

Abstract
Background and purpose:
Human and rodent P2X7 receptors exhibit differences in their sensitivity to antagonists. In this study we have cloned and characterized the dog P2X7 receptor to determine if its antagonist sensitivity more closely resembles the human or rodent orthologues.
Experimental approach:
A cDNA encoding the dog P2X7 receptor was isolated from a dog heart cDNA library, expressed in U-2 OS cells using the BacMam viral expression system and characterized in electrophysiological, ethidium accumulation and radioligand binding studies. Native P2X7 receptors were examined by measuring ATP-stimulated interleukin-1β release in dog and human whole blood.
Key results:
The dog P2X7 receptor was 595 amino acids long and exhibited high homology (>70%) to the human and rodent orthologues although it contained an additional threonine at position 284 and an amino acid deletion at position 538. ATP possessed low millimolar potency at dog P2X7 receptors. 2′-&3′-O-(4benzoylbenzoyl) ATP had slightly higher potency but was a partial agonist. Dog P2X7 receptors possessed relatively high affinity for a number of selective antagonists of the human P2X7 receptor although there were some differences in potency between the species. Compound affinities in human and dog blood exhibited a similar rank order of potency as observed in studies on the recombinant receptor although absolute potency was considerably lower.
Conclusions and implications:
Dog recombinant and native P2X7 receptors display a number of pharmacological similarities to the human P2X7 receptor. Thus, dog may be a suitable species for assessing target-related toxicity of antagonists intended for evaluation in the clinic.
Keywords: P2X7 receptor, human, dog, whole blood, BzATP, KN62, CBB, GW791343, compound-17
Introduction
The P2X7 receptor for extracellular ATP is proving to be a highly attractive target for both academic and pharmaceutical industrial research. The intriguing and increasingly complex biophysical and cell signalling properties of the receptor have prompted extensive research into this receptor and shown that the receptor-associated ion channel appears to change its permeability to cations during prolonged agonist exposure either by channel dilation (Surprenant et al., 1996) or by coupling to the pannexin hemichannel (Pelegrin and Surprenant, 2007). The receptor also activates an increasingly divergent number of intracellular signalling pathways (North, 2002).
From a therapeutic standpoint, the P2X7 receptor is a particularly attractive target given its unique and pivotal role in controlling the release of the inflammatory cytokine interleukin-1β (IL-1β) (Ferrari et al., 2006). The attractiveness of this target has been further enhanced by preclinical studies in P2X7 knockout mice demonstrating that the receptor may be involved in a number of inflammatory conditions such as rheumatoid arthritis and pain (Chessell et al., 2005). This has lead to the development of selective P2X7 receptor antagonists, several of which have proved effective in preclinical animal studies (Honore et al., 2006; Carroll et al., 2007). However, one of the major limitations of this target is that there are considerable species differences in antagonist sensitivity with many compounds displaying higher affinity for human than for rodent receptors (Humphreys et al., 1998; Stokes et al., 2006). This presents problems in the development of antagonists for use in humans as their low affinity for rodent receptors may preclude detecting target-related toxicity in rodents, the most commonly used preclinical species.
In this study we have cloned and characterized the dog P2X7 receptor and found that it possesses relatively high homology to the human receptor and that its antagonist sensitivity more closely resembles the human than the rodent receptors making the dog a more suitable species than rodents for determining the target-related toxicity of P2X7 receptor antagonists. A recent publication has also highlighted the close similarity in antagonist sensitivity of dog and human P2X7 receptors (Sluyter et al., 2007).
Methods
Cloning of dog P2X7 receptor
The receptor nomenclature used here follows Alexander et al. (2008). The dog P2X7 receptor was cloned from heart cDNA template using standard methods. Briefly, the dog P2X7 receptor, including the 5′- and 3′-un-translated regions, was amplified from dog heart cDNA by nested PCR using Pfu Turbo HotStart (Stratagene, La Jolla, CA, USA). The dog P2X7 receptor coding sequence obtained was confirmed from four templates (brain, heart and two different testis and ovary cDNA templates). The 1792 basepair product was then cloned into pENTR/D-TOPO (Invitrogen, LaJolla, CA, USA) to obtain the plasmid pENTR/D-DogP2X7 used for expression of the receptor.
Construction of pFastBac-Mam-1 expression plasmids and BacMam-expression viruses
The dog P2X7 receptor cDNA was subcloned into the BacMam baculovirus transfer vector, pFastBac-Mam-1, and BacMam baculovirus stocks were generated. Briefly, dog P2X7 cDNA was subcloned as a 1813 basepair Not1-to-Asc1 fragment from pENTR/D-DogP2X7 into the Not1 and Asc1 sites of pFastBac-Mam-NotAsc, which is a derivative vector of pFastBac-Mam-1 in which the polylinker region has been replaced by unique Not1 and Asc1 sites alone. The BacMam baculovirus transfer vector pFastBac-Mam-1 has been previously described (Condreay et al., 1999). The resulting plasmid, pFBM-NA/DogP2X7, was then used to generate the BacMam baculovirus BacMam-DogP2X7. The BacMam baculovirus stocks were generated from the transfer plasmid in Sf9 insect cells using standard methods, described previously (Clay et al., 2003).
Generation of a stable cell line expressing dog P2X7 receptor
Stable cell lines expressing the dog P2X7 receptor were generated in human osteosarcoma U-2 OS cells or human embryonic kidney HEK293 cells (obtained from ATCC) using the BacMam viral transduction method (Ames et al., 2004; Fonfria et al., 2008) followed by clonal selection with G418 (Invitrogen, Paisley, UK) using standard procedures.
U-2 OS or HEK293 cells were maintained in adherent culture conditions in the presence of Dulbecco's modified Eagle's medium: nutrient mixture F-12 supplemented with Glutamax (Invitrogen) and 10% foetal bovine serum (Invitrogen) at 37°C, 5% CO2. For ethidium accumulation studies, cells were harvested from the culture flasks using 0.05% trypsin/EDTA (Invitrogen) and resuspended at a concentration of ∼750 × 103 cells·mL−1 in culture media. Cells (70–80 000) were plated into individual wells of poly-D-lysine pretreated 96-well plates (Costar, High Wycombe, UK), and the plates were incubated at 37°C, 5% CO2 overnight.
In studies to measure antagonist effects in radioligand binding studies, U-2 OS cells expressing the dog P2X7 receptor were grown to confluence in T175 cm2 flasks, harvested using Versene (Invitrogen) and membranes prepared as described previously (Michel et al., 2007).
Whole-cell patch-clamp electrophysiology
Electrophysiological studies were conducted according to previously described methods (Michel et al., 2006), using standard whole-cell patch-clamp methods (Hamill et al., 1981). All recordings were performed on well-isolated, individual-phase bright cells at room temperature (20–23°C) using an Axopatch 200B amplifier controlled via the pClamp software (Axon Instruments Inc., Union City, CA, USA). The recording chamber was continuously perfused with an extracellular solution consisting of (in mM) 135 NaCl, 2 KCl, 0.5 MgCl2, 1 CaCl2, 10 HEPES and 12 glucose (pH 7.3 with NaOH; osmolarity 290–300 mOsm). Recording electrodes were fabricated on a horizontal electrode puller (Sutter Instruments P87; Sutter Instrument Company, Novato, CA, USA) from thick-walled borosilicate glass (GC120F10; Harvard Apparatus, Holliston, MA, USA) and filled with the following solution (in mM) 150 NaCl, 10 BAPTA and 10 HEPES (pH 7.3 with NaOH, osmolarity 320 mOsm). Electrode resistances using these solutions were between 2 and 5 MΩ. Once whole cell recording had been established, cells were lifted up from the coverslips to speed solution exchange. Data were only obtained from cells with a residual series resistance of less than 20 MΩ. In all experiments, cells were voltage clamped at −60 mV. ATP and 2′- & 3′-O-(4benzoylbenzoyl) ATP (BzATP) were prepared as 100 mM stocks in buffer, and drug applications were controlled using an automated fast-switching solution exchange system (SF-77B; Warner Instruments, Hamden, CT, USA). Data were acquired at 10 kHz and filtered at 2 kHz, and series resistance compensation of up to 80% was used, where appropriate. Agonist concentration–response curves were obtained by an individual cell being exposed to two concentrations of agonist, one test concentration and one standard concentration. Responses were normalized to the standard concentration for each cell. For ATP the standard concentration was 3, 10 and 30 mM, respectively, for studies on rat, human and dog receptors. For BzATP the standard concentration was 0.3, 1 and 3 mM, respectively, for studies on rat, human and dog receptors. This approach was used to reduce the problems of current growth that can occur with multiple agonist additions during electrophysiological studies on the P2X7 receptor (Hibell et al., 2001). For studies on the rat and human P2X7 receptors, agonists were applied for 2 s while for studies on the dog P2X7 receptor a 4 s agonist application was used.
Cellular ethidium accumulation measurements
Studies were performed as described previously (Michel et al., 2006; Fonfria et al., 2008) using assay buffers comprising (in mM): 10 HEPES, 5 N-methyl-D-glucamine, 5.6 KCl, 10 D-glucose, 0.5 CaCl2 (pH 7.4) and supplemented with either 280 mM sucrose (sucrose buffer) or 140 mM NaCl (NaCl buffer). Before use, growth media was completely removed from the cells and they were rinsed with 350 µL of the appropriate assay buffer that was also removed before performing assay additions. All solutions were aspirated using 25-gauge bevelled syringe needles to provide complete solution removal. In all studies the final assay volume was 100 µL, and studies were performed at room temperature of 19–21°C.
Cells were incubated with antagonist for 40 min before addition of a mixture containing the agonists, ATP or BzATP, and ethidium bromide (100 µM final assay concentration). After agonist addition, incubations were continued until approximately 10–30% of maximal agonist-stimulated dye accumulation occurred. Reactions were rapidly terminated by addition of 25 µL of 1.3 M sucrose assay buffer containing 5 mM reactive black 5, and cellular accumulation of ethidium was determined by immediately measuring fluorescence (excitation wavelength of 530 nm and emission wavelength of 620 nm) from below the plate with a 96-well plate fluorescence reader (FlexStation, Molecular Devices, Wokingham, UK).
Radioligand binding studies
The radioligand binding studies using [3H]-GSK1271360 [2-(1-adamantyl)-N-[2-({2-[bis(2 hydroxyethyl)amino]ethyl}amino)quinolin-5-yl]acetamide] were performed as described previously for [3H]-compound-17 [N-[2-({2-[(2-hydroxyethyl)amino]ethyl}amino)-5-quinolinyl]-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide] (Michel et al., 2007). GSK1271360 is a close analogue of compound-17 (example 158 from WO2003080579A1) and in its radiolabelled form has lower levels of non-specific binding and was more suitable for the present studies than [3H]-compound-17. Briefly, studies were performed using membranes prepared from U-2 OS cells stably transfected with the recombinant P2X7 dog receptors. The radioligand, [3H]-GSK1271360, was used at a concentration of 2–3 nM. Incubations were for 60 min at room temperature in a final assay volume of 200 µL of 50 mM Tris HCl buffer containing 0.01% bovine serum albumin (pH 7.4 at room temperature) and were terminated by vacuum filtration. Non-specific binding was defined using 10 µM compound-17.
IL-1β release studies
Blood was collected from healthy human volunteers or beagle dogs into citrate buffer (15% v/v) following standard ethical procedures and incubated for 80 min in the absence or presence of 1 µg·mL−1 lipopolysaccharide (LPS: Escherichia coli serotype 7136, Sigma, St. Louis, MO) at 37°C. Thereafter, 50 µL aliquots of blood were added to each well of a 96-well plate together with 30 µL of phosphate-buffered saline or antagonist and the plates incubated for 40 min at 37°C before adding 20 µL of ATP. The plates were mixed and the mixtures incubated at 37°C for 30 min (antagonist studies) or 0–100 min (agonist time course studies). Reactions were terminated by the addition of ice cold RPMI-1640 HEPES buffer (Invitrogen). The 96-well plates were centrifuged at 250×g for 5 min, and the resulting supernatants were harvested, diluted and their IL-1β content determined using a bioassay described previously (Buell et al., 1998). None of the agonists and antagonists appeared to cause lysis of red blood cells as evidenced by the absence of visible haemoglobin within the plasma supernatants.
THP-1 IL-1β release studies were performed as described previously (Michel et al., 2006). For these studies we measured BzATP-stimulated IL-1β release from LPS-stimulated THP-1 cells and determined antagonist effects following a 40 min antagonist pre-incubation and 30 min agonist incubation.
Data analysis
Data are the mean ± SEM of three to six independent experiments. Curve fitting and statistical analyses were performed using GraphPad Prism 3 (GraphPad Software Inc., San Diego, CA, USA). Individual concentration–effect or inhibition curves from each experiment were fitted to a four-parameter logistic function to determine the maximum and minimum responses and to calculate the EC50 or IC50 values and the Hill slope. For graphical purposes, most concentration–effect and inhibition curves are presented as a percentage of the maximal response obtained in the control group.
As the compounds produced non-competitive antagonist effects in the Schild studies (e.g. see Figures 4 and 5), the data from the Schild studies were also analysed to calculate antagonist pIC50 values at each agonist concentration, as this provided some quantitative estimate of antagonist potency. Statistical comparisons were made using Student's t-test or one-way anova followed by Tukey's post hoc test. Differences were assessed as significant when P < 0.05.
Figure 4.
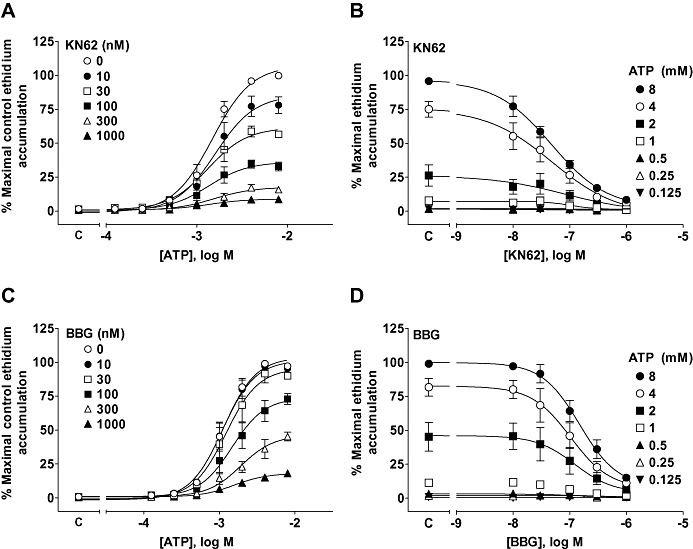
Antagonism of ATP-stimulated ethidium accumulation in cells expressing the dog P2X7 receptor. HEK293 cells expressing the dog P2X7 receptor were pre-incubated for 40 min with antagonist before measuring ATP-induced ethidium accumulation. Studies were performed in NaCl buffer. Antagonists were pre-incubated with cells for 40 min before measuring ATP responses. (A) The effect of 1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine (KN62) on ATP responses. (B) Transposition of the data in (A) to illustrate the effect of KN62 on responses to ATP. (C) The effect of brilliant blue G (BBG) on ATP responses. (D) Transposition of the data in (C) to illustrate the effect of BBG on responses to ATP. Basal ethidium accumulation in the absence of agonist is indicated on the X-ordinate as C in (A and C). The data are the mean ± SEM of 3–4 separate experiments.
Figure 5.
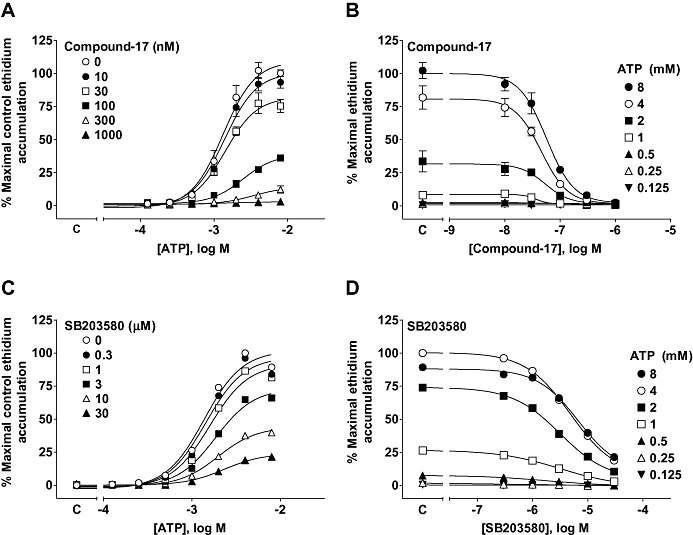
Antagonism of ATP-stimulated ethidium accumulation in cells expressing the dog P2X7 receptor. HEK293 cells expressing the dog P2X7 receptor were pre-incubated for 40 min with antagonist before measuring agonist-induced ethidium accumulation. Studies were performed in NaCl buffer. Antagonists were pre-incubated with cells for 40 min before measuring ATP responses. (A) The effect of N-[2-({2-[(2-hydroxyethyl)amino]ethyl}amino)-5-quinolinyl]-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide (compound-17) on ATP responses. (B) Transposition of the data in (A) to illustrate the effect of compound-17 on responses to ATP. (C) The effect of SB203580 on ATP responses. (D) Transposition of the data in (C) to illustrate the effect of SB203580 on responses to ATP. Basal ethidium accumulation in the absence of agonist is indicated on the X-ordinate as C in (A and C). The data are the mean ± SEM of 3–4 separate experiments.
Materials
ATP, BzATP, ethidium bromide, 1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine (KN62), brilliant blue G (BBG), pyridoxal phosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS), suramin, lipopolysaccharide from Salmonella typhosa (LPS), para-nitrophenyl phosphate, diethanolamine, N-methyl-D-glucamine and reactive black 5 were obtained from Sigma (Poole, UK). Human recombinant IL-1β was obtained from Calbiochem (Darmstadt, Germany). A neutralizing antibody for human IL-1β, a pan caspase fmk inhibitor (z-VAD) and an IL-1 receptor antagonist (IL-1ra) were all purchased from R&D Systems, Abingdon, UK.
All culture media were obtained from Invitrogen while other reagents were obtained from VWR (Loughborough, UK). [3H]-GSK1271360 was from Tritec, Switzerland (specific activity was 112 Ci·mmol−1 and purity was >99% by HPLC).
GSK314181, GW791343, compound-17, GSK1271360, GSK361390 and SB203580 were synthesized in the Chemistry Department of GSK, Harlow, UK: GSK314181, 5-{[(3R)-3-amino-1-pyrrolidinyl]methyl}-2-chloro-N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)benzamide, is the single isomer form of example 11 in WO0061569 (Alcaraz et al., 2000). GW791343 is N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride (Michel et al., 2008). Compound-17 is N-[2-({2-[(2-hydroxyethyl)amino]ethyl}amino)-5-quinolinyl]-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide (Michel et al., 2007). GSK1271360 is 2-(1-adamantyl)-N-[2-({2-[bis(2 hydroxyethyl)amino]ethyl}amino)quinolin-5-yl]acetamide (Ford et al., 2003). GSK361390 is N-adamantan-1-ylmethyl-2-chloro-5-(3,5-dioxo-4,5-dihydro-3H-[1,2,4]triazin-2-yl)-benzamide (Duplantier, 2003).
Results
Cloning of the dog P2X7 receptor
The dog P2X7 receptor exhibited the highest homology to the human receptor (85%) and slightly lower homology to the rodent orthologues (73–76%, Table 1). The receptor was 595 amino acids in length, similar to the human, rat and mouse species orthologues (Figure 1), although the dog receptor contained an additional threonine between asparagine 284 and valine 285 and was missing valine 538. For all further comparisons between the dog and other orthologues, the amino acid numbering for the human receptor is used. The guinea-pig receptor has a missing amino acid at position 77 but this was not the case with the dog receptor. A dog P2X7 receptor exists in the NCBI/GenBank database (accession number XM_534669), and the structure of this receptor is identical to the one we have cloned (Figure 1).
Table 1.
Homology between P2X7 receptor orthologues
| Human | Rat | Mouse | Guinea-pig | Dog | |
|---|---|---|---|---|---|
| Human | 100 | 80 | 80 | 77 | 85 |
| Rat | 100 | 84 | 74 | 76 | |
| Mouse | 100 | 75 | 76 | ||
| Guinea-pig | 100 | 73 | |||
| Dog | 100 |
Percent similarity (homology) was determined from pair distances using the ClustalW method (settings = slow/accurate, Gonnet).
Figure 1.

Sequence alignment and comparison of the predicted protein sequence for human and dog P2X7 receptors, marked at every 10 amino acids. Residues that differ between the sequences are shaded in grey. Dog (EU) is the sequence from the present study (Accession Number EU334661) while Dog (EX) is the sequence from Accession number XM_534669.
In other respects the dog P2X7 receptor was similar to the other species orthologues in containing the same conserved cysteine residues at positions 119, 129, 135, 152, 162, 168, 216, 226, 260 and 269 in the extracellular domain. The dog P2X7 receptor, like the other orthologues, has potential N-linked glycosylation sites (N-X-S/T) on the conserved asparagines at positions 187, 202, 213 and 241. Similar to the human and guinea-pig P2X7 receptors, the dog receptor did not possess the potential glycosylation site at position 74 that is present in rat and mouse receptors. It also did not possess the glycosylation site at asparagine 284 that is present in the human and rat P2X7 receptors, but not the mouse or guinea-pig P2X7 receptors. However, it did have a potential N-linked glycosylation site on asparagine 282.
Several single nucleotide polymorphisms (SNPs) of the human P2X7 receptor have been identified. Mutation of histidine 155 to tyrosine is a reported gain of function mutation in human P2X7 receptors (Cabrini et al., 2005) and, in the dog receptor, the corresponding residue was tyrosine as in the rat, mouse and guinea-pig receptors (see Figure 1, Young et al., 2007; Fonfria et al., 2008). The human SNPs arginine 307 to glutamine (Gu et al., 2004), threonine 357 to serine (Shemon et al., 2006), glutamate 496 to alanine (Gu et al., 2001), isoleucine 568 to asparagine (Wiley et al., 2003) and arginine 574 to histidine (Fernando et al., 2005) are reported to affect function of human P2X7 receptors either by altering the trafficking to the plasma membrane or producing defective pore formation. In dog, as in guinea-pig, rat and mouse P2X7 receptors, the corresponding residues were arginine at position 307, threonine at position 357, glutamate at position 496, isoleucine at position 568 and arginine at position 574 (Figure 1 and see Young et al., 2007).
Several splice variants were identified among the dog clones analysed. An alternative splice site within exon 13 would result in the deletion of residues 432–522 of the full-length protein in one of the clones. These residues are believed to form part of the long intracellular C-terminus that may be involved in a signal transduction process unique to the P2X7 receptor (Liang and Schwiebert, 2005). Another clone was missing exon 7. The deletion results in a frame-shift and introduces a premature stop codon. This message may be degraded by nonsense mediated decay. However, if translated, the protein product would comprise a truncated P2X7 receptor consisting of residues 1–205 plus an additional 13 residues at the C-terminus that are unique to this variant and so would retain the first transmembrane domain and part of the extracellular loop.
Characterization of the dog P2X7 receptor in electrophysiological studies
ATP and BzATP evoked inward currents in HEK293 cells stably expressing the dog P2X7 receptor (Figure 2). The onset of inward currents elicited by BzATP and ATP appeared to be slower at the dog P2X7 receptor than at the human or rat P2X7 receptors. This was particularly evident in the case of ATP where inward currents had not reached a clear plateau after a 2 s agonist application, thereby necessitating a 4 s agonist application for studies on the dog P2X7 receptor (Figure 2Ai). The decline in inward currents after cessation of agonist application also appeared slower with the dog P2X7 receptor than in studies of the human P2X7 receptor (Figure 2A and Ai with Figure 2C and Ci) although further more quantitative studies would be required to confirm this.
Figure 2.
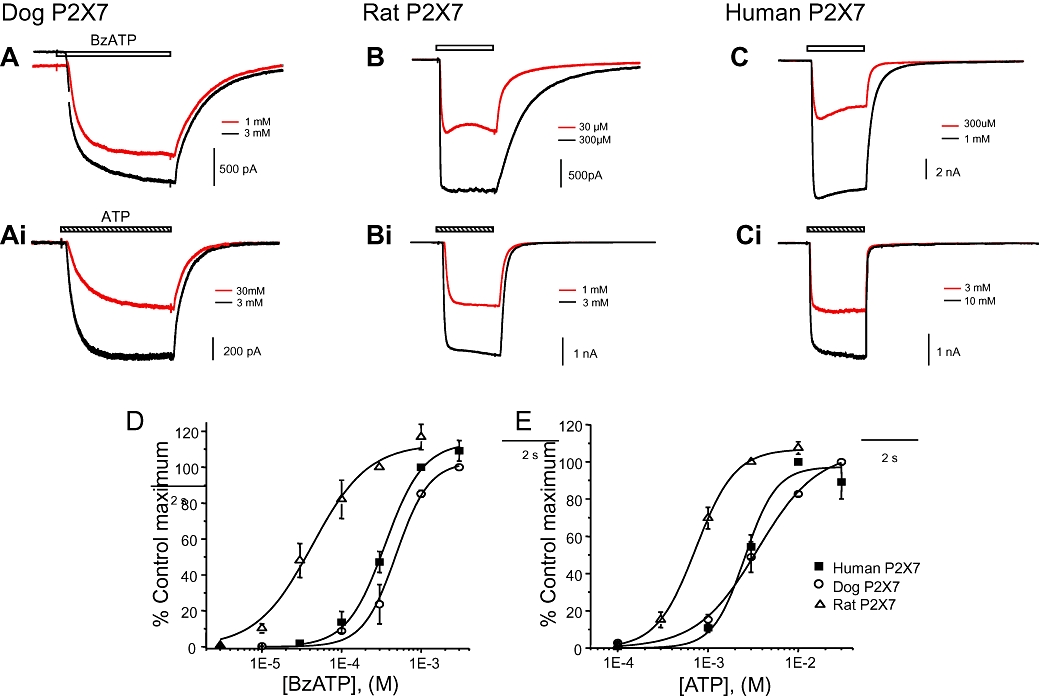
Electrophysiological characterization of agonist responses in HEK293 cells stably expressing dog, rat or human P2X7 receptors. (A–C) Representative current traces from dog, rat or human P2X7 expressing HEK293 cells in response to a 2 (rat and human) or 4 (dog) second application of 2′- & 3′-O-(4benzoylbenzoyl) ATP (BzATP). The clear bar denotes the duration of agonist application, and the response to the standard concentration of agonist is shown by the black line and the response to a test concentration of agonist by the grey line. (Ai–Ci) Similar studies performed using ATP as agonist. (D). Graph illustrating the mean concentration response curve to BzATP for dog, rat and human P2X7 receptors (n= 3–12). (E). Graph illustrating the mean concentration response curve to ATP for dog, rat and human P2X7 receptors (n= 3–11).
The EC50 values for ATP at the dog and human P2X7 receptors were not significantly different from each other but were fourfold lower than at the rat P2X7 receptor (Table 2). Similarly, the EC50 values for BzATP at the dog and human P2X7 receptors were not significantly different but were 10–15-fold lower than at the rat P2X7 receptor. The maximal currents for both ATP and BzATP did not differ at any of the species orthologues (P > 0.05, Dunnett's test) although we could not directly compare maximal effects to ATP and BzATP in the same cells due to the methods used.
Table 2.
Effect of ATP and 2′- & 3′-O-(4benzoylbenzoyl) ATP (BzATP) at rat, human and dog P2X7 receptors in electrophysiological studies
|
BzATP |
ATP |
|||||
|---|---|---|---|---|---|---|
| pEC50 | Hill slope | Max current (nA) | pEC50 | Hill slope | Max current (nA) | |
| Rat | 4.4 ± 0.10 | 1.2 ± 0.3 | 6.1 ± 1.5 | 3.1 ± 0.03 | 2.0 ± 0.3 | 4.3 ± 0.4 |
| Human | 3.4 ± 0.04* | 1.8 ± 0.3 | 3.6 ± 0.6 | 2.6 ± 0.03* | 2.5 ± 0.5 | 2.7 ± 0.6 |
| Dog | 3.3 ± 0.03† | 2.1 ± 0.2 | 3.5 ± 0.8 | 2.5 ± 0.05† | 0.8 ± 0.1 | 2.5 ± 1.5 |
The maximal currents for ATP and BzATP were determined at the standard concentration for each orthologue. For ATP this was 3, 10 and 30 mM, respectively, for studies on rat, human and dog P2X7 receptors. For BzATP the standard concentration was 0.3, 1 and 3 mM, respectively, for studies on rat, human and dog P2X7 receptors.
Significantly different (P < 0.05) from value at rat P2X7 receptor but not significantly different to value at dog P2X7 receptor.
Significantly different (P < 0.05) from value at rat P2X7 receptor. Data are mean ± SEM, n= 3–11 experiments.
Characterization of the dog P2X7 receptor in ethidium accumulation studies
When the dog P2X7 receptor was transiently expressed in U-2 OS cells BzATP (Figure 3A) and ATP (data not shown) both readily stimulated ethidium accumulation when measured in a sucrose assay buffer. However, in NaCl buffer, no response to BzATP could be detected and only a doubling of ethidium accumulation occurred with ATP (data not shown).
Figure 3.
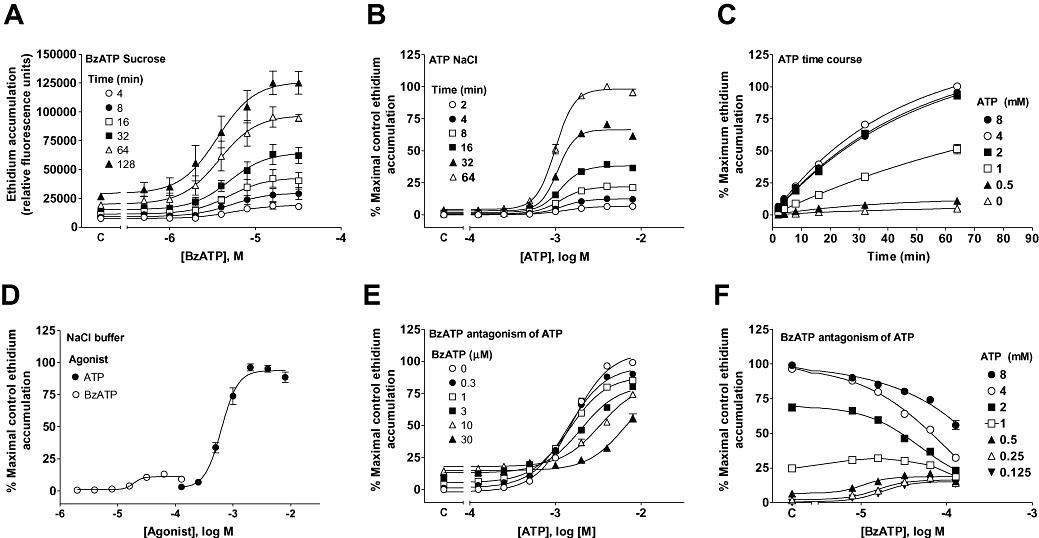
Characterization of the dog P2X7 receptor in ethidium accumulation studies. Studies were performed using HEK293 cells expressing the dog recombinant P2X7 receptor and measured agonist-stimulated cellular ethidium accumulation. (A) 2′- & 3′-O-(4benzoylbenzoyl) ATP (BzATP) concentration–effect curve obtained in sucrose buffer at the indicated time points. (B) ATP concentration effect curve obtained in NaCl buffer at the indicated time points. (C) Time course for ATP responses in NaCl buffer derived from the data in (B). (D) Comparison of the effects of BzATP and ATP at the dog receptor measured after a 16 min agonist exposure. (E) Antagonism of ATP responses by BzATP. Cells were pre-equilibrated with BzATP for 10 min prior to addition of ATP and ethidium, and ethidium accumulation was measured 16 min later. (F) Transposition of the data in (E) to illustrate the interaction between ATP and BzATP. Basal ethidium accumulation in the absence of agonist is indicated on the X-ordinate as C in (A and B). In (E) the response to the indicated concentrations of BzATP in the absence of ATP is shown as C while in (F) the response to the indicated concentrations of ATP in the absence of BzATP is shown as C. The data are the mean ± SEM of 3–4 separate experiments.
In contrast, in both U-2 OS and HEK293 cell lines stably expressing the dog P2X7 receptor, ATP stimulated appreciable ethidium accumulation (>fourfold basal) in both sucrose and NaCl buffer. In the HEK293 cell line, the response to ATP in NaCl buffer was rapid in onset, and maximal responses were obtained after 30 min (Figure 3B and C). Agonist potency did not vary greatly with time with pEC50 values of 2.89 ± 0.02, 2.92 ± 0.01, 2.94 ± 0.01, 2.94 ± 0.01, 2.97 ± 0.01 and 3.00 ± 0.01 at 2, 4, 8, 16, 32 and 64 min respectively (Figure 3B). The pEC50 values are similar to those obtained at the mouse (2.92 ± 0.05), human (3.06 ± 0.04) and guinea-pig (3.22 ± 0.10) receptor but fourfold lower than at the rat receptor (3.66 ± 0.02) as determined in a separate study conducted at the same time as these studies on the dog P2X7 receptor (Fonfria et al., 2008). BzATP only produced a small response in NaCl buffer (7.4 ± 1.3% of the maximal response to ATP) and this represented only a 37.4% increase over basal levels of ethidium accumulation (Figure 3D). The potency of BzATP at the dog receptor (pEC50= 4.67 ± 0.04) was similar to its EC50 at the human receptor (pEC50= 4.43 ± 0.05) but 4.3-fold lower than at the rat receptor (pEC50= 5.30 ± 0.08) and 4.1-fold higher than at the mouse receptor (pEC50= 4.06 ± 0.03) as determined in a separate study conducted at the same time as these studies on the dog P2X7 receptor (Fonfria et al., 2008).
To confirm if BzATP was a true partial agonist of the dog P2X7 receptor, responses to ATP were measured in the absence and presence of varying concentrations of BzATP. This resulted in a rightward shift in the ATP concentration–effect curve although we could not test high enough concentrations of ATP to determine if this shift was competitive (Figure 3E and F). However, the data were consistent with BzATP being a partial agonist of the dog P2X7 receptor.
Antagonist sensitivity of the dog P2X7 receptor in ethidium accumulation studies
As the dog receptor was functional in the NaCl buffer we chose to characterize antagonist effects in this more physiological buffer. KN62 and BBG were potent non-competitive inhibitors of the dog P2X7 receptor reducing the maximal response to ATP with little effect on the EC50 for ATP (Figure 4). Compound-17 was a relatively potent non-competitive antagonist of the dog receptor (Figure 5A and Table 3). GSK1271360 was of similar potency to compound-17 at the dog P2X7 receptor and also blocked this receptor in a non-competitive manner (data not shown and Table 3). The MAP kinase inhibitor SB203580 is a low-affinity non-competitive antagonist of the human P2X7 receptor but has no effect at rat or mouse receptors (Michel et al., 2006). SB203580 was a low-affinity non-competitive antagonist of the dog receptor (Figure 5B) although it did not produce the complete inhibition of responses observed in studies on the human receptor (Michel et al., 2006).
Table 3.
Relative potencies (pIC50) of P2X7 antagonists at the rat, human and dog native and recombinant P2X7 orthologues
| Human ethidium | Dog ethidium | Rat ethidium | Human blood | Dog blood | |
|---|---|---|---|---|---|
| KN62 | 7.64 ± 0.10 | 7.98 ± 0.1 | <5* | 5.75 ± 0.10 | 6.76 ± 0.1 |
| GW791343 | 7.03 ± 0.02 | 6.70 ± 0.07 | Potentiate | <5 | ND |
| BBG | 6.49 ± 0.1 | 7.31 ± 0.07 | 6.8 ± 0.10* | <5 | <5 |
| Compound-17 | 8.58 ± 0.03 | 7.24 ± 0.03 | 7.55 ± 0.13 | 6.28 ± 0.08 | 5.63 ± 0.18 |
| GSK1271360 | 8.03 ± 0.08 | 7.31 ± 0.13 | 7.70 ± 0.10 | ND | ND |
| GSK314181 | 8.39 ± 0.02 | 7.31 ± 0.10 | 7.06 ± 0.02 | 6.39 ± 0.23 | 5.90 ± 0.03 |
| GSK361390 | 8.09 ± 0.06 | 7.43 ± 0.12 | 5.52 ± 0.03 | 5.99 ± 0.15 | 6.09 ± 0.35 |
Ethidium studies were performed in NaCl buffer at room temperature, and IL-1β release studies were in whole blood at 37°C using ATP as the agonist. Data are mean ± SEM, n= 3–4 experiments.
BBG, brilliant blue G; compound-17, N-[2-({2-[(2-hydroxyethyl)amino]ethyl}amino)-5-quinolinyl]-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide; GSK1271360, 2-(1-adamantyl)-N-[2-({2-[bis(2 hydroxyethyl)amino]ethyl}amino)quinolin-5-yl]acetamide; GSK314181, 5-{[(3R)-3-amino-1-pyrrolidinyl]methyl}-2-chloro-N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)benzamide; GSK361390, N-adamantan-1-ylmethyl-2-chloro-5-(3,5-dioxo-4,5-dihydro-3H-[1,2,4]triazin-2-yl)-benzamide; GW791343, N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride; IL-1β, interleukin-1β; KN62, 1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine; ND, not determined.
Data from Hibell et al. (2001).
Given the non-competitive nature of the antagonists, which is also observed in studies on other species orthologues (Hibell et al., 2001; Fonfria et al., 2008), we examined other remaining antagonists against a single dose of ATP (2 mM, dog and human rat P2X7 receptors or 0.5 mM, rat P2X7 receptor) that was close to the EC50 at each orthologue. PPADS was a relatively potent antagonist, but suramin was a weak antagonist (Figure 6). GW791343 is an antagonist of human receptors but has no effect at mouse receptors and potentiates responses at the rat receptor (Michel et al., 2008). At the dog P2X7 receptor, GW791343 was an antagonist with similar potency to that determined at the human receptor (Table 3). GSK314181, an adamantyl antagonist of the human P2X7 receptor (Alcaraz et al., 2000), also blocked the dog P2X7 receptor with relatively high potency (Figure 6 and Table 3). GSK361390, an adamantyl antagonist of the human P2X7 receptor (Duplantier, 2003), with very low affinity for the rat receptor blocked the dog P2X7 receptor with relatively high potency (Table 3). A human monoclonal antibody that effectively blocks the human receptor had no effect at the dog receptor at concentrations up to 1 µg·mL−1 (data not shown). In addition, the monoclonal antibody did not label the dog receptor in immunohistochemical studies (S. Roman, unpubl. obs.).
Figure 6.
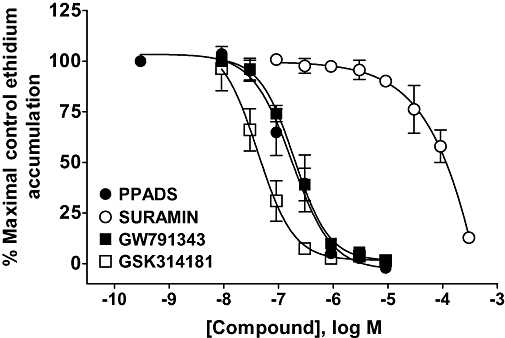
Antagonism of ATP-stimulated ethidium accumulation in cells expressing the dog P2X7 receptor. HEK293 cells expressing the dog P2X7 receptor were pre-incubated for 40 min with antagonist before measuring ATP-induced ethidium accumulation. Antagonists were pre-incubated with cells for 40 min before measuring the response to 2 mM ATP. The data are the mean ± SEM of 3–4 separate experiments. GSK314181, 5-{[(3R)-3-amino-1-pyrrolidinyl]methyl}-2-chloro-N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)benzamide; GW791343, N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride; PPADS, pyridoxal phosphate-6-azophenyl-2′,4′-disulphonic acid.
Radioligand binding studies
There was no detectable specific binding (defined using ATP or compound-17) of [3H]-compound-17 or [3H]-GSK1271360 in membranes prepared from wild type U-2 OS or HEK293 cells nor in U-2 OS cells transiently transduced with the dog P2X7 receptor. However, in membranes prepared from the HEK293 cell line stably expressing the dog P2X7 receptor, specific binding of both radioligands could be measured. The ratio of specific to total binding was greatest with [3H]-GSK1271360 (data not shown), and so we chose to use this radioligand for further characterization of the dog P2X7 receptor. We could not utilize high enough concentrations of radioligand to measure the radioligand KD or receptor density (data not shown). In competition studies, ATP and compound-17 inhibited binding by 81.3 ± 1.2% and 100%, respectively, although low concentrations of compound-17 also increased binding (Figure 7). KN62 and BBG increased binding up to 3.3 ± 0.3 and 7.1 ± 0.4-fold, respectively, although there were clear signs of a bell shaped curve in the effect of BBG.
Figure 7.
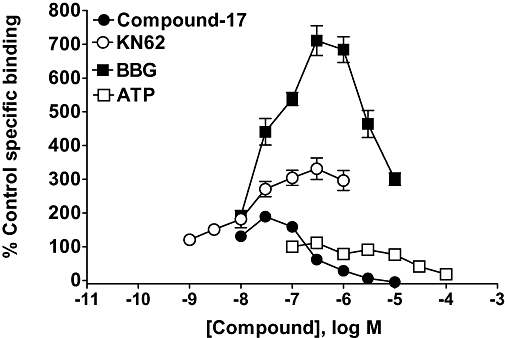
The effect of P2X7 receptor ligands on [3H]-GSK1271360 binding to membranes prepared from HEK293 cells stably expressing the dog P2X7 receptor. The radioligand concentration was 2 nM, and specific binding was defined with 10 µM compound-17. The data are the mean ± SEM of three separate experiments. BBG, brilliant blue G; compound-17, N-[2-({2-[(2-hydroxyethyl)amino]ethyl}amino)-5-quinolinyl]-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide; GSK1271360, 2-(1-adamantyl)-N-[2-({2-[bis(2 hydroxyethyl)amino]ethyl}amino)quinolin-5-yl]acetamide; KN62, 1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine.
Characterization of native tissue P2X7 receptors in human whole blood
To measure P2X7 function in a native tissue assay we measured ATP-stimulated IL-1β release from whole blood. For assay development and comparative purposes we first evaluated compound effects in human whole blood.
For these studies we used a previously described reporter gene-based bioassay to measure IL-1β release (Buell et al., 1998). To validate this assay for measuring IL-1β release in whole blood we first confirmed that the reporter gene assay response from 0.25–8 mM ATP-stimulated human blood was blocked by a neutralizing antibody for human IL-1β and an IL-1 receptor antagonist (IL-1ra). The neutralizing antibody (16 µg·mL−1) and the IL-ra (0.05 µg·mL−1) completely inhibited the reporter gene responses. To provide further evidence that the response to ATP measured in the reporter gene bioassay reflected release of IL-1β we examined the effect of the caspase inhibitor z-VAD in the whole blood assay. The response to ATP was inhibited in a non-competitive manner by z-VAD (Figure 8A) with complete inhibition of responses to 8 mM ATP at a concentration of 30 µM (pIC50 values were 5.41, 5.87, 5.60, 5.59 and 5.58 at ATP concentrations of 0.5, 1, 2, 4 and 8 mM respectively). Finally, absolute levels of IL-1β detected using the reporter gene bioassay were similar to those determined using an elisa kit (M. Lucas, pers. comm.).
Figure 8.
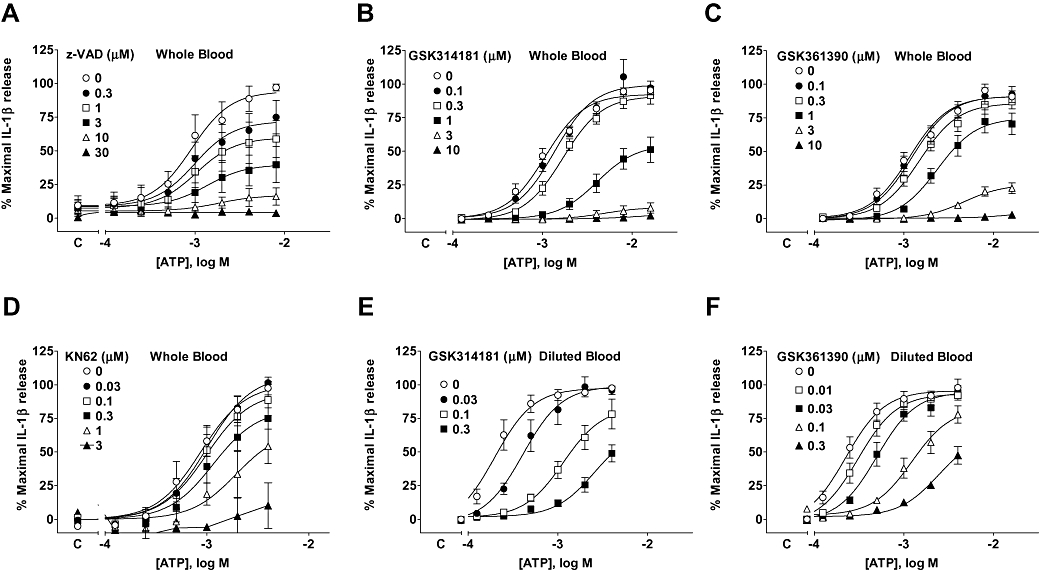
Antagonism of ATP-stimulated IL-1β release from human whole and diluted blood. (A–D) Human whole blood or (E and F) human blood diluted 10-fold with phosphate-buffered saline was stimulated with LPS (1 µg·mL−1) for 80 min and then pre-incubated for 40 min with antagonist before measuring ATP-induced IL-1β release over a 30 min period. Studies were conducted at 37°C. The figures show the effect of (A) z-VAD, (B) GSK314181, (C) GSK361390 and (D) KN62 on ATP responses in whole blood. The effect of (E) GSK314181 or (F) GSK361390 is also shown in human blood diluted 10-fold with phosphate-buffered saline. Basal IL-1β release in the absence of antagonist is indicated on the X-ordinate as C. The data are the mean ± SEM of 3–6 separate experiments. GSK314181, 5-{[(3R)-3-amino-1-pyrrolidinyl]methyl}-2-chloro-N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)benzamide; GSK361390, N-adamantan-1-ylmethyl-2-chloro-5-(3,5-dioxo-4,5-dihydro-3H-[1,2,4]triazin-2-yl)-benzamide; IL-1β, interleukin-1β; KN62, 1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine; LPS, lipopolysaccharide.
In human whole blood, ATP stimulated IL-1β release with a relatively rapid time course (Figure 9A and B). Maximal release was achieved in 10–20 min and thereafter remained relatively stable for 40 min although there was a slight decrease in levels detected after 60 min. The pEC50 values for ATP were quite similar between 20 and 60 min although the pEC50 was lower when measured after 10 min. For subsequent studies we used an ATP exposure time of 30 min.
Figure 9.
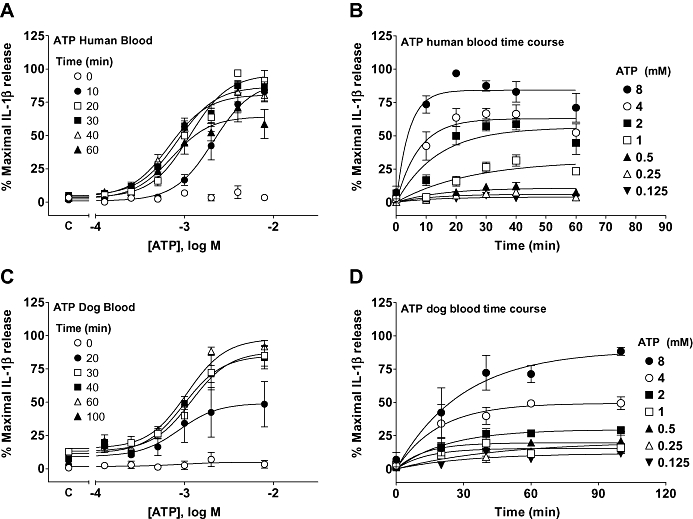
The effect of ATP on interleukin-1β (IL-1β) release from human or dog whole blood. (A) Concentration–effect curve for the ability of ATP to stimulate IL-1β release from lipopolysaccharide- (1 µg·mL−1) stimulated human whole blood at the indicated time points. (B) Transposition of the data from (A) to illustrate the time course for ATP-stimulated IL-1β release in human whole blood. (C) Concentration–effect curve for the ability of ATP to stimulate IL-1β release from dog whole blood at the indicated time points. (D) Transposition of the data from (C) to illustrate the time course for ATP-stimulated IL-1β release in dog whole blood. Basal IL-1β release in the absence of agonist is indicated on the X-ordinate as C in (A and C). The data are the mean ± SEM of 3–6 separate experiments.
A number of P2X7 receptor antagonists including GSK314181, GSK361390 and KN62 inhibited ATP-stimulated IL-1β release in human whole blood but appeared to be non-competitive antagonists (Figure 8B–D).
The potency of the antagonists in whole blood was substantially lower than that measured in the ethidium accumulation studies on the recombinant receptor (Table 3). The reduction in potency may reflect loss of compounds due to binding to blood cells or serum proteins as the potency of GSK361390 and GSK314181 were higher in whole blood diluted 10-fold with phosphate-buffered saline (Figure 8E and F) than in whole blood (Figures 8B and C). Furthermore, when similar studies were performed using THP-1 cells in the absence of serum, antagonist potency was higher than in whole blood or diluted blood for both compounds (Figure 10).
Figure 10.
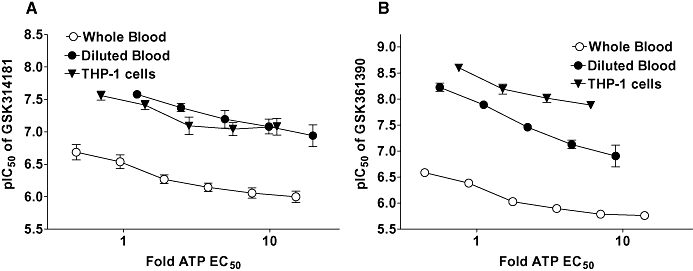
Antagonism of agonist-stimulated IL-1β release from human whole blood, human diluted blood and THP-1 cells. Human whole blood or human blood diluted 10-fold with phosphate-buffered saline was stimulated with LPS (1 µg·mL−1) for 80 min and then pre-incubated for 40 min with antagonist before measuring ATP-induced IL-1β release over a 30 min period. THP-1 cells were stimulated overnight with LPS (1 µg·mL−1), harvested, pre-incubated for 40 min with antagonist before measuring BzATP-induced IL-1β release over a 30 min period. The pIC50 of the antagonists was calculated at each concentration of agonist and is shown as a function of the agonist concentration. Agonist concentration is expressed relative to agonist EC50 at each receptor such that log (Fold EC50) represents logarithm (agonist concentration/agonist EC50). (A) The effect of GSK314181. (B) The effect of GSK361390. The data are the mean ± SEM of 4–6 separate experiments. BzATP, 2′- & 3′-O-(4benzoylbenzoyl) ATP; GSK314181, 5-{[(3R)-3-amino-1-pyrrolidinyl]methyl}-2-chloro-N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)benzamide; GSK361390, N-adamantan-1-ylmethyl-2-chloro-5-(3,5-dioxo-4,5-dihydro-3H-[1,2,4]triazin-2-yl)-benzamide; IL-1β, interleukin-1β; LPS, lipopolysaccharide.
Several other antagonists were evaluated in the whole blood assay. These included BBG and GW791343, but neither compound produced any antagonist effect at concentrations up to 10 µM (Table 3).
Characterization of native tissue P2X7 receptors in dog whole blood
We could use the same reporter gene assay as used for the studies on human blood to measure release of dog IL-1β in dog blood as dog recombinant IL-1β activated the human IL-1β receptor in the A549 cells used, although with approximately 10-fold lower potency.
The time course of ATP-stimulated IL-1β release in dog blood was similar to that in human blood with release detectable within 20 min of stimulation and release not differing greatly between 20 and 90 min (Figure 9C and D). As in human blood, the response to ATP was also inhibited by the caspase inhibitor z-VAD (1–30 µM, data not shown) providing further evidence that the response to ATP measured in the reporter gene bioassay reflected release of IL-1β.
The effects of ATP in dog blood were inhibited in a non-competitive manner by KN62 (Figure 11), and its potency was slightly higher than in human whole blood (Table 3). We evaluated other compounds (BBG, compound-17, GSK314181 and GSK361390) against 2 mM ATP on dog and human recombinant receptors and the corresponding native receptors in blood (Table 3). As in human blood, all of the compounds had lower affinity than in the ethidium accumulation studies on the recombinant receptor. We could not study IL-1β release in diluted dog blood to determine if this reflected loss of compounds due to binding to cellular constituents as the levels of IL-1β in diluted blood were below the level of detection of the reporter gene assay used to measure Il-1β.
Figure 11.
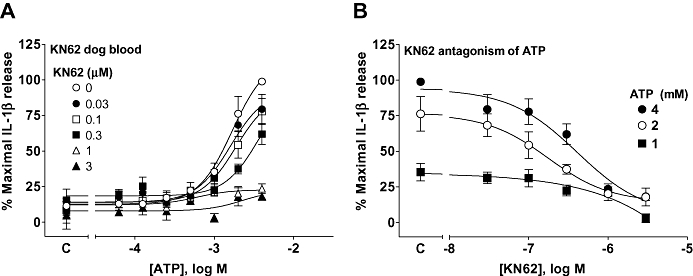
Antagonism by KN62 of ATP-stimulated IL-1β release from dog whole blood. Dog blood was stimulated with LPS (1µg·mL−1) for 80 min and then pre-incubated for 40 min with antagonist before measuring ATP-induced IL-1β release over a 30 min period. Studies were conducted at 37°C. (A) The effect of KN62 on ATP responses. (B) Transposition of the data from (A) to further illustrate the effect of KN62 on responses to ATP. Basal IL-1β release in the absence of antagonist is indicated on the X-ordinate as C. The data are the mean ± SEM of four separate experiments. KN62, 1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine; IL-1β, interleukin-1β; LPS, lipopolysaccharide.
Discussion
In this study we have cloned and characterized the dog P2X7 receptor and also characterized native human and dog receptors in whole blood. The main finding of this study is that the pharmacological properties of the dog P2X7 receptor more closely resemble those of the human receptor and differ considerably from rodent receptors in this respect.
The dog P2X7 receptor we cloned is identical to that already described in the Genbank database (accession number XM_534669). The dog P2X7 receptor was around 73–76% homologous to the rodent receptors and 85% homologous to the human receptor and possessed many of the common structural features previously identified in this family. However, the dog P2X7 receptor did possess two differences in sequence from all other P2X7 receptor species orthologues, as it had an additional threonine residue after asparagine at position 284 and an amino acid was deleted at position 538 (human numbering). The deletion at position 538 is in the C-terminus where several residues have been identified as being important in controlling expression of the receptor, but position 538 is not close to any regions that have been previously shown to affect expression or function. Thus, the residue is midway between residues at positions 460 (Ohlendorff et al., 2007; McQuillin et al., 2008) and 496 (Gu et al., 2001) and residues 551–582 (Smart et al., 2003), 568 (Wiley et al., 2003) and 573–590 (Denlinger et al., 2003; Denlinger et al., 2006) that have been identified as affecting agonist responses. In contrast, the additional threonine inserted after position 284 is close to a region important for agonist potency and rat mouse species differences in agonist sensitivity (Young et al., 2006; Young et al., 2007). Thus, the mouse P2X7 receptor, unlike the rat receptor, does not possess a glycosylation site at position 284, and mutational studies in which one was introduced into the mouse receptor by replacing aspartate 284 with asparagine resulted in a threefold increase in BzATP and sixfold to sevenfold increase in ATP potency at the mouse receptor (Young et al., 2006). The exact reason for the changes in agonist potency at the mouse P2X7 receptor was not determined, but it seemed plausible that it was due to an increase in glycosylation status. In this respect the dog P2X7 receptor did not possess the same glycosylation site as in the human receptor at position 284 but did have a site at 282 resulting in the same net number of five glycosylation sites as the human and mouse receptors. Interestingly, ATP and BzATP potency were quite similar at human, guinea-pig, mouse and dog P2X7 receptors where there are four to five glycosylation sites but lower than at the rat P2X7 receptor that possesses six glycosylation sites.
The human receptor contains a considerable number of SNPs. These do not appear to be present in other species with the residue at the corresponding residues to the position of the human SNP being conserved across mouse, rat, guinea-pig and now dog P2X7 receptors. Obviously, the sequences for the non-human species represent just a single animal or strain so it remains to be definitively determined if the human SNPs also exist across other species. The dog P2X7 receptor also appears to exist as a number of splice variants. We did not study this in any great detail, but the presence of splice variants has been noted before (Cheewatrakoolpong et al., 2005; Feng et al., 2006) and in some cases they are expressed at high levels and there is evidence that they can form hetero-oligomers with the full-length protein and block P2X7 receptor-mediated apoptosis (Feng et al., 2006).
The agonist sensitivity of the dog P2X7 receptor was similar to that at the other species in general terms of possessing very low affinity for ATP (1 mM) and slightly higher affinity for BzATP. We have previously found that the P2X7 receptor species orthologues differ in their maximal responses to BzATP in ethidium accumulation studies performed using NaCl buffer (Fonfria et al., 2008). For rat P2X7 receptors, the intrinsic activity of BzATP is greater than ATP while for the human receptor the intrinsic activity of BzATP is slightly lower than for ATP. At mouse P2X7 receptors BzATP has much lower intrinsic activity than ATP while at the guinea-pig receptor BzATP has negligible intrinsic activity and acts as an antagonist. In this respect the dog P2X7 receptor was intermediate between the mouse and guinea-pig receptors as the intrinsic activity of BzATP was only 7–8% of ATP. This did not appear to be an artefact of the methodology as BzATP acted as an antagonist of ATP responses. The lower intrinsic activity of BzATP may be partly related to the glycosylation status of the receptors as the rat P2X7 receptor has six, the human, dog and mouse P2X7 receptors have five, and the guinea-pig P2X7 receptor has only four potential glycosylation sites. However, other features may also be responsible as intracellular residues can affect expression and it has not been clearly demonstrated that there is any relationship between receptor expression and agonist potency. Furthermore, our observations on the intrinsic activity of BzATP and ATP efficacy differ from other studies where BzATP was identified as a full agonist compared with ATP (Sluyter et al., 2007). Similarly, when we measured IL-1β release from human THP-1 cells, the intrinsic activity of BzATP was greater than ATP that contrasts with observations on the recombinant receptor measured using ethidium accumulation studies (Michel and Fonfria, 2007). Furthermore, we did not observe significant differences in maximal responses to ATP and BzATP in the present electrophysiological studies, although these studies were difficult to perform and interpret due to the problems that arise when studying the P2X7 receptor using electrophysiological studies (Hibell et al., 2001). Clearly, further study will be required to resolve these discrepancies.
The antagonist sensitivity of the dog P2X7 receptor most closely resembled that of the human receptor and was clearly different to that observed at the rodent P2X7 receptors where KN62 has much lower potency. Indeed, KN62 possessed even higher potency at the dog than at the human P2X7 receptor. The effect of GW791343 at the dog P2X7 receptor was also similar to that observed at the human receptor. In contrast, GW791343 had no effect at mouse or guinea-pig P2X7 receptors and increased responses at the rat receptor. Finally, SB203580 has no effect at rat, mouse or guinea-pig P2X7 receptors but blocked the dog receptor in a similar manner to that observed at the human receptor. The dog P2X7 receptor also possessed relatively high affinity for compound-17, GSK1271360 and GSK314181 although the potency of these compounds at the dog receptor was not as high as at the human receptor. We have also found that AZ11645373 (Stokes et al., 2006), which is a highly selective antagonist of human, as compared with rodent, P2X7 receptors also blocks the dog P2X7 receptor and its affinity is similar to that at the human receptor (A. D. Michel, unpubl. obs.).
We attempted to characterize the dog P2X7 receptor using radioligand binding studies and, although it proved possible to directly label the dog receptor, the binding properties were complex. Thus, although binding was inhibited by ATP and compound-17, other compounds such as KN62 and BBG markedly increased binding at concentrations effective in functional studies. We previously noted this behaviour in studies on the human receptor when using low concentrations of radioligand relative to the radioligand affinity and suggested that this reflected an action at a distinct site on the receptor or that binding of compounds to one subunit of the P2X7 multimeric receptor resulted in a conformational change in the other sites on adjacent subunits in the receptor complex and an increase in radioligand affinity. In the present studies for technical reasons we could only use low concentrations of radioligand relative to the ligand KD due to its overall low affinity for the receptor so the same situation may prevail at the dog P2X7 receptor. Further study would be required to resolve this, but the method did not prove suitable for comparing antagonist potency between species orthologues.
Finally, we attempted to evaluate the pharmacological properties of the native dog P2X7 receptor. For these studies we developed an assay in whole blood that enabled a comparison of dog and human receptors. For this assay we used and further validated a bioassay for measuring IL-1β release in human blood. We also found that this assay could be adapted to dog blood that enabled the study of the native dog receptor. This was an important finding as there is no available immunoassay-based method for measuring dog IL-1β.
In agreement with previous studies, the response to ATP in whole blood appeared to be P2X7 receptor-mediated due to the low affinity of ATP although the stability of ATP in whole blood is a clear issue. However, the P2X7 receptor is the only member of the P2 receptor family that has been clearly shown to simulate IL-1β release from immune cells, and the response was sensitive to P2X7 receptor antagonists, providing confidence that the response measured was mediated through the P2X7 receptor. These studies provided some additional evidence for similarities in the dog and human P2X7 receptors although the potency estimates of antagonists were much lower than at the recombinant receptor in both cases. For studies on the human receptor, this may reflect degradation of compounds by human blood or most likely binding of compounds to blood cells or serum albumin as potency estimates increased several fold when studies were conducted in diluted blood and were even higher when effects on IL-1β release from THP-1 cells were measured in the absence of serum.
Nevertheless, the native tissue data demonstrated that KN62 potency in dog blood was slightly higher than in human blood, in agreement with studies on the recombinant receptor. Furthermore, the relative potencies of compound-17 and GSK314181 in dog and human blood were similar to their potencies at dog and human P2X7 receptors in ethidium accumulation studies. A recently study on P2X7 receptors in dog erythrocytes (Sluyter et al., 2007) found that KN62 was a relatively high-affinity antagonist providing additional evidence that dog and human P2X7 receptors are pharmacologically similar.
Overall, there were some discrepancies in affinity estimates between the dog and human P2X7 receptors, but these were much less than observed between rodent and human P2X7 receptors and so dog is the closest species to human in terms of its antagonist sensitivity. This should greatly facilitate the development of P2X7 antagonists as dog should provide a more appropriate species for detecting target-related toxicological effects than rodents and increase the confidence of safely progressing P2X7 antagonists into human clinical trials.
In summary, these studies describe the further characterization of an additional species analogue of the P2X7 receptor family. The receptor has some structural differences to other family members, but its homology and antagonist sensitivity more closely resembles the human than the rodent orthologues.
Acknowledgments
The authors would like to thank DK Dean, M Ponce, J Allen, M Deschamps, SC Pearce, CL Bartlett and L Taylor for their valuable assistance in providing reagents for this study.
Glossary
Abbreviations:
- BBG
brilliant blue G
- BzATP
2′- & 3′-O-(4benzoylbenzoyl) ATP
- compound-17
N-[2-({2-[(2-hydroxyethyl)amino]ethyl}amino)-5-quinolinyl]-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide
- GSK1271360
2-(1-adamantyl)-N-[2-({2-[bis(2 hydroxyethyl)amino]ethyl}amino)quinolin-5-yl]acetamide
- GSK314181
5-{[(3R)-3-amino-1-pyrrolidinyl]methyl}-2-chloro-N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)benzamide
- GSK361390
N-adamantan-1-ylmethyl-2-chloro-5-(3,5-dioxo-4,5-dihydro-3H-[1,2,4]triazin-2-yl)-benzamide
- GW791343
N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride
- IL-1β
interleukin-1β
- KN62
1-(N,O-bis-[5-isoquinoline-sulphonyl]-N-methyl-L-tyroyl)-4-phenyl-piperazine
- SNP
single nucleotide polymorphism
Statement of conflict of interest
The authors are employed by GlaxoSmithKline.
References
- Alcaraz L, Furber M, Mortimore M. Adamantane derivatives. PCTInt Appl 160 pp WO 00/61569.
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames R, Nuthulaganti P, Fornwald J, Shabon U, van der Keyl H, Elshourbagy N. Heterologous expression of G protein-coupled receptors in U-2 OS osteosarcoma cells. Receptors Channels. 2004;10:117–124. doi: 10.1080/10606820490515012. [DOI] [PubMed] [Google Scholar]
- Buell G, Chessell IP, Michel AD, Collo G, Salazzo M, Herren S, et al. Blockade of human P2X7 receptor function with a monoclonal antibody. Blood. 1998;92:3521–3528. [PubMed] [Google Scholar]
- Cabrini G, Falzoni S, Forchap SL, Pellegatti P, Balboni A, Agostini P, et al. A His-155 to Tyr polymorphism confers gain-of-function to the human P2X7 receptor of human leukemic lymphocytes. J Immunol. 2005;175:82–89. doi: 10.4049/jimmunol.175.1.82. [DOI] [PubMed] [Google Scholar]
- Carroll WA, Kalvin DM, Perez MA, Florjancic AS, Wang Y, Donnelly-Roberts DL, et al. Novel and potent 3-(2,3-dichlorophenyl)-4-(benzyl)-4H-1,2,4-triazole P2X7 antagonists. Bioorg Med Chem Lett. 2007;17:4044–4048. doi: 10.1016/j.bmcl.2007.04.075. [DOI] [PubMed] [Google Scholar]
- Cheewatrakoolpong B, Gilchrest H, Anthes JC, Greenfeder S. Identification and characterization of splice variants of the human P2X7 ATP channel. Biochem Biophys Res Commun. 2005;332:17–27. doi: 10.1016/j.bbrc.2005.04.087. [DOI] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Clay WC, Condreay JP, Moore LB, Weaver SL, Watson MA, Kost TA, et al. Recombinant baculoviruses used to study estrogen receptor function in human osteosarcoma cells. Assay Drug Dev Technol. 2003;1:801–810. doi: 10.1089/154065803772613435. [DOI] [PubMed] [Google Scholar]
- Condreay JP, Witherspoon SM, Clay WC, Kost TA. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc Natl Acad Sci USA. 1999;96:127–132. doi: 10.1073/pnas.96.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denlinger LC, Sommer JA, Parker K, Gudipaty L, Fisette PL, Watters JW, et al. Mutation of a dibasic amino acid motif within the C terminus of the P2X7 nucleotide receptor results in trafficking defects and impaired function. J Immunol. 2003;171:1304–1311. doi: 10.4049/jimmunol.171.3.1304. [DOI] [PubMed] [Google Scholar]
- Denlinger LC, Coursin DB, Schell K, Angelini G, Green DN, Guadarrama AG, et al. Human P2X7 pore function predicts allele linkage disequilibrium. Clin Chem. 2006;52:995–1004. doi: 10.1373/clinchem.2005.065425. [DOI] [PubMed] [Google Scholar]
- Duplantier AJ. N-alkyl-adamantyl derivatives as P2X7-receptor antagonists. PCTInt Appl 57 pp WO 03/042190 A1.
- Feng YH, Li X, Zeng R, Gorodeski GI. Endogenously expressed truncated P2X7 receptor lacking the C-terminus is preferentially upregulated in epithelial cancer cells and fails to mediate ligand-induced pore formation and apoptosis. Nucleosides Nucleotides Nucleic Acids. 2006;25:1271–1276. doi: 10.1080/15257770600890921. [DOI] [PubMed] [Google Scholar]
- Fernando SL, Saunders BM, Sluyter R, Skarratt KK, Wiley JS, Britton WJ. Gene dosage determines the negative effects of polymorphic alleles of the P2X7 receptor on adenosine triphosphate-mediated killing of mycobacteria by human macrophages. J Infect Dis. 2005;192:149–155. doi: 10.1086/430622. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Clay WC, Levy DS, Goodwin JA, Roman S, Smith GD, et al. Cloning and pharmacological characterization of the guinea pig P2X7 receptor orthologue. Br J Pharmacol. 2008;153:544–556. doi: 10.1038/sj.bjp.0707596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford R, Leroux F, Stocks M. Novel adamantane derivatives. PCTInt Appl 171 pp WO 2003080579 A1.
- Gu BJ, Zhang W, Worthington RA, Sluyter R, Dao-Ung P, Petrou S, et al. A Glu-496 to Ala polymorphism leads to loss of function of the human P2X7 receptor. J Biol Chem. 2001;276:11135–11142. doi: 10.1074/jbc.M010353200. [DOI] [PubMed] [Google Scholar]
- Gu BJ, Sluyter R, Skarratt KK, Shemon AN, Dao-Ung LP, Fuller SJ, et al. An Arg307 to Gln polymorphism within the ATP-binding site causes loss of function of the human P2X7 receptor. J Biol Chem. 2004;279:31287–31295. doi: 10.1074/jbc.M313902200. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hibell AD, Thompson KM, Simon J, Xing M, Humphrey PP, Michel AD. Species- and agonist-dependent differences in the deactivation-kinetics of P2X7 receptors. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:639–648. doi: 10.1007/s002100100412. [DOI] [PubMed] [Google Scholar]
- Honore P, Donnelly-Roberts D, Namovic MT, Hsieh G, Zhu CZ, Mikusa JP, et al. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J Pharmacol Exp Ther. 2006;319:1376–1385. doi: 10.1124/jpet.106.111559. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Virginio C, Surprenant A, Rice J, Dubyak GR. Isoquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus rat receptor homologues. Mol Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- Liang L, Schwiebert EM. Large pore formation uniquely associated with P2X7 purinergic receptor channels. Focus on ‘Are second messengers crucial for opening the pore associated with P2X7 receptor? Am J Physiol Cell Physiol. 2005;288:C240–C242. doi: 10.1152/ajpcell.00532.2004. [DOI] [PubMed] [Google Scholar]
- McQuillin A, Bass NJ, Choudhury K, Puri V, Kosmin M, Lawrence J, et al. Case-control studies show that a non-conservative amino-acid change from a glutamine to arginine in the P2RX7 purinergic receptor protein is associated with both bipolar- and unipolar-affective disorders. Mol Psychiatry. 2008;14:614–620. doi: 10.1038/mp.2008.6. [DOI] [PubMed] [Google Scholar]
- Michel AD, Fonfria E. Agonist potency at P2X7 receptors is modulated by structurally diverse lipids. Br J Pharmacol. 2007;152:523–537. doi: 10.1038/sj.bjp.0707417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Thompson KM, Simon J, Boyfield I, Fonfria E, Humphrey PP. Species and response dependent differences in the effects of MAPK inhibitors on P2X(7) receptor function. Br J Pharmacol. 2006;149:948–957. doi: 10.1038/sj.bjp.0706938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Chambers LJ, Clay WC, Condreay JP, Walter DS, Chessell IP. Direct labelling of the human P2X7 receptor and identification of positive and negative cooperativity of binding. Br J Pharmacol. 2007;151:103–114. doi: 10.1038/sj.bjp.0707196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Chambers LJ, Walter DS. Negative and positive allosteric modulators of the P2X(7) receptor. Br J Pharmacol. 2008;153:737–750. doi: 10.1038/sj.bjp.0707625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Ohlendorff SD, Tofteng CL, Jensen JE, Petersen S, Civitelli R, Fenger M, et al. Single nucleotide polymorphisms in the P2X7 gene are associated to fracture risk and to effect of estrogen treatment. Pharmacogenet Genomics. 2007;17:555–567. doi: 10.1097/FPC.0b013e3280951625. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J Biol Chem. 2007;282:2386–2394. doi: 10.1074/jbc.M610351200. [DOI] [PubMed] [Google Scholar]
- Shemon AN, Sluyter R, Fernando SL, Clarke AL, Dao-Ung LP, Skarratt KK, et al. A Thr357 to Ser polymorphism in homozygous and compound heterozygous subjects causes absent or reduced P2X7 function and impairs ATP-induced mycobacterial killing by macrophages. J Biol Chem. 2006;281:2079–2086. doi: 10.1074/jbc.M507816200. [DOI] [PubMed] [Google Scholar]
- Sluyter R, Shemon AN, Hughes WE, Stevenson RO, Georgiou JG, Eslick GD, et al. Canine erythrocytes express the P2X7 receptor: greatly increased function compared with human erythrocytes. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2090–R2098. doi: 10.1152/ajpregu.00166.2007. [DOI] [PubMed] [Google Scholar]
- Smart ML, Gu B, Panchal RG, Wiley J, Cromer B, Williams DA, et al. P2X7 receptor cell surface expression and cytolytic pore formation are regulated by a distal C-terminal region. J Biol Chem. 2003;278:8853–8860. doi: 10.1074/jbc.M211094200. [DOI] [PubMed] [Google Scholar]
- Stokes L, Jiang LH, Alcaraz L, Bent J, Bowers K, Fagura M, et al. Characterization of a selective and potent antagonist of human P2X(7) receptors, AZ11645373. Br J Pharmacol. 2006;149:880–887. doi: 10.1038/sj.bjp.0706933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- Wiley JS, Dao-Ung LP, Li C, Shemon AN, Gu BJ, Smart ML, et al. An Ile-568 to Asn polymorphism prevents normal trafficking and function of the human P2X7 receptor. J Biol Chem. 2003;278:17108–17113. doi: 10.1074/jbc.M212759200. [DOI] [PubMed] [Google Scholar]
- Young MT, Pelegrin P, Surprenant A. Identification of Thr283 as a key determinant of P2X7 receptor function. Br J Pharmacol. 2006;149:261–268. doi: 10.1038/sj.bjp.0706880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MT, Pelegrin P, Surprenant A. Amino acid residues in the P2X7 receptor that mediate differential sensitivity to ATP and BzATP. Mol Pharmacol. 2007;71:92–100. doi: 10.1124/mol.106.030163. [DOI] [PubMed] [Google Scholar]