C. is the recipient of Michael Smith Foundation for Health Research/St's Paul Hospital Foundation Trainee Award. H.H.C.Y. is the recipient of Michael Smith Foundation for Health Research Junior Trainee Award and NSERC: Alexander Graham Bell Canada Graduate Scholarship.
Glossary
Abbreviations:
- eNOS
endothelial nitric oxide synthase
- FBN-1
fibrillin-1
- MMP
matrix metalloproteinase
- TGF
transforming growth factor
- TIMP
tissue inhibitor of matrix metalloproteinase
Conflicts of interest
None.
References
- Baumgartner D, Baumgartner C, Schermer E, Engl G, Schweigmann U, Matyas G, et al. Different patterns of aortic wall elasticity in patients with Marfan syndrome: a noninvasive follow-up study. J Thorac Cardiovasc Surg. 2006;132:811–819. doi: 10.1016/j.jtcvs.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., 3rd Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew DK, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–1010. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Chung AW, Au Yeung K, Cortes SF, Sandor GG, Judge DP, Dietz HC, et al. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br J Pharmacol. 2007a;150:1075–1083. doi: 10.1038/sj.bjp.0707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC, van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007b;101:512–522. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- Chung AW, Yang HH, van Breemen C. Imbalanced synthesis of cyclooxygenase-derived thromboxane A(2) and prostacyclin compromises vasomotor function of the thoracic aorta in Marfan syndrome. Br J Pharmacol. 2007c;152:305–312. doi: 10.1038/sj.bjp.0707391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Yang HHC, Au Yeung K, van Breemen C. Mechanical and pharmacological approaches to investigate the pathogenesis of Marfan syndrome in the abdominal aorta. J Vasc Res. 2008a;45:314–322. doi: 10.1159/000113603. [DOI] [PubMed] [Google Scholar]
- Chung AW, Yang HHC, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008b;102:e73–e85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER, 3rd, et al. Transforming growth factor-beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg. 2007;83:946–953. doi: 10.1016/j.athoracsur.2006.10.026. [DOI] [PubMed] [Google Scholar]
- Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arrigo C, Burl S, Withers AP, Dobson H, Black C, Boxer M. TGF-beta1 binding protein-like modules of fibrillin-1 and -2 mediate integrin-dependent cell adhesion. Connect Tissue Res. 1998;37:29–51. doi: 10.3109/03008209809028898. [DOI] [PubMed] [Google Scholar]
- Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- Davis EC. Immunolocalization of microfibril and microfibril-associated proteins in the subendothelial matrix of the developing mouse aorta. J Cell Sci. 1994;107:727–736. doi: 10.1242/jcs.107.3.727. [DOI] [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Han YP, Tuan TL, Hughes M, Wu H, Garner WL. Transforming growth factor-beta- and tumor necrosis factor-alpha-mediated induction and proteolytic activation of MMP-9 in human skin. J Biol Chem. 2001;276:22341–22350. doi: 10.1074/jbc.M010839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersony DR, McClaughlin MA, Jin Z, Gersony WM. The effect of beta-blocker therapy on clinical outcome in patients with Marfan's syndrome: a meta-analysis. Int J Cardiol. 2007;114:303–308. doi: 10.1016/j.ijcard.2005.11.116. [DOI] [PubMed] [Google Scholar]
- Grainger DJ, Kemp PR, Metcalfe JC, Liu AC, Lawn RM, Williams NR, et al. The serum concentration of active transforming growth factor-beta is severely depressed in advanced atherosclerosis. Nat Med. 1995;1:74–79. doi: 10.1038/nm0195-74. [DOI] [PubMed] [Google Scholar]
- Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. sci. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, Kaplan BS, et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation. 2006;114:I365–I370. doi: 10.1161/CIRCULATIONAHA.105.000810. [DOI] [PubMed] [Google Scholar]
- Inoue N, Venema RC, Sayegh HS, Ohara Y, Murphy TJ, Harrison DG. Molecular regulation of the bovine endothelial cell nitric oxide synthase by transforming growth factor-beta 1. Arterioscler Thromb Vasc Biol. 1995;15:1255–1261. doi: 10.1161/01.atv.15.8.1255. [DOI] [PubMed] [Google Scholar]
- Jones KB, Myers L, Judge DP, Kirby PA, Dietz HC, Sponseller PD. Toward an understanding of dural ectasia: a light microscopy study in a murine model of Marfan syndrome. Spine. 2005;30:291–293. doi: 10.1097/01.brs.0000152166.88174.1c. [DOI] [PubMed] [Google Scholar]
- Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge DP, Dietz HC. Marfan's syndrome. Lancet. 2005;366:1965–1976. doi: 10.1016/S0140-6736(05)67789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa A, Guo JP, Ma XL, Lefer AM. Beneficial effects of transforming growth factor-beta and tissue plasminogen activator in splanchnic artery occlusion and reperfusion in cats. J Cardiovasc Pharmacol. 1991;18:95–105. doi: 10.1097/00005344-199107000-00013. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- Kenny D, Coughlan MG, Pagel PS, Kampine JP, Warltier DC. Transforming growth factor beta 1 preserves endothelial function after multiple brief coronary artery occlusions and reperfusion. Am Heart J. 1994;127:1456–1461. doi: 10.1016/0002-8703(94)90370-0. [DOI] [PubMed] [Google Scholar]
- Lacro RV, Dietz HC, Wruck LM, Bradley TJ, Colan SD, Devereux RB, et al. Pediatric Heart Network Investigators Rationale and design of a randomized clinical trial of beta-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J. 2007;154:624–631. doi: 10.1016/j.ahj.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt P, Habashi J, Carrel T, Cameron DE, Van Eyk JE, Dietz HC. Recent advances in understanding Marfan syndrome: should we now treat surgical patients with losartan? J Thorac Cardiovasc Surg. 2008;135:389–394. doi: 10.1016/j.jtcvs.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Nataatmadja M, West M, West J, Summers K, Walker P, Nagata M, et al. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation. 2003;108:II329–II334. doi: 10.1161/01.cir.0000087660.82721.15. [DOI] [PubMed] [Google Scholar]
- Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff M, Reinhardt DP, Sakai LY, Timpl R. Cell adhesion and integrin binding to recombinant human fibrillin-1. FEBS Lett. 1996;384:247–250. doi: 10.1016/0014-5793(96)00325-0. [DOI] [PubMed] [Google Scholar]
- Rios AS, Silber EN, Bavishi N, Varga P, Burton BK, Clark WA, et al. Effect of long-term beta-blockade on aortic root compliance in patients with Marfan syndrome. Am Heart J. 1999;137:1057–1061. doi: 10.1016/s0002-8703(99)70362-5. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Broekelmann T, Cheresh DA, Ramirez F, Rosenbloom J, Mecham RP. Cell-type specific recognition of RGD- and non-RGD-containing cell binding domains in fibrillin-1. J Biol Chem. 1996;271:4916–4922. [PubMed] [Google Scholar]
- Saura M, Zaragoza C, Cao W, Bao C, Rodríguez-Puyol M, Rodríguez-Puyol D, et al. Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ Res. 2002;91:806–813. doi: 10.1161/01.res.0000040397.23817.e5. [DOI] [PubMed] [Google Scholar]
- Schleicher M, Sessa WC. Are the mechanisms for NO-dependent vascular remodeling different from vasorelaxation in vivo? Arterioscler Thromb Vasc Biol. 2008;28:1207–1208. doi: 10.1161/ATVBAHA.108.167403. [DOI] [PubMed] [Google Scholar]
- Sharma K, Deelman L, Madesh M, Kurz B, Ciccone E, Siva S, et al. Involvement of transforming growth factor-beta in regulation of calcium transients in diabetic vascular smooth muscle cells. Am J Physiol Renal Physiol. 2003;285:F1258–F1270. doi: 10.1152/ajprenal.00145.2003. [DOI] [PubMed] [Google Scholar]
- Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan's syndrome. N Engl J Med. 1994;330:1335–1341. doi: 10.1056/NEJM199405123301902. [DOI] [PubMed] [Google Scholar]
- Simpson CF, Kling JM, Palmer RF. The use of propranolol for the protection of turkeys from the development of beta-aminopropionitrile-induced aortic ruptures. Angiology. 1968;19:414–418. doi: 10.1177/000331976801900705. [DOI] [PubMed] [Google Scholar]
- Tong XK, Hamel E. Transforming growth factor-beta 1 impairs endothelin-1-mediated contraction of brain vessels by inducing mitogen-activated protein (MAP) kinase phosphatase-1 and inhibiting p38 MAP kinase. Mol Pharmacol. 2007;72:1476–1483. doi: 10.1124/mol.107.039602. [DOI] [PubMed] [Google Scholar]
- Viñals F, Pouysségur J. Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-alpha signaling. Mol Cell Biol. 2001;21:7218–7230. doi: 10.1128/MCB.21.21.7218-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Viappiani S, Sawicka J, Schulz R. Inhibition of endogenous nitric oxide in the heart enhances matrix metalloproteinase-2 release. Br J Pharmacol. 2005;145:43–49. doi: 10.1038/sj.bjp.0706144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, et al. Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol. 2006;26:1503–1509. doi: 10.1161/01.ATV.0000225777.58488.f2. [DOI] [PubMed] [Google Scholar]
- Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]
- Yin FC, Brin KP, Ting CT, Pyeritz RE. Arterial hemodynamic indexes in Marfan's syndrome. Circulation. 1989;79:854–862. doi: 10.1161/01.cir.79.4.854. [DOI] [PubMed] [Google Scholar]