

Abstract
Background and purpose:
Disturbances in pulmonary vascular reactivity are important components of inflammatory lung disease. Haem oxygenase-1 (HO-1) is an important homeostatic enzyme upregulated in inflammation. Here we have investigated the potentially protective effect of HO-1 against cytokine-induced impairment in pulmonary artery relaxation.
Experimental approach:
Haem oxygenase-1 protein levels were assessed by immunofluorescence. HO activity was assessed by conversion of haemin to bilirubin. Rings of rat isolated pulmonary artery in organ baths were used to measure relaxant responses to the endothelium-dependent agent ACh and the endothelium-independent agent sodium nitroprusside (SNP). Production of nitric oxide (NO) and reactive oxygen species (ROS) was assessed by confocal fluorescence microscopy and fluorescent probes.
Key results:
Haem oxygenase-1 protein expression was strongly induced in pulmonary artery after 24-h incubation with either haemin (5 µM) or curcumin (2 µM), accompanied by a significant increase in HO activity. Incubation with tumour necrosis factor α (TNFα, 1 ng·mL−1, 2 h) significantly decreased relaxation of arterial rings to ACh, without affecting responses to SNP. Induction of HO-1 by curcumin or haemin protected against TNFα-induced hyporesponsiveness to ACh. The competitive HO inhibitor, tin protoporphyrin (20 µM), abolished the protective effect of haemin. HO-1 induction prevented a TNFα-induced increase in NO generation without affecting the TNFα-induced increase in ROS generation. HO-1 induction prevented the TNFα-induced decrease in ACh-stimulated NO generation.
Conclusions and implications:
Induction of HO-1 protected against TNFα impairment of endothelium-dependent relaxation in pulmonary artery, by a mechanism involving a reduction in inducible NO synthase-derived NO production.
Keywords: pulmonary artery, endothelial-dependent relaxation, haem oxygenase, cytokines, tumour necrosis factor, immunofluorescence, immunoblotting, nitric oxide
Introduction
Vascular endothelial cells have an essential role in the control of the human pulmonary circulation. Endothelial cells release potent vasodilators that regulate the vascular reactivity to various stimuli (Hida et al., 2002). Many studies have implicated endothelial dysfunction in the development of chronic obstructive pulmonary disease (Peinado et al., 1998), a disease associated with local and systemic inflammation (Wouters, 2005). Lung inflammation was found to induce pulmonary vascular hyporesponsiveness to vasodilators (Sheridan et al., 1996). Tumour necrosis factor (TNFα) is a major cytokine that is elevated in many diseases and seems to be involved in the associated disturbances in vascular reactivity (Vila and Salaices, 2005).
Some of the proinflammatory actions of cytokines are mediated by inducible nitric oxide synthase (iNOS)-derived nitric oxide (NO) production. Three isoforms of NOS have been described: constitutive-type isoforms like neuronal NOS and endothelial NOS (eNOS), and the inducible type of the enzyme (see Ricciardolo et al., 2004). Also, overproduction of reactive oxygen species (ROS) under pathophysiological conditions is believed to be integral to the development of cardiovascular diseases (see Madamanchi et al., 2005). One of the most important ROS in the vasculature is superoxide, which is formed by the univalent reduction of oxygen (Droge, 2002). Although superoxide can itself exert effects on vascular function, it is also pivotal in generating other reactive species. Reaction of superoxide with NO generates peroxynitrite, a potentially deleterious ROS. Dismutation of superoxide by superoxide dismutase (SOD) produces the more stable ROS, hydrogen peroxide. H2O2 can also react with reduced transition metals to be converted to the highly reactive hydroxyl radical, or it can be metabolized by myeloperoxidase to form hypochlorous acid. Virtually all types of vascular cells produce superoxide and H2O2 (Taniyama and Griendling, 2003).
Of the three known isoforms of the enzyme haem oxygenase (HO), HO-1 is inducible. HO-1 is highly expressed in erythropoietic tissues, where its function is haem degradation, but it is also expressed in vascular smooth muscle cells, where its role may include regulation of vascular tone (Soares and Bach, 2009). Strong expression of HO-1 has been associated with a major anti-inflammatory response and, as it is induced in most organs in the body, it has been implicated in protection against several disease states (reviewed by Ryter et al., 2006). HO is the rate-limiting enzyme in the degradation of haem. In breaking down haem, HO releases iron, biliverdin (which is subsequently converted to bilirubin) and carbon monoxide (CO). Each of these products of this enzymic activity plays unique and often protective roles in the human body (Morse and Choi, 2005).
The present work aimed to investigate the potentially protective effect of HO-1 against TNFα-induced impairment of relaxation in isolated pulmonary arteries.
Methods
Animals and tissues
All animals were housed and maintained in compliance with the University of Bath and the UK Home Office Guidelines and Standards of Care. Male Wistar rats (270–290 g, University of Bath) were killed by cervical dislocation and the lungs and the heart were rapidly isolated. The pulmonary artery branches were dissected and 2–3 mm rings from each animal were divided between the different treatment groups. The isolated tissue was then incubated for 24 h with or without haemin or curcumin in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Paisley, UK) supplemented with 10% foetal calf serum, penicillin/streptomycin (100 iu·mL−1/100 µg·mL−1) and HEPES buffer (25 mM) at 37°C in a humidified 5% CO2 atmosphere. For assessment of HO-1 induction, haemin or curcumin was added at different times before the end of the 24-h incubation period.
HO-1 immunofluorescence
Immunofluorescence was assessed as previously described (Hemmings et al., 2004) after optimization for HO-1 detection. Briefly, at the end of 24-h incubation, pulmonary artery rings were embedded in OCT compound (Raymond A Lamb, East Sussex, UK) and snap-frozen in liquid nitrogen. The frozen sections were cut on a cryostat and consecutive 7 µm sections adhered to Superfrost Plus glass slides (VWR, Leics., UK) and then fixed with 4% paraformaldhyde for 40 min. Perforation was carried out by incubation with cold methanol for 5 min at −20°C. Non-specific binding was blocked by incubating the sections with 70 µL of fluorescence-enhanced blocking buffer (Molecular Probes, Paisley, UK) for 30 min. The sections were then incubated with 70 µL of the rabbit anti-HO-1 primary antibody (dilution 1:200 in 0.2% BSA; StressGen, Ann Arbor, MI, USA) for 1 h at room temperature followed by incubation with 70 µL of the Alexa fluor conjugated goat anti-rabbit secondary antibody (dilution 1:1000 in 0.2% BSA; Molecular Probes, Paisley, UK) for 1 h in the dark. A coverslip was mounted with 20 µL of fluorescence mounting media (Molecular Probes, Paisley, UK) and slides were left in dark for 20 h before examination by Zeiss LSM510 confocal microscope (Carl Zeiss, Göttingen, Germany) with argon laser excitation at 488 nm and a 505 nm longpass emission filter. Sections treated with the secondary antibody alone did not show specific staining.
HO activity assay
Activity of HO was assessed by conversion of haemin to bilirubin as previously described (McNally et al., 2004) with slight modifications. After 24-h incubation, chopped lung tissues were homogenized on ice with 0.1 M phosphate buffer supplemented with 2 mM MgCl2 and the homogenate centrifuged at 1000×g for 10 min at 4°C. The supernatant was added to 400 µL reaction mixtures containing 1 mg of rat liver cytosol, 50 mM haemin, 2 mM glucose 6-phosphate, 0.2 units of glucose-6-phosphate dehydrogenase and 0.8 mM NADPH and incubated for 1 h at 37°C in the dark. Liver cytosol was prepared by homogenizing a rat liver in one volume of 20 mM Tris HCl buffer plus 154 mM KCl, pH 7.4. The homogenate was centrifuged at 10 000×g for 20 min 4°C, and the supernatant was further centrifuged at 100 000×g for 1 h at 4°C. The formed bilirubin was extracted with chloroform and the absorption measured as the difference between 464 and 530 nm (extinction coefficient, 40 mM−1·cm−1 for bilirubin). HO activity is expressed as pmol of bilirubin formed per mg protein per h.
Pulmonary artery relaxation
At the end of 24-h incubation, pulmonary artery rings were suspended under 8 mN resting tension in individual 21 mL organ chambers containing Krebs–Henseleit buffer (containing in mM: NaCl 118.1, KCl 4.69, KH2PO4 1.2, NaHCO3 25.0, glucose 11.7, MgSO4 0.5, CaCl2 2.5) at 37°C and aerated with 95% oxygen, 5% carbon dioxide. Ring tension was determined by use of an isometric force transducer (K30, Hugosachs Elektronik, March, Germany). Force displacement was recorded with a PowerLab Data Interface Module connected to a PC running Chart software (v4.2, ADInstruments, Oxon, UK). Rings were equilibrated for 60 min, during which time the bath solution was changed every 15 min. Before beginning the studies, vessel viability was assessed by exposing arteries to KCl (80 mM). This was repeated until stable responses were achieved (usually two exposures). Rings were then precontracted with a submaximal phenylephrine concentration (3 × 10−7 M, as determined in preliminary experiments). The contraction was allowed to reach plateau and then cumulative concentrations of ACh or sodium nitroprusside (SNP) (10−8 M to 10−5 M) were tested. After washing, rings were then incubated with or without rat TNFα (1 ng·mL−1) in the organ bath for 2 h before repeating the cumulative response curve to ACh. In some experiments, tetrahydrobiopterin (BH4) (3 µM), N[[3(aminomethyl)phenyl]methyl]ethanimidamide dihydrochloride (1400 W) (1 µM) or tempol (1 mM) were added for the final 30 min of the TNFα treatment period. The change in Emax and EC50 in each tissue because of the 2-h incubation was used to compare the effect of treatments.
Determination of intracellular NO and ROS levels
The intracellular levels of NO and ROS were determined by the fluorescent probe 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM) and dihydroethidium (DHE) (Molecular Probes, Paisley, UK) according to the method described by Somers et al. (2000). DAF-FM is virtually nonfluorescent until it reacts with NO to form a fluorescent benzotrizole, while ROS oxidize DHE forming ethidium which forms a highly fluorescent complex with DNA. After 24-h incubation, artery rings were transferred into fresh phenol red-free DMEM media and incubated with or without rat recombinant TNFα (1 ng·mL−1). Rings were then embedded in OCT Compound and snap-frozen in liquid nitrogen. The frozen sections were cut on a cryostat in consecutive 30 µm sections and adhered to a Superfrost Plus glass slide (VWR, Leics., UK). The cryostat sections were then allowed to thaw at room temperature and double loaded for 40 min at room temperature with 5 µM of both DAF-FM and DHE in Krebs–Henseleit buffer containing 25 mM HEPES and 0.1% Pluronic F-127 (Molecular Probes, Paisley, UK). Segments were then washed twice with Krebs–Henseleit buffer, covered with a coverslip and immediately examined by Zeiss LSM510 confocal microscope (Carl Zeiss, Göttingen, Germany) with excitation at 488 nm and a 505–530 nm or 590–615 nm bandpass emission filter systems.
Fluorescence measurements from confocal images
Images were acquired with identical acquisition parameters, with minimum gain to avoid interference by tissue autofluorescence. Quantitative comparisons of fluorescence were made with Image J software (National Institutes of Health, Bethesda, MD, USA). For printing purposes, the contrast and the brightness of the confocal pictures were adjusted equally after the fluorescence measurements were carried out on unmanipulated pictures.
Real-time measurement of intracellular NO and ROS generation
The real time generation of intracellular NO and ROS were investigated with the fluorescence probes DAF-FM diacetate (Itoh et al., 2000) and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, Szabados et al., 1999) respectively. Briefly, at the end of 24-h incubation, artery segments were fixed with stainless steel pins in a 96 black multiwell plate coated with 2 mm thickness layer of Sylgard 184 (Dow Corning, MI, USA). The segments were then loaded for 40 min at room temperature with 5 µM of either DAF-FM diacetate or H2DCFDA plus 0.1% Pluronic F-127 in Krebs–Henseleit buffer containing 25 mM HEPES. Segments were then washed twice and incubated with 200 µL of Krebs–Henseleit buffer containing HEPES for 30 min at 37°C before addition of TNFα (1 ng·mL−1). Readings (λex = 490 nm and λem = 510 − 570 nm) were taken before and every 15 min after cytokine addition in a Fluoroskan II plate reader (ICN-Flow Laboratories, High Wycombe, Oxon, UK). For studying the formation of intracellular nitric oxide because of ACh stimulation, cumulative ACh concentrations (10−6 to 10−4 M) were added after 2-h incubation with TNFα. Readings were taken before and 3 min after each addition of ACh. The NO donor diethylamine NONOate (100 nM) and the ROS stimulus ATP (3 mM) were used as a positive control for DAF and DCF respectively.
Statistical analysis
All data are expressed as mean ± SEM. Statistical analysis was performed by analysis of variance followed by Newman–Keuls' post hoc test. Sigmoidal curve fitting was applied using 3-parameter non-linear regression using Prism 4 software (Graphpad, La Jolla, CA, USA) on a PC.
Materials
The following chemicals were used: rat recombinant TNFα (Peprotec, London, UK), haemin and tin protoporphyrin (SnPP, Frontier, Aberdeen, UK), PE, ACh, BH4, tempol and curcumin (Sigma-Aldrich, Poole, Dorset, UK), 1400 W (Tocris, Bristol, UK). 1400 W, BH4, tempol PE, ACh, ATP and diethylamine NONOate were dissolved in distilled water. Haemin, curcumin and SnPP were freshly dissolved in 0.5 N NaOH in dark and quickly diluted 200-fold with phosphate buffer saline before adding to the media to reach the required concentration.
Results
Assessment of HO-1 induction and inhibition
Haemin (5 µM) and curcumin (2 µM) were able to induce HO-1 protein in pulmonary artery segments (Figure 1) with a significant increases from control values of 3.03 ± 0.03 to 12.38 ± 2.04 (P < 0.01, n = 3) and 7.08 ± 0.55 fluorescence units per pixel at 24 h (P < 0.05, n = 3) for haemin and curcumin-treated tissues respectively. The induction of protein expression was accompanied by an increase in HO enzyme activity, which was apparently increased at 4 h and significantly increased after 24-h incubation with haemin (5 µM, P < 0.01) or curcumin (2 µM, P < 0.05; Figure 2). The induction of HO-1 by haemin and curcumin over this time-course was confirmed by Western blotting (data not shown). The ability of SnPP to inhibit HO was confirmed by incubation of spleen tissue lysate with 20 µM SnPP, which inhibited the HO enzyme activity from 1958 ± 150 to 1083 ± 83 pmole (mg spleen tissue)−1·h−1 in the absence or presence of SnPP respectively.
Figure 1.
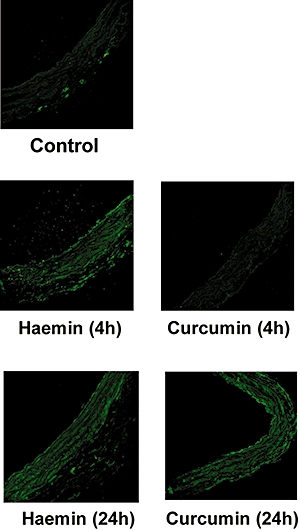
Effect of in vitro incubation with haemin (5 µM) or curcumin (2 µM) on haem oxygenase-1 protein expression after 4- or 24-h incubation, as indicated on the figure, in isolated rat pulmonary artery cryostat sections. Micrographs (325 × 325 µm frame) are representative of data from three independent experiments each using tissue from different animals.
Figure 2.
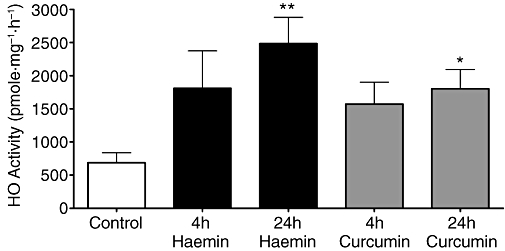
Induction of haem oxygenase activity in rat lung tissue incubated with haemin (5 µM) or curcumin (2 µM). Bars indicate mean ± SEM for n = 3–4 independent determinations. *P < 0.05 and **P < 0.01; significantly different from control. HO, haem oxygenase.
Pulmonary artery relaxation
Addition of cumulative concentrations of ACh to the organ bath resulted in concentration-related decreases in the tension of pulmonary artery rings precontracted with phenylephrine (Figure 3). Incubation of tissues with TNFα (1 ng·mL−1, 2 h) or haemin (5 µM, 24 h) had no effect on the basal tension (data not shown) and similarly these treatments had no significant effect on the contractile response to the submaximal concentration (3 × 10−7 M) of phenylephrine (2.25 ± 0.16 mN and 1.85 ± 0.25 mN compared with 2.28 ± 0.18 mN in controls respectively, n = 7–13). In contrast, incubation with TNFα (1 ng·mL−1; 2 h) resulted in a large reduction in pulmonary artery responsiveness to ACh, reflected by significant changes in apparent Emax (P < 0.001) and pEC50 (P < 0.001) (Figure 3, Table 1).
Figure 3.
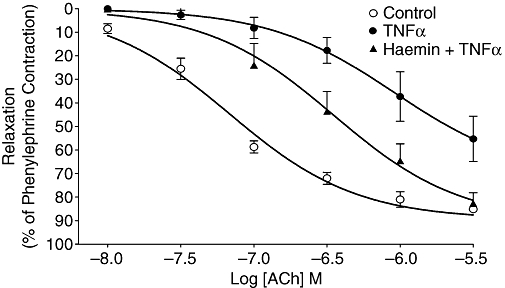
ACh-induced relaxation of phenylephrine-precontracted pulmonary artery rings after 2-h incubation with Krebs–Henseleit buffer (Control), TNFα (1 ng·mL−1), or TNFα (1 ng·mL−1) after 24 h treatment with haemin (5 µM). Symbols indicate mean ± SEM for n = 8–9 tissues from different animals for each treatment group. TNFα, tumour necrosis factor α.
Table 1.
Effect of haemin (5 µM, 24 h), SnPP (20 µM, 24 h), 1400 W (1 µM, 30 min), tempol (1 mM, 30 min) or BH4 (3 µM, 30 min) on ACh Emax and pEC50 in TNFα-treated isolated rat pulmonary artery rings
| Treatment | Δ Emax (% of PE contraction) | Δ pEC50 (log M) |
|---|---|---|
| Control | 2.6 ± 4.3 | 0.35 ± 0.07 |
| TNFα (1 ng·mL−1, 2 h) | 34.4 ± 6.1*** | 1.03 ± 0.17*** |
| TNFα + curcumin | 18.1 ± 3.0† | 0.35 ± 0.07†† |
| TNFα + haemin | 9.7 ± 2.1††† | 0.63 ± 0.17†† |
| TNFα + haemin + SnPP | 28.8 ± 3.2**# | 0.45 ± 0.11† |
| TNFα + 1400 W | 17.3 ± 2.7† | 0.45 ± 0.06†† |
| TNFα + tempol | 5.1 ±3.7††† | 0.50 ± 0.11† |
| TNFα + BH4 | 7.2 ± 5.0††† | 0.51 ± 0.04† |
Values are expressed as the mean ± s.e mean change (Δ) compared with initial ACh curves prior to 2-h control or TNFα incubation in tissues from n = 5–8 different animals.
P < 0.001,
P < 0.01, compared with control tissue responsiveness.
P < 0.05,
P < 0.01,
P < 0.001; compared with tissues treated with TNFα alone.
P < 0.05 compared with tissues treated with TNFα plus haemin; by Student–Newman–Keuls test.
1400 W, N[[3(aminomethyl)phenyl]methyl]ethanimidamide dihydrochloride; BH4, tetrahydrobiopterin; SnPP, tin protoporphyrin; TNFα, tumour necrosis factor α.
The TNFα-induced hyporesponsiveness to ACh was inhibited by preincubation with haemin (5 µM, 24 h; Figure 3 and Table 1). Similarly, HO-1 induction by curcumin (2 µM) was also able to inhibit TNFα-induced hyporesponsiveness significantly (Table 1). Conversely, co-incubation with the HO-1 inhibitor SnPP (20 µM) blocked the protective effect of haemin against the TNFα-induced ACh hyporesponsiveness (Table 1). When added to the organ bath 30 min before the second cumulative ACh response curve, the selective iNOS inhibitor 1400 W (1 µM), the SOD mimetic tempol (1 mM) or the NOS cofactor BH4 (3 µM) inhibited TNFα-induced hyporesponsiveness (Table 1). In the absence of TNFα treatment, incubation of tissues with haemin, curcumin, SnPP (20 µM, 24 h), 1400 W (1 µM, 30 min), tempol (1 mM, 30 min) or BH4 (3 µM, 30 min) had no effect on the response of the pulmonary artery to cumulative concentrations (3 × 10−8−3 × 10−6 M) of ACh. Also, TNFα incubation did not affect the sensitivity of pulmonary artery to SNP (data not shown).
Determination of intracellular NO level and ROS levels
Incubation of pulmonary arterial rings with TNFα (1 ng·mL−1, 2 h) was able to increase the intracellular level of NO in cryostat sections from control by about threefold (Figure 4). In the same sections, ROS levels were also increased, to about double control values (Figure 4). Induction of HO-1, by pre-incubation with haemin (5 µM, 24 h), totally prevented this TNFα-induced increase in intracellular NO (P < 0.05; Figure 4) but did not have a significant effect on the TNFα-induced increase in intracellular ROS.
Figure 4.
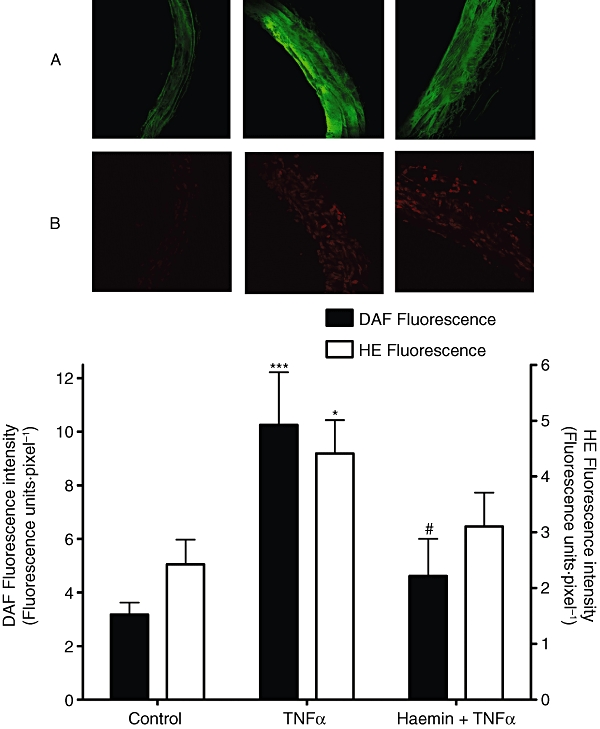
Effect of preincubation with haemin (5 µM, 24 h) on TNFα (1 ng·mL−1, 2 h) induced increases in A. intracellular NO and B. ROS levels. Sections are shown with the lumen on the left side of the frames. HE is oxidized by intracellular ROS forming a fluorescent complex with the DNA in the nuclei while DAF fluorescent cells were distributed throughout the endothelial and smooth muscle layers of the tissue sections. Bars indicate mean ± SEM for n = 3–5 tissues from different animals. *P < 0.05, ***P < 0.001; significantly different from control, #P < 0.05; significantly different from TNFα-treated tissue. DAF, 4-amino-5-methylamino-2′,7′-difluorofluorescein; HE, hydroethidium; NO, nitric oxide; ROS, reactive oxygen species; TNFα, tumour necrosis factor α.
Real-time measurement of intracellular NO generation
Tumour necrosis factor α (1 ng·mL−1) significantly increased NO generation in pulmonary artery segments (Figure 5; P < 0.001), and this increase was completely blocked by either 1400 W (1 µM, 30 min; P < 0.001) or HO-1 induction by haemin (5 µM, 24 h, P < 0.001). In addition, TNFα significantly increased the ROS generation in the pulmonary artery segments (Figure 5; P < 0.001), but HO-1 induction by haemin had no effect on this TNFα-induced increase in ROS generation.
Figure 5.
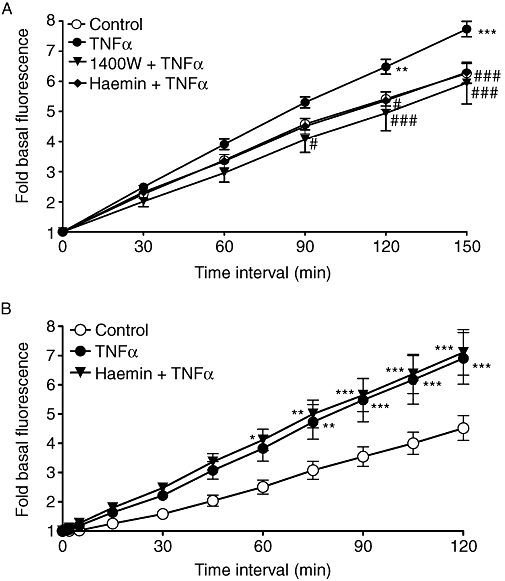
Effect of TNFα (1 ng·mL−1) on the rate of (A) NO and (B) ROS formation in isolated rat pulmonary artery segments, and inhibition of the TNFα response by 1400 W (1 µM) or haemin (5 µM). *P < 0.05; **P < 0.01; ***P < 0.001; significantly different from the corresponding control values; #P < 0.01; ###P < 0.001; significantly different from the corresponding values in TNFα-treated tissue; n = 8–12 tissues from different animals. 1400 W, N[[3(aminomethyl)phenyl]methyl]ethanimidamide dihydrochloride; NO, nitric oxide; ROS, reactive oxygen species; TNFα, tumour necrosis factor α.
Although TNFα induced NO generation (above), it inhibited NO generation stimulated by 3-min exposure to 10−4 M ACh (Figure 6; P < 0.001). 1400 W (1 µM, 30 min, P < 0.05), BH4 (3 µM, 30 min, P < 0.01) or HO-1 induction by haemin (5 µM, 24 h, P < 0.05) was able to prevent this inhibition (Figure 6). However, tempol (1 mM, 30 min) did not have a significant effect on the TNFα-induced decrease in intracellular NO generation in response to ACh stimulation.
Figure 6.
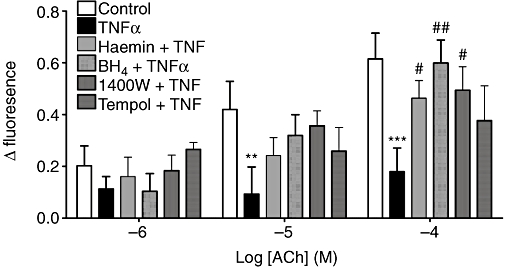
Effect of TNFα (1 ng·mL−1, 2 h) on NO formation in isolated rat pulmonary artery segments with or without haemin (5 µM, 24 h), BH4 (3 µM, 2 h), 1400 W (1 µM, 2 h), or tempol (1 mM, 2 h) in response to cumulative concentrations of ACh (10−6 to 10−4 M). The Δ fluorescence is the fluorescence 2 min after each ACh addition after subtracting the fluorescence at 0 ACh (as assessed after 2 h cytokine pretreatment) and dividing by the basal fluorescence of each vessel for normalization. **P < 0.01; ***P < 0.001; significantly different from the corresponding values in control tissue; #P < 0.05; ##P < 0.01; significantly different from the corresponding values after TNFα; n = 6–10 tissues from different animals. 1400 W, N[[3(aminomethyl)phenyl]methyl]ethanimidamide dihydrochloride; BH4, tetrahydrobiopterin; NO, nitric oxide; TNFα, tumour necrosis factor α.
Discussion
The purpose of this study was to investigate the potentially protective effect of HO-1 against TNFα-induced impairment in pulmonary artery relaxation. We have succeeded in inducing HO-1 enzyme in rat pulmonary artery in vitro. We have shown, for the first time, that induction of HO-1 protein and activity was associated with protection against cytokine-induced dysfunction of pulmonary artery relaxation and that this protection was mediated by modulation of NO production in arterial tissue. These findings suggest upregulation of HO-1 as a novel potential therapy in the management of vascular disturbances associated with inflammatory lung disease.
HO-1 enzyme was induced in our system by haemin or curcumin. Haemin is a strong inducer of HO-1 expression in a variety of cell types both in vitro (Lang et al., 2005) and in vivo (Attuwaybi et al., 2004). The present study indicated that in vitro treatment of pulmonary artery with haemin leads to a time-dependent increase in HO-1 expression and activity. Curcumin has been described as a potent HO-1 inducer in bovine aortic endothelial cells (Motterlini et al., 2000), astrocytes (Scapagnini et al., 2002) and in vivo (Farombi et al., 2008).
After optimizing the conditions for HO-1 induction and inhibition in our system, we investigated the potentially protective effect of HO-1 against TNFα-induced impairment of endothelium-dependent relaxation in pulmonary artery. HO-1 induction by haemin significantly inhibited the TNFα-induced hyporesponsiveness to ACh without affecting responses of the pulmonary artery to SNP. These results indicate that HO-1 induction can protect against the impairment of endothelium-dependent relaxation in pulmonary artery, without modifying the sensitivity of pulmonary artery smooth muscle to NO.
The HO-1 enzyme can be inhibited by a variety of synthetic metalloporphyrins (Ryter et al., 2006), including SnPP. SNP is a potent competitive inhibitor of HO that has been widely used in vivo (Gueler et al., 2007) and in vitro (Evans et al., 2007). In our study, we found that SnPP at concentration of 20 µM was able to inhibit HO activity in spleen tissue lysate, and this concentration was employed in the pulmonary artery experiments. Co-incubation with SnPP blocked the protective effect of haemin against TNFα-induced hyporesponsiveness, while neither haemin nor SnPP had any direct effect on the relaxation when applied in control experiments. These data confirm that the observed protection was due to the HO-1 activity and was not a consequence of a direct effect of haemin on smooth muscle contraction.
In chronic hypoxia models, induction of HO-1 prevents the development of pulmonary hypertension in the rat (Christou et al., 2000), and attenuates lipopolysaccharide (LPS)-induced acute lung injury in mice (Gong et al., 2008). Increases of vascular HO-1 expression have been observed in several experimental models of hypertension including angiotensin II-induced hypertension (Li et al., 2004), spontaneously hypertensive rats (SHRs) (Cheng et al., 2004), Dahl-Rapp salt-sensitive rats (Teran et al., 2005), and deoxycorticosterone salt-sensitive rats (Johnson et al., 2004). The HO-1/CO system has been associated with antihypertensive effects (Ndisang et al., 2004) as well as pathological roles in the case of salt-induced hypertension models (Teran et al., 2005). Administration of exogenous CO or haem derivatives reduces blood pressure in SHR (Johnson et al., 1996) and human HO-1 gene delivery by vector-directed gene therapy reduces blood pressure in young SHR (Sabaawy et al., 2001). HO-1 induction alleviates the high glucose-induced, endothelium-dependent, acute vascular dysfunction in rat aortic rings (Meng et al., 2009). Conversely, application of metalloporphyrins (zinc proporphyrin IX, chromium mesoporphyrin) reverse the antihypertensive effects of haem and also elevated systolic blood pressure in SHR (Cheng et al., 2004). Taken together, these findings strongly suggest that induction of HO-1 expression is a compensatory response during the development of hypertension (Ryter et al., 2006).
The roles of NO and ROS in TNFα-induced impairment of endothelium-dependent relaxation in pulmonary artery were assessed to help explain the observed HO-1 protection. We found that the intracellular levels and rate of generation of both NO and ROS were significantly increased by TNFα. The highly selective iNOS inhibitor 1400 W and the superoxide scavenger, tempol, blocked the TNFα-induced attenuation of the vascular response to ACh and also ACh-stimulated NO release. These results suggest that generation of NO and ROS mediated the impairment in endothelium-dependent relaxation. Previously a role for iNOS-derived NO in the impairment of endothelium-dependent relaxation has been demonstrated in rat middle cerebral arteries following LPS treatment (Hernanz et al., 2004). Also, an increase in superoxide production after LPS has been reported in rat aorta (Brandes et al., 1999) and middle cerebral artery (Hernanz et al., 2003). Wimalasundera et al. (2003) reported that preincubation with SOD prevents the TNFα attenuation of endothelium-dependent relaxation in rat mesenteric artery.
Preincubation with 1400 W completely blocked the TNFα-induced increase in the rate of NO generation, indicating a role for iNOS. We did not directly assess iNOS protein expression in these experiments, because we did not anticipate increased expression levels over the 2-h treatment period used in these experiments. Previous studies have indicated that iNOS mRNA level starts to increase only 4 h after stimulation (Lowenstein et al., 1992). However, the effect of iNOS-derived NO production starts to affect the relaxation before a significant expression of the iNOS protein can be detected by immunoblotting. Hernanz et al. (2003) found that the iNOS inhibitor aminoguanidine increased serotonin-induced contraction from the first hour (statistically significant at hour 3) of LPS incubation while increased iNOS protein expression was only detected after 5 h.
Generated superoxide interacts with NO forming peroxynitrite (Laursen et al., 2001). Peroxynitrite is a potent oxidant that can switch eNOS, via the oxidation of BH4, from a NO-generating to a superoxide-generating enzyme (a process termed NOS uncoupling) (Landmesser et al., 2003). This is supported by the observation that the NOS cofactor BH4 blocked the TNFα-induced attenuation in the vascular response to ACh and ACh-stimulated NO generation.
The mechanism(s) by which HO-1 induction protects against the impairment in endothelium-dependent relaxation could be mediated, at least in part, by modulation of NO production. Chemical inhibition of HO activity by zinc protoporphyrin in macrophages (Turcanu et al., 1998) results in increased NO production, suggesting that HO may exert an inhibitory effect on NOS. These findings are supported by the observation that induction of HO-1 in human renal epithelial cells suppressed cytokine-induced NO generation (Svensson et al., 2009) and inhibited NO generation in macrophages (Kim et al., 2008). As NOS is a haemoprotein, it is reasonable to postulate that CO generated by HO activity could bind to NOS, causing inactivation. CO is capable of binding to NOS (McMillan et al., 1992), and exogenously administered CO inhibits NOS activity (White and Marletta, 1992). In this study, we found that HO-1 induction by haemin was able to prevent the TNFα-induced increase in the intracellular level and rate of generation of NO as well as the TNFα-induced decrease in ACh-stimulated NO generation. However, HO-1 induction did not affect the TNFα-induced increase in the intracellular ROS level or the rate of ROS generation.
In summary, HO-1 induction protects against inflammatory cytokine-induced impairment of endothelium-dependent relaxation. Inhibition of iNOS is a likely underlying mechanism of the protective effect of HO-1. These findings suggest therapeutic strategies that increase HO-1 activity are likely to be beneficial in inflammatory lung disease
Acknowledgments
We are grateful to Dr Adrian Rogers and Dr James Hewinson for technical assistance with confocal microscopy and real-time measurement of intracellular NO and ROS generation. Hany M. El-Bassossy was funded by an Egyptian Government Channel Scholarship.
Glossary
Abbreviations:
- 1400 W
N[[3(aminomethyl)phenyl]methyl]ethanimidamide dihydrochloride
- BH4
tetrahydrobiopterin
- DAF-FM
4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate
- DHE
dihydroethidium
- eNOS
endothelial nitric oxide synthase
- HO-1
haem oxygenase-1
- iNOS
inducible nitric oxide synthase
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rat
- SNP
sodium nitroprusside
- SnPP
tin protoporphyrin
- SOD
superoxide dismutase
- TNFα
tumour necrosis factor α
Conflict of interest
The authors state no conflict of interest.
References
- Attuwaybi BO, Kozar RA, Moore-Olufemi SD, Sato N, Hassoun HT, Weisbrodt NW, et al. Heme oxygenase-1 induction by hemin protects against gut ischemia/reperfusion injury. J Surg Res. 2004;118:53–57. doi: 10.1016/j.jss.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Koddenberg G, Gwinner W. Role of increased production of superoxide anions by NAD(P)H oxidase and xanthine oxidase in prolonged endotoxemia. Hypertension. 1999;33:243–1249. doi: 10.1161/01.hyp.33.5.1243. [DOI] [PubMed] [Google Scholar]
- Cheng PY, Chen JJ, Yen MH. The expression of heme oxygenase-1 and inducible nitric oxide synthase in aorta during the development of hypertension in spontaneously hypertensive rats. Am J Hypertens. 2004;17:1127–1134. doi: 10.1016/j.amjhyper.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Christou H, Morita T, Hsieh C, Koike H, Arkonac B, Perrella MA, et al. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ Res. 2000;86:1224–1229. doi: 10.1161/01.res.86.12.1224. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Evans JP, Xu FY, Sirisawad M, Miller R, Naumovski L, de Montellano PRO. Motexafin gadolinium-induced cell death correlates with heme oxygenase-1 expression and inhibition of P450 reductase-dependent activities. Mol Pharmacol. 2007;71:193–200. doi: 10.1124/mol.106.028407. [DOI] [PubMed] [Google Scholar]
- Farombi EO, Shrotriya S, Na HK, Kim SH, Surh YJ. Curcumin attenuates dimethylnitrosamine-induced liver injury in rats through Nrf2-mediated induction of heme oxygenase-1. Food Chem Toxicol. 2008;46:1279–1287. doi: 10.1016/j.fct.2007.09.095. [DOI] [PubMed] [Google Scholar]
- Gong Q, Yin H, Fang M, Xiang Y, Yuan CL, Zheng GY, et al. Heme oxygenase-1 upregulation significantly inhibits TNF-α and Hmgb1 releasing and attenuates lipopolysaccharide–induced acute lung injury in mice. Int Immunopharmacol. 2008;8:792–798. doi: 10.1016/j.intimp.2008.01.026. [DOI] [PubMed] [Google Scholar]
- Gueler F, Park JK, Rong S, Kirsch T, Lindschau C, Zheng W, et al. Statins attenuate ischemia-reperfusion injury by inducing heme oxygenase-1 in infiltrating macrophages. Am J Pathol. 2007;170:1192–1199. doi: 10.2353/ajpath.2007.060782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings DG, Xu Y, Davidge ST. Sphingosine 1-phosphate-induced vasoconstriction is elevated in mesenteric resistance arteries from aged female rats. Br J Pharmacol. 2004;143:276–284. doi: 10.1038/sj.bjp.0705752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernanz R, Alonso MJ, Briones AM, Vila E, Simonsen U, Salaices M. Mechanisms involved in the early increase of serotonin contraction evoked by endotoxin in rat middle cerebral arteries. Br J Pharmacol. 2003;140:671–680. doi: 10.1038/sj.bjp.0705501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernanz R, Briones AM, Alonso MAJ, Vila E, Salaices M. Hypertension alters role of iNOS, COX-2, and oxidative stress in bradykinin relaxation impairment after LPS in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2004;287:H225–H234. doi: 10.1152/ajpheart.00548.2003. [DOI] [PubMed] [Google Scholar]
- Hida W, Tun Y, Kikuchi Y, Okabe S, Shirato K. Pulmonary hypertension in patients with chronic obstructive pulmonary disease: recent advances in pathophysiology and management. Respirology. 2002;7:3–13. doi: 10.1046/j.1440-1843.2002.00366.x. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Ma FH, Hoshi H, Oka M, Noda K, Ukai Y, et al. Determination and bioimaging method for nitric oxide in biological specimens by diaminofluorescein fluorometry. Anal Biochem. 2000;287:203–209. doi: 10.1006/abio.2000.4859. [DOI] [PubMed] [Google Scholar]
- Johnson FK, Durante W, Peyton KJ, Johnson RA. Heme oxygenase-mediated endothelial dysfunction in DOCA-salt, but not in spontaneously hypertensive, rat arterioles. Am J Physiol Heart Circ Physiol. 2004;286:H1681–H1687. doi: 10.1152/ajpheart.00409.2003. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Lavesa M, DeSeyn K, Scholer MJ, Nasjletti A. Heme oxygenase substrates acutely lower blood pressure in hypertensive rats. Am J Physiol Heart Circ Physiol. 1996;40:H1132–H1138. doi: 10.1152/ajpheart.1996.271.3.H1132. [DOI] [PubMed] [Google Scholar]
- Kim KM, Pae HO, Zhung M. Involvement of anti-inflammatory heme oxygenase-1 in the inhibitory effect of curcumin on the expression of pro-inflammatory inducible nitric oxide synthase in RAW264.7 macrophages. Biomed Pharmacother. 2008;62:630–636. doi: 10.1016/j.biopha.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D, Reuter S, Buzescu T, August C, Heidenreich S. Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 up-regulation. Int Immunol. 2005;17:155–165. doi: 10.1093/intimm/dxh196. [DOI] [PubMed] [Google Scholar]
- Laursen J, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- Li P, Jiang HL, Yang LM, Quan S, Dinocca S, Rodriguez F, et al. Angiotensin II induces carbon monoxide production in the perfused kidney: relationship to protein kinase C activation. Am J Physiol Renal Physiol. 2004;287:F914–F920. doi: 10.1152/ajprenal.00073.2004. [DOI] [PubMed] [Google Scholar]
- Lowenstein CJ, Glatt CS, Bredt DS, Snyder SH. Cloned and expressed macrophage nitric-oxide synthase contrasts with the brain enzyme. Proc Nail Acad Sci USA. 1992;89:6711–6715. doi: 10.1073/pnas.89.15.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- McMillan K, Bredt DS, Hirsch DJ, Snyder SH, Clark JE, Masters BSS. Cloned, expressed rat cerebellar nitric-oxide synthase contains stoichiometric amounts of heme, which binds carbon-monoxide. Proc Natl Acad Sci USA. 1992;89:11141–11145. doi: 10.1073/pnas.89.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally SJ, Ross JA, James Garden O, Wigmore SJ. Optimization of the paired enzyme assay for heme oxygenase activity. Anal Biochem. 2004;332:398–400. doi: 10.1016/j.ab.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Meng Xh, Ni C, Zhu L, Shen Yl, Wang Ll, Chen Y. Puerarin protects against high glucose-induced acute vascular dysfunction: role of heme oxygenase-1 in rat thoracic aorta. Vasc Pharmacol. 2009;50:110–115. doi: 10.1016/j.vph.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Morse D, Choi AMK. Heme oxygenase-1 – from bench to bedside. Am J Respir Crit Care Med. 2005;172:660–670. doi: 10.1164/rccm.200404-465SO. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Foresti R, Bassi R, Green CJ. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic Biol Med. 2000;28:1303–1312. doi: 10.1016/s0891-5849(00)00294-x. [DOI] [PubMed] [Google Scholar]
- Ndisang JF, Tabien HEN, Wang R. Carbon monoxide and hypertension. J Hypertens. 2004;22:1057–1074. doi: 10.1097/00004872-200406000-00002. [DOI] [PubMed] [Google Scholar]
- Peinado VI, Barbera JA, Ramirez J, Gomez FP, Roca J, Jover L, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol Lung Cell Mol Physiol. 1998;274:L908–L913.. doi: 10.1152/ajplung.1998.274.6.L908. [DOI] [PubMed] [Google Scholar]
- Ricciardolo FLM, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84:731–765. doi: 10.1152/physrev.00034.2003. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- Sabaawy HE, Zhang F, Nguyen XD, ElHosseiny A, Nasjletti A, Schwartzman M, et al. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension. 2001;38:210–215. doi: 10.1161/01.hyp.38.2.210. [DOI] [PubMed] [Google Scholar]
- Scapagnini G, Foresti R, Calabrese V, Stella AMG, Green CJ, Motterlini R. Caffeic acid phenethyl ester and curcumin: A novel class of heme oxygenase-1 inducers. Mol Pharmacol. 2002;61:554–561. doi: 10.1124/mol.61.3.554. [DOI] [PubMed] [Google Scholar]
- Sheridan BC, Mcintyre RC, Agrafojo J, Meldrum DR, Meng XZ, Fullerton DA. Neutrophil depletion attenuates endotoxin-induced dysfunction of cGMP-mediated pulmonary vasorelaxation. Am J Physiol Lung Cell Mol Physiol. 1996;15:L820–L828. doi: 10.1152/ajplung.1996.271.5.L820. [DOI] [PubMed] [Google Scholar]
- Soares MP, Bach FH. Heme oxygenase-1: from biology to therapeutic potential. Trends Mol Med. 2009;15:50–58. doi: 10.1016/j.molmed.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Somers MJ, Mavromatis K, Galis ZS, Harrison DG. Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation. 2000;101:1722–1728. doi: 10.1161/01.cir.101.14.1722. [DOI] [PubMed] [Google Scholar]
- Svensson L, Mohlin C, Persson K. Upregulation of heme oxygenase-1 as a host mechanism for protection against nitric oxide-induced damage in human renal epithelial cells. Urology. 2009;73:1150–1155. doi: 10.1016/j.urology.2008.02.027. [DOI] [PubMed] [Google Scholar]
- Szabados E, Fischer GM, Toth K, Csete B, Nemeti B, Trombitos K, et al. Role of reactive oxygen species and poly–ADP–ribose polymerase in the development of AZT–induced cardiomyopathy in rat. Free Radical Biol Med. 1999;26:309–317. doi: 10.1016/s0891-5849(98)00199-3. [DOI] [PubMed] [Google Scholar]
- Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- Teran FJ, Johnson RA, Stevenson BK, Peyton KJ, Jackson KE, Appleton SD, et al. Heme oxygenase-derived carbon monoxide promotes arteriolar endothelial dysfunction and contributes to salt-induced hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R615–R622. doi: 10.1152/ajpregu.00123.2004. [DOI] [PubMed] [Google Scholar]
- Turcanu V, Dhouib M, Poindron P. Nitric oxide synthase inhibition by haem oxygenase decreases macrophage nitric-oxide-dependent cytotoxicity: a negative feedback mechanism for the regulation of nitric oxide production. Res Immunol. 1998;149:741–744. doi: 10.1016/s0923-2494(99)80050-9. [DOI] [PubMed] [Google Scholar]
- Vila E, Salaices M. Cytokines and vascular reactivity in resistance arteries. Am J Physiol Heart Circ Physiol. 2005;288:H1016–H1021. doi: 10.1152/ajpheart.00779.2004. [DOI] [PubMed] [Google Scholar]
- White KA, Marletta MA. Nitric-oxide synthase is a cytochrome-P-450 type hemoprotein. Biochemistry (Mosc) 1992;31:6627–6631. doi: 10.1021/bi00144a001. [DOI] [PubMed] [Google Scholar]
- Wimalasundera R, Fexby S, Regan L, Hughes AD. Effect of tumour necrosis factor-alpha and interleukin 1 beta on endothelium-dependent relaxation in rat mesenteric resistance arteries in vitro. Br J Pharmacol. 2003;138:1285–1294. doi: 10.1038/sj.bjp.0705168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters EFM. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:26–33. doi: 10.1513/pats.200408-039MS. [DOI] [PubMed] [Google Scholar]