

Abstract
Background and purpose:
Marfan syndrome, a connective tissue disorder caused by mutations in FBN1 encoding fibrillin-1, results in life-threatening complications in the aorta, but little is known about its effects in resistance vasculature.
Experimental approach:
Second-order mesenteric arteries from mice at 3, 6 and 10 months of age (n= 30) heterozygous for the Fbn1 allele encoding a cysteine substitution (Fbn1C1039G/+) were compared with those from age-matched control littermates.
Key results:
Stress–strain curves indicated that arterial stiffness was increased at 6 and 10 months of age in Marfan vessels. Isometric force measurement revealed that contraction in response to potassium (60 mM)-induced membrane depolarization was decreased by at least 28% in Marfan vessels at all ages, while phenylephrine (3 µM)-induced contraction was reduced by at least 40% from 6 months. Acetylcholine-induced relaxation in Marfan vessels was reduced to 70% and 45% of control values, respectively, at 6 and 10 months. Sensitivity to sodium nitroprusside was reduced at 6 months (pEC50= 5.64 ± 0.11, control pEC50= 7.34 ± 0.04) and 10 months (pEC50= 5.99 ± 0.07, control pEC50= 6.99 ± 0.14). Pretreatment with Nω-Nitro-L-arginine methyl ester (200 µM) had no effect on acetylcholine-induced relaxation in Marfan vessels, but reduced vasorelaxation in control vessels to 57% of control values. Addition of indomethacin (10 µM) and catalase (1000 U·mL−1) further inhibited vasorelaxation in Marfan vessels to a greater degree compared with control vessels.
Conclusions and implications:
Pathogenesis of Marfan syndrome in resistance-sized arteries increases stiffness and impairs vasomotor function.
Keywords: Marfan syndrome, mesenteric artery, vascular smooth muscle, elasticity, endothelium-dependent relaxation
Introduction
Marfan syndrome is an autosomal dominant disorder caused by mutations in the gene encoding for fibrillin-1 and affects many tissues including those of the cardiovascular, skeletal, ocular and pulmonary systems (Dietz et al., 1991; Pyeritz, 2000; Judge and Dietz, 2005). Fibrillin-1 is the structural glycoprotein for microfibrils, which act as scaffolding proteins for elastin deposition and formation of elastic fibres (Reinhardt et al., 1995). Abnormalities in the formation and integrity of elastic fibres in Marfan syndrome cause weakening of the blood vessel walls and are especially pronounced in the aorta due to its high (∼50%) elastin content, which normally allows it to buffer pressure variations during the cardiac cycle and permit constant blood flow and organ perfusion (Rosenbloom, 1993; Safar and London, 1994). Weakening of the aortic wall leads to root dilatation, dissection and eventual rupture, the major cause of death in patients with Marfan syndrome (Murdoch et al., 1972).
Marfan syndrome is associated not only with extensive degeneration of elastic fibres, but also with endothelial dysfunction and reduction of smooth muscle contractility in the vasculature (Chung et al., 2007a,b;). The alteration of the structural integrity of elastic fibres leads to reduced distensibility and elasticity (Bunton et al., 2001). Furthermore, alteration of fibrillin-1 may also disrupt the attachment of elastic fibres to the cells in the endothelial layer and impair endothelial permeability (Davis, 1994; Sheremet'eva et al., 2004). Although elastic fibre composition is gradually reduced along the arterial tree, elastin remains an important determinant of passive mechanical properties in mesenteric arteries (Dobrin, 1978; Milnor, 1989; Mulvany and Aalkjaer, 1990; Briones et al., 2003; González et al., 2005). However, little is known about how Marfan syndrome affects vessel elasticity and vasomotor function in the resistance vasculature, although dysfunction of these vessels may have important clinical consequences. For example, aneurysms in peripheral and resistance vessels have been reported in patients with Marfan syndrome (Savolainen et al., 1993; Hatrick et al., 1998; Goffi et al., 2000; Lay et al., 2006), although no clear link has been established between resistance artery dysfunction and aortic dilatation and rupture (Jondeau et al., 1999). Furthermore, maximum forearm blood flow in response to acetylcholine (ACh) is reduced in patients with Marfan syndrome (Nakamura et al., 2000), and impairment in flow-mediated vasodilation is also observed (Wilson et al., 1999).
In the present study, we compared the stiffness and vascular function of resistance-sized mesenteric arteries from a mouse model of Marfan syndrome with those from their wild-type littermates. We conclude that during the progression of Marfan syndrome, mesenteric arteries show signs of increased stiffness. Furthermore, the contractile function of smooth muscle cells and endothelium-dependent and endothelium-independent vasorelaxation are all markedly impaired. Therefore, we suggest that Marfan syndrome should be considered as a disorder not only in the aorta, but also in the peripheral resistance vasculature.
Methods
Experimental animals and tissue preparation
All animal care and experimental procedures were approved by the institutional Animal Ethics Board. Heterozygous (Fbn1C1039G/+) mice were mated to C57BL/6 mice to produce equal numbers of Fbn1C1039G/+ Marfan subjects and wild-type controls as described previously (Judge et al., 2004; Ng et al., 2004; Habashi et al., 2006; Chung et al., 2007a,b;). Both strains were housed in the institutional animal facility (Child and Family Research Institute, University of British Columbia) under standard animal room conditions (12 h light–12 h dark, at 25°C, 2–5 in a cage, Purina Chow diet). Mice at ages 3 (n= 30), 6 (n= 30), and 10 (n= 30) months were anaesthetized with a mixture of ketamine hydrochloride (80 mg·kg−1) and xylazine hydrochloride (12 mg·kg−1) given i.p. for experiments. The mesenteric arcade was excised and placed in ice-cold oxygenated (95% O2–5% CO2) HEPES-PSS solution (see below for composition). Second-order branches of the mesenteric artery with diameters 130–150 µm were dissected and cut into 2 mm segments.
Mechanical properties
‘Vessel elasticity’ was deduced from the stress–strain curves. In a small-vessel myograph (A/S Danish Myotechnology, Aarhus, Denmark), a 2 mm mesenteric artery segment was stretched by increasing the distance between the two stainless steel wires (=increase in length of vascular smooth muscle cell) and held at each length for 1 min. The chambers were kept at 37°C and bubbled continuously with 95% O2–5% CO2 in HEPES-PSS solution. Initially, the two wires were adjusted to Lo, at which the vessel was not stretched. The inside circumference of the mesenteric segment was measured as twice the distance between the two wires, plus the wire circumference, plus two wire radii (2 × 40 µm). The distance between the two wires was then increased by 25 µm, and the new length was denoted as ‘L’. The developed force (mN) was divided by the surface area (=inside circumference of the segment × length of the segment) of the blood vessel segment (mm2) to calculate the wall stress (mN·mm−2). The procedure was repeated until the vessel was unable to maintain its tone. The ΔL/L0 and the wall stress were fitted on an exponential curve. ‘Passive force’ was measured by repeating the above procedures in a calcium-free HEPES-PSS solution prepared by replacing CaCl2 with 320 µM EGTA to eliminate smooth muscle cell contractility. ‘Total force’ was determined by assessing the active contractility at each level of stretch in response to depolarization (60 mM KCl).
To study the ‘reversibility of vessel contractility’ after stretching, the mesenteric segment was stimulated with 60 mM KCl at the optimal tension (ΔL/L0 is approximately equal to 2.0, a value that gives the maximal force generation in response to KCl), then stretched to either ΔL/L0= 2.5, 3.0, 3.5 or 4.0 for 3 min, and restored to optimal tension. Contraction was induced after 3 min, and the percentage of developed force change compared with the optimal tension was calculated.
Measurement of isometric force
Mesenteric artery segments were mounted isometrically in a small-vessel wire myograph (A/S Danish Myotechnology, Aarhus N, Denmark) for measuring generated force. The chambers were kept at 37°C and bubbled continuously with 95% O2–5% CO2 in HEPES-PSS solution. Optimal tension was determined in preliminary experiments by subjecting arterial segments to different resting tensions and stimulating with 60 mM KCl. The vessels were stretched to the optimal tension (the maximal force generation given in response to 60 mM KCl, which was the same for control and Marfan mouse mesenteric arteries; 4 mN) for 60 min. The vessels were challenged twice with 60 mM KCl before experiments were continued.
Concentration–response curves of phenylephrine-induced contraction were constructed. The negative logarithm (pD2) of the concentration of phenylephrine giving half-maximum response (EC50) was assessed by linear interpolation on the semi-logarithm concentration–response curve [pD2=−log(EC50)]. To determine endothelium-dependent and endothelium-independent relaxations, vessels were pre-contracted with 3 µM phenylephrine before making cumulative applications of ACh or sodium nitroprusside (SNP; 1 nM–10 µM) respectively. Control concentration response curves to ACh were produced and compared with those in vessels pretreated with Nω-Nitro-L-arginine methyl ester (L-NAME) (200 µM), indomethacin (10 µM) or catalase (1000 U·mL−1) for 30 min. Other vessels were pretreated with L-NAME (200 µM) and indomethacin (10 µM) for 30 min and concentration–response curves to ACh were produced. These responses were then repeated in the presence of catalase (1000 U·mL−1) or carbenoxolone (100 µM). Per cent relaxation was calculated as the per cent decrease in force with respect to the initial phenylephrine (3 µM)-induced precontraction, and the per cent relaxation was used to construct the concentration–response curves of ACh-induced relaxation.
Detection of H2O2 production from endothelial cells
Mesenteric artery segments of control and Marfan mice were cut into rings and then opened longitudinally. The vascular strip was loaded with dichlorodihydrofluorescein diacetate (DCF; 5 µM), a peroxide-sensitive fluorescence dye (Ohba et al., 1994), for 10 min at 25°C. Images were acquired on an upright Olympus BX50WI microscope with a 60× water-dipping objective (NA 0.9) equipped with an Ultraview confocal imaging system (Perkin-Elmer, USA). The tissue was illuminated using the 488 nm line of an argon-krypton laser and a high-gain photomultiplier tube collected the emission at wavelengths between 505 and 550 nm. The tissue was preincubated with indomethacin (10 µM) and L-NAME (200 µM) for 30 min, then stimulated with ACh (10 µM). Additionally, the effect of catalase (1000 U·mL−1) on the ACh-induced increase in fluorescence intensities was also determined.
Statistics
Values are expressed as mean ± standard error from at least four independent experiments. Statistical analysis and construction of concentration–response curves were performed using GraphPad Prism 4.0 software (San Diego, CA, USA). Differences between control and Marfan groups were analysed by Student's two-tailed t-test. Differences between concentration–response curves were analysed by two-way anova. Statistical significance was defined as P-values <0.05.
Materials
HEPES-PSS containing (in mM) NaCl 130, HEPES 10, glucose 6, KCl 4, NaHCO3 4, CaCl2 1.8, MgSO4 1.2, KH2PO4 1.18 and EDTA 0.03 (pH 7.4) was used for all studies. High-K+ (60 mM extracellular K+) PSS was identical in composition to normal PSS with the exception of (in mM) NaCl 74 and KCl 60. Phenylephrine, ACh, SNP, L-NAME, catalase, carbenoxolone and indomethacin were obtained from Sigma-Aldrich (Oakville, Ontario, Canada). DCF was obtained from Molecular Probes (Eugene, OR, USA). Superoxide dismutase was obtained from Calbiochem (San Diego, CA, USA).
Results
Vessel stiffening and weakness in Marfan mesenteric artery
In the measurement of stiffness, stress increases exponentially as a function of vessel diameter. At 3 months of age, the fitted curves for the stress–strain relationship from control and Marfan vessels were not significantly different (Figure 1A). However, at 6 and 10 months of age, the slope of the stress–strain curves from the Marfan vessels was increased compared with that of the control vessels, which indicates increased stiffness (Figure 1B and C).
Figure 1.
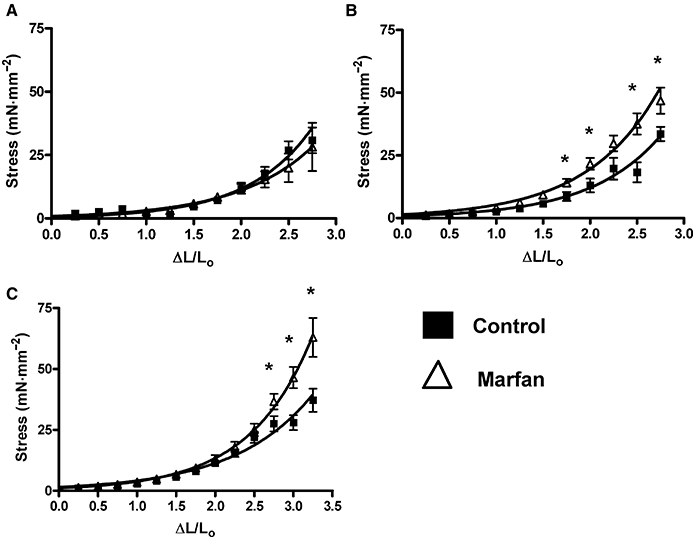
Vessel elasticity during ageing in mesenteric arteries from Marfan and control mice from (A) 3, (B) 6 and (C) 10 months of age (*P < 0.05 vs. control, n= 8–12).
However, true elasticity also implies the capability to return to the original conformation or length, a situation analogous to an elastic band. To test this elasticity, we measured the ‘reversibility of mesenteric artery elasticity’ by comparing two stress–strain curves from control and Marfan vessels at 6 and 10 months of age. As we did not observe any increase in stiffness in the Marfan vessels at 3 months of age, these measurements were not done in this group. We found that the apparent vessel elasticity in the second measurement remained for the most part unchanged in the control vessels, but was highly increased in the Marfan vessels (Figure 2). This may suggest a weakening of the vessel wall at 10 months of age, and a similar observation was also found at 6 months of age (data not shown). We also measured the effects of stretch on contractility in 6- and 10-month-old mice. After distending to ΔL/L0= 2.5, the KCl-induced contraction in the Marfan vessels at both 6 and 10 months of age (normalized to the optimal contraction recorded at ΔL/L0= 2.0) was not significantly decreased. However, distending to ΔL/L0= 3.0 reduced the contraction in the Marfan vessels, while contractions in the control vessels were less affected, at 6 and 10 months of age respectively (Figure 3). This Figure also shows that increasing ΔL/L0 to 3.5 and then to 4.0 induced further successive reductions in contractility in both Marfan and control vessels, but with clearly greater effect in the Marfan tissues.
Figure 2.
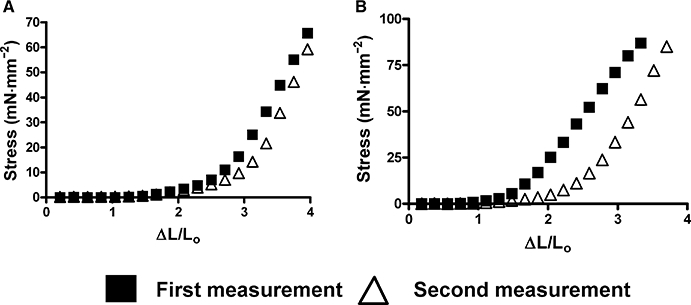
Reversibility of mesenteric artery elasticity at 10 months of age in (A) control and (B) Marfan mice. Reversibility of elasticity was tested by performing two consecutive stress–strain measurements. Vessel elasticity from the first and second measurement was compared in each group. Representative results are shown from three independent experiments.
Figure 3.
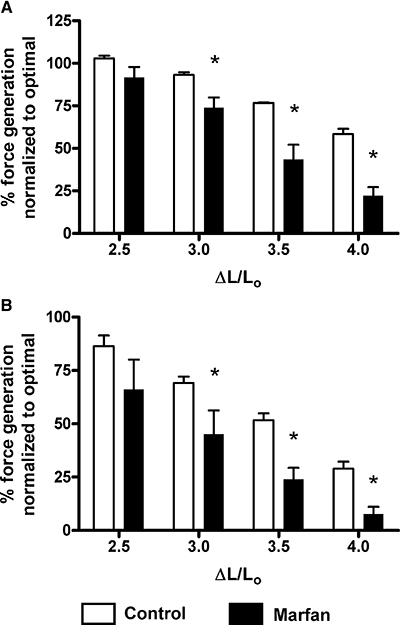
Reversibility of contractile function of mesenteric artery from (A) 6 and (B) 10 months of age after stretching at ΔL/L0= 2.5, 3.0, 3.5 and 4.0. After being stretched for 3 min and restored to optimal tension, vessels were stimulated with 60 mM KCl (*P < 0.05 vs. control, n= 6–8). Values (%) are changes of force generation normalized to that at the optimal tension.
Reduced contractile function of smooth muscle cells in Marfan mesenteric artery
To determine if smooth muscle contractile function is also affected in Marfan mesenteric arteries, we stimulated the vessels with both KCl depolarization and phenylephrine. From 3 months of age onward, vasoconstriction in response to KCl-induced depolarization in Marfan vessels was significantly less than that in age-matched controls, a 28% reduction (Figure 4A). At 6 and 10 months of age, contractility of the Marfan vessels was reduced to by 30% and by 52%, respectively, relative to the corresponding responses of the control tissues.
Figure 4.
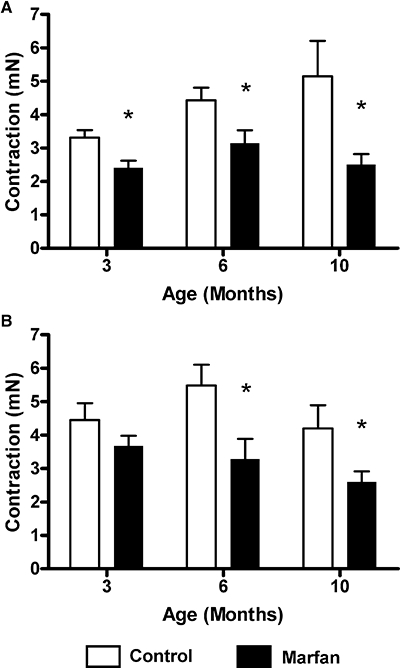
Isometric force measurement. Maximal force generated in response to (A) 60 mM KCl and (B) phenylephrine (3 µM) was compared between control and Marfan vessels (*P < 0.05 vs. control, n= 8–12).
Vasoconstriction in response to receptor-mediated stimulation using phenylephrine was also studied. Although there was no significant difference at 3 months of age, maximal contraction was significantly reduced at 6 and 10 months of age in the Marfan vessels, compared with the age-matched controls (Figure 4B). However, there were no significant differences in pEC50 values at all ages (3 months: control 6.37 ± 0.17, Marfan 6.02 ± 0.25; 6 months: control 5.83 ± 0.21, Marfan 5.72 ± 0.21; 10 months: control 5.95 ± 0.23, Marfan 5.66 ± 0.42).
To further examine smooth muscle contractile function, we measured the active force in response to depolarization with KCl. Control arteries at 3 months of age generated active force over a range of strain ΔL/L0 0.2–1.7, while control arteries at 6 and 10 months of age generated active force over a range of strain ΔL/L0 0.2–2.0 (Figure 5). However, this range was markedly reduced in the Marfan vessels at all age groups. Furthermore, the maximum active force generated in the Marfan mesenteric vessels was also markedly reduced at all age groups by 52% (3 months), 56% (6 months) and 66% (10 months) compared with the age-matched controls.
Figure 5.
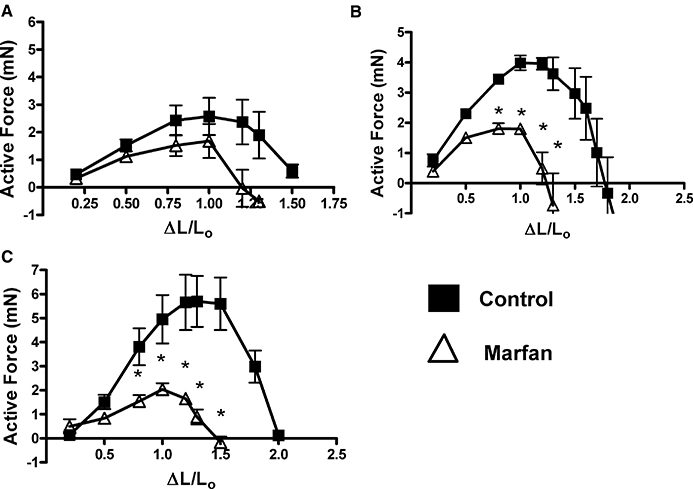
Active force, the difference between total and passive force, was compared between control and Marfan vessels from (A) 3, (B) 6 and (C) 10 months of age (*P < 0.05 vs. control, n= 8–12).
Reduced endothelium-dependent and -independent relaxation in Marfan mesenteric artery
In phenylephrine (3 µM)-precontracted vessels from control and Marfan mice, addition of ACh resulted in a concentration-dependent relaxation at all age groups (Figure 6). There was no difference in the maximal response (Emax) to ACh (10 µM)-induced relaxation between Marfan and control vessels in mice at 3 months of age, although Emax in Marfan vessels at 6 and 10 months of age were 69.7% and 44.9% of the controls respectively. It should be noted that the maximal relaxation values did not tend to change with increasing age in the control animals. Values for pEC50 for ACh indicated that at 6 months of age, the Marfan vessels were less sensitive to ACh than the controls (6.49 ± 0.21 vs. 7.17 ± 0.30 respectively). This difference was not seen at 3 months (control pEC50, 7.17 ± 0.12; Marfan pEC50, 7.23 ± 0.11) or at 10 months of age (control pEC50, 6.03 ± 0.30; Marfan pEC50, 6.23 ± 0.16).
Figure 6.
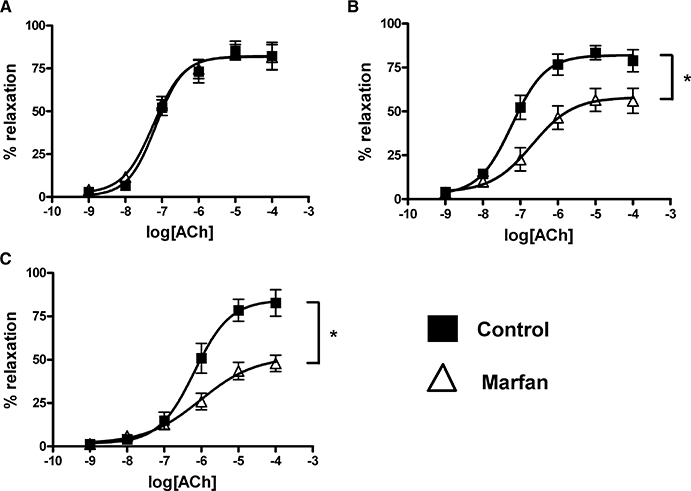
Concentration–response curve of acetylcholine (ACh)-induced relaxation in phenylephrine-precontracted mesenteric arteries from control and Marfan mice at (A) 3, (B) 6 and (C) 10 months of age (*P < 0.05 vs. control, n= 8–12).
Endothelium-independent vasodilatation was studied by the addition of SNP, a NO donor that bypasses endogenous NO production by endothelial cells, and resulted in complete dilatation in phenylephrine-precontracted control and Marfan mesenteric vessels at 6 and 10 months of age (Figure 7). Although there was no difference in Emax of SNP-relaxation between control and Marfan vessels, there was a significant increase in the pEC50 in Marfan vessels at 6 months (pEC50= 5.64 ± 0.11, control pEC50= 7.34 ± 0.04) and 10 months (pEC50= 5.99 ± 0.07, control pEC50= 6.99 ± 0.14), indicating that the Marfan vessels are less sensitive to NO at these ages. However, there was no significant difference in pEC50 at 3 months of age (data not shown).
Figure 7.
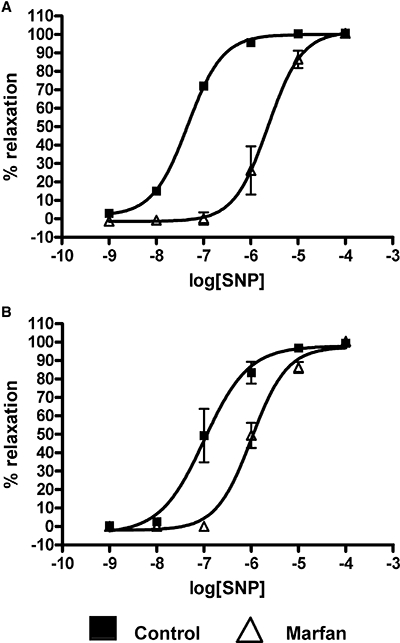
Concentration–response curve of sodium nitroprusside (SNP)-induced relaxation in phenylephrine-precontracted mesenteric arteries from control and Marfan mice at (A) 6 and (B) 10 months of age (*P < 0.05 vs. control, n= 8–12).
We then assessed the ACh response in the presence of L-NAME (200 µM), an inhibitor of NO synthase. Because impairment of ACh-induced vasorelaxation was most evident in the Marfan mice at 10 months of age, we focused our experiments on this age group. L-NAME preincubation inhibited maximal relaxation in the control vessels to ACh but did not change potency (see Table 1). However, L-NAME affected neither maximal relaxation to ACh nor its potency in Marfan vessels. To determine the contribution of the cyclooxygenase (COX) pathway, vessels were preincubated with indomethacin (10 µM), a non-selective COX inhibitor. In control vessels, indomethacin significantly increased maximal relaxation and potency (Table 1), while in vessels from Marfan mice, maximal relaxation and potency were decreased (Figure 8A and B; Table 1).
Table 1.
Effects of different treatments on potency (pEC50) and maximal (% of maximal response,Emax) ACh-induced relaxation in control and Marfan mice at 10 months of age
| Strain | Control | Marfan |
|---|---|---|
| Emax | ||
| No treatment | 84.4 ± 4.8 | 44.9 ± 3.2 |
| L-NAME | 57.1 ± 4.6* | 38.7 ± 3.0 |
| L-NAME + Indo | 57.8 ± 4.3 | 25.3 ± 4.1* |
| L-NAME + Indo + catalase | 40.3 ± 7.8# | 14.3 ± 2.3# |
| L-NAME + Indo + carbenoxolone | 55.1 ± 5.2 | 24.3 ± 2.8 |
| L-NAME + catalase | 42.3 ± 3.2* | 18.6 ± 1.7 |
| Catalase | 81.5 ± 5.6* | 30.2 ± 2.4* |
| Indo | 99.4 ± 2.2* | 36.0 ± 3.3* |
| pEC50 | ||
| No treatment | 6.03 ± 0.30 | 6.23 ± 0.16 |
| L-NAME | 5.88 ± 0.16 | 5.95 ± 0.20 |
| L-NAME + Indo | 5.73 ± 0.18 | 5.50 ± 0.12* |
| L-NAME + Indo + catalase | 6.13 ± 0.32 | 5.27 ± 0.31# |
| L-NAME + Indo + carbenoxolone | 6.23 ± 0.24 | 5.58 ± 0.17 |
| L-NAME + catalase | 5.82 ± 0.23 | 6.38 ± 0.24 |
| Catalase | 5.99 ± 0.20 | 5.80 ± 0.14* |
| Indo | 6.58 ± 0.16* | 5.56 ± 0.17* |
P < 0.05 without inhibitors in corresponding groups;
P < 0.05 with L-NAME + Indo in corresponding groups.
L-NAME, Nω-Nitro-L-arginine methyl ester; Indo, indomethacin.
Figure 8.
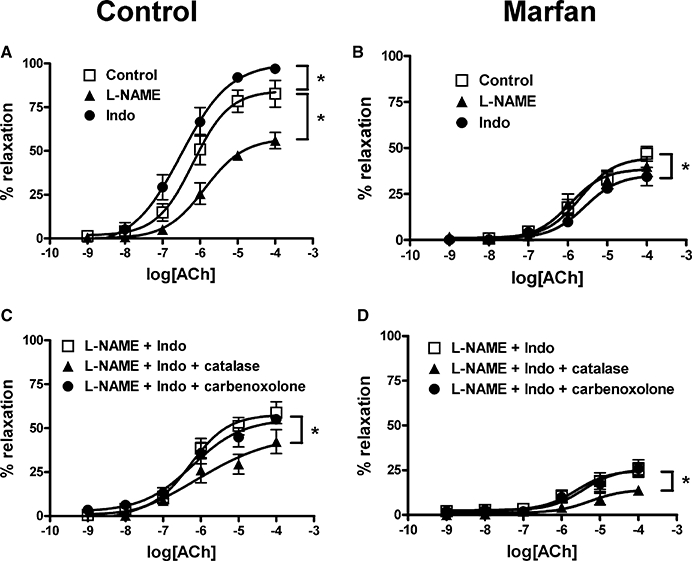
Effects of Nω-nitro-L-arginine methyl ester (L-NAME), indomethacin (Indo) and catalase on acetylcholine (ACh)-induced relaxation, from 10-month-old control and Marfan mice. The concentration–response curves of ACh are shown. (A) Control and (B) Marfan vessels were preincubated with L-NAME (200 µM) or Indo (10 µM) for 30 min and then contracted with phenylephrine (3 µM). (C) Control and (D) Marfan vessels were preincubated with catalase (1000 U·mL−1) or carbenoxolone (100 µM) in the presence of L-NAME (200 µM) and Indo (10 µM) for 30 min and then contracted with phenylephrine (3 µM) (n= 8–12).
Neither maximal contraction to phenylephrine (Emax; without indomethacin: 4.20 ± 0.70 mN; with indomethacin: 3.92 ± 0.41 mN) nor its potency (pEC50; without indomethacin: 5.95 ± 0.23; with indomethacin: 6.12 ± 0.20) was affected by indomethacin in control mice. However, maximal contraction was significantly increased in the Marfan mice (Emax; without indomethacin: 2.60 ± 0.31 mN; with indomethacin: 4.62 ± 0.35 mN) but did not significantly change potency (pEC50; without indomethacin: 5.66 ± 0.42; with indomethacin: 5.78 ± 0.31).
Nature of EDHF
The contribution of H2O2 to the EDHF-mediated relaxation was examined by the inhibitory effect of catalase (1000 U·mL−1), an enzyme that dismutates H2O2 to form water and oxygen. In the presence of indomethacin (10 µM) and L-NAME (200 µM), the addition of catalase to control vessels markedly reduced maximal relaxation to ACh but did not affect its potency (Table 1). Similarly, the addition of catalase to Marfan vessels markedly reduced Emax and decreased potency (Figure 8C and D; Table 1). To determine the role of gap junctions in the EDHF-mediated relaxation response, we used carbenoxolone (100 µM), a derivative of glycyrrhetinic acid and uncoupler of gap junctions (Tare et al., 2002). In both control and Marfan mice, carbenoxolone had no inhibitory effect on the EDHF-mediated relaxation (Figure 8C and D).
In the presence of L-NAME, catalase reduced Emax but did not change potency in either control or Marfan vessels (Table 1). Catalase alone did not change potency or maximal relaxation in control mice, although in Marfan mice, this treatment did decrease potency to ACh and decreased maximal relaxation (Table 1).
Role of superoxide
To determine the role of reactive oxygen species, vessels were preincubated with superoxide dismutase (150 U·mL−1), an enzyme that converts superoxide to H2O2. In the control mice, superoxide dismutase had no significant effect on either the maximal contraction to phenylephrine (3 µM)or its potency. Moreover, ACh-induced relaxation was also not affected. In contrast, superoxide dismutase potentiated the phenylephrine (3 µM)-induced contraction in the Marfan vessels and increased maximal contraction to control levels. Maximal relaxation to ACh was also improved and potency was also increased (Figure 9).
Figure 9.
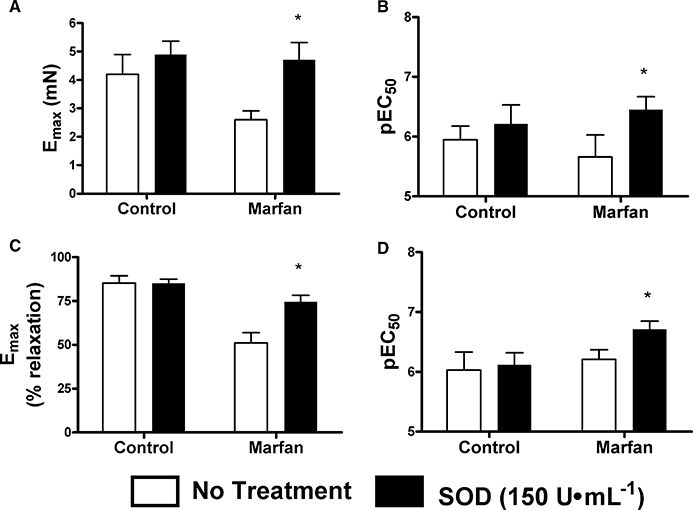
Effect of superoxide dismutase (SOD, 150 U·mL−1) on phenylephrine (3 µM)-stimulated contraction and acetylcholine (ACh) (10 µM)-mediated relaxation in mesenteric arteries from control and Marfan mice at 10 months of age. Bar graphs show (A) Emax and (B) pEC50 in response to phenylephrine in the presence and absence of SOD, while (C) Emax and (D) pEC50 show responses to ACh-mediated relaxation (*P < 0.05 vs. control, n= 5–8).
H2O2 production by endothelial cells
H2O2 production by endothelial cells was detected in the experiments using a laser confocal microscope with DCF, a peroxide-sensitive fluorescence dye. Exposure to ACh (10 µM) caused a significant increase in the DCF fluorescence intensity in endothelial cells, which was unaffected by pretreatment with indomethacin (10 µM) and L-NAME (200 µM). When the vessel was pretreated with catalase (1000 U·mL−1) in the presence of both indomethacin and L-NAME, the ACh-induced increase in the fluorescence intensity was abolished (Figure 10).
Figure 10.
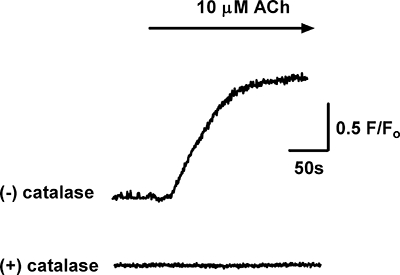
Acetylcholine (ACh, 10 µM)-induced production of H2O2 by the endothelium, detected as an increase in fluorescence intensity in dichlorodihydrofluorescein diacetate-loaded endothelial cells in small mesenteric arteries of mice. The ACh-induced increase in fluorescence intensity was abolished when the artery was preincubated with catalase (1000 U·mL−1). All experiments were performed in the presence of Nω-nitro-L-arginine methyl ester (200 µM) and indomethacin (10 µM). Representative traces shown are typical of the responses obtained in 23 cells from four mice.
Discussion
Using a genetically defined and validated mouse model of Marfan syndrome, we have demonstrated increased vessel stiffness, reduced smooth muscle contractile function, decreased resistance to mechanical stress and impaired endothelium-dependent and endothelium-independent relaxation in the small mesenteric arteries. We used appropriate control littermates to distinguish between observations derived from the pathogenesis of Marfan syndrome from those due to the physiological process of ageing. We have concluded that Marfan syndrome is a genetic disorder that affects not only the aorta and other large blood vessels, but also the smaller resistance vessels.
Degeneration of elastic fibres during the progression of Marfan syndrome is expected to decrease blood vessel elasticity. Indeed, vessel elasticity was decreased in Marfan vessels compared with the age-matched controls, although this was not apparent until 6 months of age (Figure 1). This delayed effect may be due to the relative paucity of elastic fibres in the resistance vessels, as decreased vessel elasticity can always be seen in aorta from the same mouse model of Marfan syndrome starting at 3 months of age (Chung et al., 2007a). Second, vessel wall weakening was also indicated by irreversible changes in vessel wall elasticity (Figure 2) and reduced contractility after exposure to stretching (Figure 3). Reduced contraction and irreversible changes in elasticity after stretching may be indicative of the ‘breakage’ of the physical linkage between smooth muscle cells and elastic fibres, which results in the changing of the phenotype in smooth muscle cells (Bunton et al., 2001). It should be noted that ageing is also associated with decreased vessel elasticity (Laurant et al., 2004). Indeed, the elasticity of control vessels progressively decreased during ageing, although the differences in elasticity between Marfan and control vessels persisted. This suggested that the increased vessel wall stiffness was due to the progression of Marfan syndrome.
The present study is the first to report aberrant contraction of smooth muscle cells in resistance vessels in Marfan syndrome. We showed that contraction in response to membrane depolarization and agonist stimulation is suppressed. The decrease in the ΔL/L0 range in which active force is generated suggests that at high distention, the association between smooth muscle cells and extracellular matrix might be disrupted (Figures 4 and 5), while reduced active force may be due to low intrinsic force generation of the contractile filaments or modifications in the coupling between the contractile elements and the cytoskeleton in smooth muscle cells (Rembold and Murphy, 1990). Additionally, decreased association between smooth muscle cells and elastic fibres would reduce the strain on the smooth muscle cells and blunt their response to agonist stimulation (Bunton et al., 2001). Furthermore, up-regulation of matrix metalloproteinase-2 and matrix metalloproteinase-9 in Marfan syndrome may inhibit calcium entry from the extracellular space and reduce vessel contraction (Chew et al., 2004; Chung et al., 2007a; 2008;), although further investigation is required to elucidate possible involvement of calcium signalling and myofilament contractile mechanisms.
The endothelium releases a variety of vasoactive mediators, including prostaglandins, NO and EDHF to regulate smooth muscle contractility and thus vascular smooth muscle tone (Ramsey et al., 1995; Boutouyrie et al., 1997; Wilkinson et al., 2002; Vanhoutte, 2004). The present study demonstrated that in Marfan mesenteric vessels, endothelium-dependent relaxation stimulated by ACh was significantly impaired at 6 and 10 months (Figure 6), suggesting an impairment of NO release. In Marfan syndrome, the endothelium is a likely target as Fbn-1-rich microfibrils are present in the connective tissue immediately subjacent to arterial endothelial cells (Davis, 1994; Kielty et al., 1996). Marfan syndrome is also associated with elevated plasma levels of homocysteine, which attenuate endothelial function and limit NO bioavailability (Giusti et al., 2003; Jiang et al., 2005). Endothelial Akt/eNOS phosphorylation and mechano-signalling can be compromised through increased vessel wall stiffness, further reducing endothelium-dependent vasorelaxation (Peng et al., 2003). However, there are conflicting data with regard to agonist-induced vasodilator responses in Marfan syndrome. Nakamura et al. (2000) showed inhibition of ACh-induced relaxation in the brachial artery of Marfan syndrome patients, while Wilson et al. (1999) showed that vasodilator responses to ACh and bradykinin were unaffected in the same artery. In addition, both groups found no impairment of the response to exogenous nitrovasodilators, while our studies have shown an approximately 100-fold reduction of sensitivity of smooth muscle cells from Marfan mice, to NO, at 6 months of age and a 10-fold reduction at 10 months of age (Figure 7). This reduced sensitivity may be attributed to a number of factors. First, bioavailability of NO is decreased due to excessive amounts of reactive oxygen species in the pathogenesis of cardiovascular diseases (Cai and Harrison, 2000; Faraci and Didion, 2004). NO bioavailability is also decreased during ageing, which may explain the difference in SNP sensitivity between control and Marfan vessels at 6 and 10 months of age (Newaz et al., 2006; Donato et al., 2007). Finally, the predisposition of the medial layer to degeneration and fibrosis in Marfan syndrome may also physically inhibit the ability of the vessel to dilate (Dietz et al., 1991; Pyeritz, 2000).
The findings from this study may have potentially important clinical implications, but should be viewed in the context of the existing in vivo data from human subjects. The apparent discrepancies highlighted above between human subjects and our studies may be explained by some of the following reasons. Relatively small groups of Marfan syndrome and non-Marfan syndrome patients were recruited for these studies (usually 20 or less). Then, a wide range of ages of Marfan patients were recruited (from 11 to 61 years old). Finally, the vessels studied were not true resistance (<500 µm) vessels. As indicated by the data from the present study and others, pathogenesis of Marfan syndrome in the vasculature with respect to the functional properties varied during ageing (Chung et al., 2007a). Therefore, combining both paediatric and adult patients in the same study may disturb the results and data interpretation.
Furthermore, the conflicting results should also be viewed in the context of our mouse model. There are over 600 genetic mutations that have been identified to cause Marfan syndrome (Williams et al., 2008). While missense mutations account for slightly over 60% of the mutations, 78% of the point mutations locate in the cbEGF modules and affect calcium binding. A further 12% of these mutations are recurrent and affect a mutation hotspot, CpG, for a cysteine residue, representing the most common mutation in classic Marfan syndrome and providing the basis of the mouse model used in our studies (Gray and Davies, 1996; Boileau et al., 2005). Therefore, although our model is useful to investigate the general pathogenesis of the most common type of Marfan syndrome, it may not be representative of all cases of Marfan syndrome.
Although advancing age is associated with derangement of endothelial cells leading to a decrease in NO production (Gerhard et al., 1996), the differences in ACh-induced relaxation between control and Marfan vessels persisted despite increasing age. It is unlikely that the effect of ageing played a significant role in this study, as changes in ACh-mediated relaxation in control mice are not apparent until around 15 months of age (Bulckaen et al., 2008). The same ACh-relaxation response in the control arteries between 3 and 10 month age groups could be related to up-regulation of alternative mechanisms to compensate for the decreasing NO bioavailability. EDHF has been shown to play a greater role in the face of reduced NO bioavailability during ageing (McCulloch et al., 1997; Nishikawa et al., 2000; Gaubert et al., 2007).
ACh-mediated vasorelaxation is primarily dependent on NO in the secondary branch of mouse mesenteric artery (McGuire et al., 2002; Ceroni et al., 2007). Preincubation with the NO synthase inhibitor, L-NAME, significantly reduced ACh-mediated vasorelaxation in control vessels, but interestingly did not have any effect on ACh-mediated vasorelaxation in the Marfan vessels, suggesting that the endothelial NO pathway is significantly compromised in Marfan syndrome. COX-derived prostanoids are also involved in the regulation of vasomotor function and prostacyclin (PGI2), produced from arachidonic acid through the COX-2 enzyme, is the most important of these (Smith et al., 2000). Up-regulation of COX-2 expression may also be induced through the loss of vessel elasticity (Vitarelli et al., 2006), which leads to increased production of PGI2 (Chung et al., 2007c). In our study, blockade of the COX pathway with indomethacin inhibited ACh-mediated vasorelaxation in Marfan vessels. The up-regulation of prostanoid-mediated relaxation may thus represent a compensatory mechanism of the Marfan endothelial cells in the apparent absence of NO-mediated vasorelaxation.
In addition to agonist-induced endothelium-dependent vasorelaxation, EDHF plays an important role in modulating vasomotor tone in the resistance vasculature through its hyperpolarization of smooth muscle cells (Shimokawa, 1999). However, its nature is still controversial; K+, gap junctions, epoxyeicosatrienoic acids and H2O2 are all thought to be potential candidates and the contribution of each appears to vary depending on the species tested and vessels used (Vanhoutte, 2004). In mouse mesenteric arteries, there are conflicting reports about the role of H2O2 as an EDHF (Matoba et al., 2000; Ellis et al., 2003). However, in our preparation the addition of catalase, an enzyme that dismutates H2O2 to form water and oxygen and thus lowers H2O2 concentration, in the presence of L-NAME and indomethacin reduced ACh-mediated vasorelaxation in the Marfan vessels to a greater degree than in control vessels. This greater inhibition may be due to the up-regulation of EDHF as a compensatory or back-up mechanism, which occurs when endothelial production of NO is impaired (Kilpatrick and Cocks, 1994; Corriu et al., 1998). Although myoepithelial gap junctions have been suggested to provide the pathway for EDHF in mouse mesenteric artery (Dora et al., 2003), we did not observe significant inhibition of ACh-mediated relaxation with carbenoxolone, an uncoupler of gap junctions, in either control or Marfan vessels. Therefore, it is unlikely that gap junctions mediated the EDHF response in our preparation.
It is well established that oxidative stress has a profound influence on vascular function, although the effect of oxidative stress on vasomotor response in the progression of Marfan syndrome has never been investigated. Oxidative stress has been reported to be involved in the pathogenesis of various cardiovascular diseases (Cai and Harrison, 2000; Faraci and Didion, 2004). Superoxide dismutase treatment was shown to reverse the hypersensitivity of the arteries in diabetic and hypertensive animal models and normalize the agonist-induced contraction to that of the control animals (Kanie and Kamata, 2000; Alvarez et al., 2008). Although the mechanism of action of reactive oxygen species on smooth muscle cell contractility is still unclear (Lyle and Griendling, 2006), reactive oxygen species have been proposed to have multiple effects on calcium signalling in both vascular endothelial and smooth muscle cells (Elmoselhi et al., 1996; Lounsbury et al., 2000; Walia et al., 2000). The impairment of the calcium signalling pathway caused by oxidative stress may consequently lead to the alteration of vascular reactivity (Sener et al., 2004). Thus, the removal of superoxide with superoxide dismutase may restore calcium signalling and thereby the contractile responses. Furthermore, oxidative stress may cause endothelial dysfunction through several direct and indirect pathways, the best known being the scavenging of NO by superoxide. Superoxide radicals bind to NO at a rate three times faster than they bind to superoxide dismutase; therefore, excess superoxide production would increase the rate of NO degradation (Cai and Harrison, 2000; Schulz et al., 2004). The improvement of endothelial function in the Marfan vessels brought about by superoxide dismutase suggests that oxidative stress may be another contributor to endothelial dysfunction in Marfan syndrome.
In conclusion, our study indicates that in Marfan syndrome, endothelium-dependent and endothelium-independent vasodilation is impaired in resistance vessels. Furthermore, we have demonstrated that smooth muscle contractility is compromised and vessel stiffness is increased. Together, these vasomotor abnormalities in the resistance vessel may have a negative and detrimental impact on the overall cardiovascular function in Marfan syndrome.
Acknowledgments
This work was supported by an operating grant from the Canadian Institutes of Health Research. HS and CY are recipients of the Michael Smith Foundation for Health Research and National Sciences and Engineering Research Council Trainee Awards, and AC is a recipient of a Michael Smith Foundation for Health Research/St Paul's Hospital Foundation Trainee Award.
Glossary
Abbreviations:
- COX
cyclooxygenase
- DCF
dichlorodihydrofluorescein diacetate
- EDHF
endothelium-derived hyperpolarizing factor
- L-NAME
Nω-Nitro-L-arginine methyl ester
- SNP
sodium nitroprusside
Conflict of interest
None.
References
- Alvarez Y, Briones AM, Hernanz R, Perez-Giron JV, Alonso MJ, Salaices M. Role of NADPH oxidase and iNOS in vasoconstrictor responses of vessels from hypertensive and normotensive rats. Br J Pharmacol. 2008;153:926–935. doi: 10.1038/sj.bjp.0707575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau C, Jondeau G, Mizuguchi T, Matsumoto N. Molecular genetics of Marfan syndrome. Curr Opin Cardiol. 2005;20:194–200. doi: 10.1097/01.hco.0000162398.21972.cd. [DOI] [PubMed] [Google Scholar]
- Boutouyrie P, Bézie Y, Lacolley P, Challande P, Chamiot-Clerc P, Benetos A, et al. In vivo/in vitro comparison of rat abdominal aorta wall viscosity. Influence of endothelial function. Arterioscler Thromb Vasc Biol. 1997;17:1346–1355. doi: 10.1161/01.atv.17.7.1346. [DOI] [PubMed] [Google Scholar]
- Briones AM, González JM, Somoza B, Giraldo J, Daly CJ, Vila E, et al. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J Physiol. 2003;552:185–195. doi: 10.1113/jphysiol.2003.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulckaen H, Prévost G, Boulanger E, Robitaille G, Roquet V, Gaxatte C, et al. Low-dose aspirin prevents age-related endothelial dysfunction in a mouse model of physiological aging. Am J Physiol Heart Circ Physiol. 2008;294:H1562, 70. doi: 10.1152/ajpheart.00241.2007. [DOI] [PubMed] [Google Scholar]
- Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Ceroni L, Ellis A, Wiehler WB, Jiang YF, Ding H, Triggle CR. Calcium-activated potassium channel and connexin expression in small mesenteric arteries from eNOS-deficient (eNOS-/-) and eNOS-expressing (eNOS+/+) mice. Eur J Pharmacol. 2007;560:193–200. doi: 10.1016/j.ejphar.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Chew DK, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–1010. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC, van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007a;101:512–522. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- Chung AW, Au Yeung K, Cortes SF, Sandor GG, Judge DP, Dietz HC, et al. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br J Pharmacol. 2007b;150:1075–1083. doi: 10.1038/sj.bjp.0707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Yang HH, van Breemen C. Imbalanced synthesis of cyclooxygenase-derived thromboxane A2 and prostacyclin compromises vasomotor function of the thoracic aorta in Marfan syndrome. Br J Pharmacol. 2007c;152:305–312. doi: 10.1038/sj.bjp.0707391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008;102:e73–e85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- Corriu C, Félétou M, Puybasset L, Bea ML, Berdeaux A, Vanhoutte PM. Endothelium-dependent hyperpolarization in isolated arteries taken from animals treated with NO-synthase inhibitors. J Cardiovasc Pharmacol. 1998;32:944–950. doi: 10.1097/00005344-199812000-00011. [DOI] [PubMed] [Google Scholar]
- Davis EC. Immunolocalization of microfibril and microfibril-associated proteins in the subendothelial matrix of the developing mouse aorta. J Cell Sci. 1994;107:727–736. doi: 10.1242/jcs.107.3.727. [DOI] [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Dobrin PB. Mechanical properties of arteries. Physiol Rev. 1978;58:397–460. doi: 10.1152/physrev.1978.58.2.397. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, et al. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100:1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- Dora KA, Sandow SL, Gallagher NT, Takano H, Rummery NM, Hill CE, et al. Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J Vasc Res. 2003;40:480–490. doi: 10.1159/000074549. [DOI] [PubMed] [Google Scholar]
- Ellis A, Pannirselvam M, Anderson TJ, Triggle CR. Catalase has negligible inhibitory effects on endothelium-dependent relaxations in mouse isolated aorta and small mesenteric artery. Br J Pharmacol. 2003;140:1193–1200. doi: 10.1038/sj.bjp.0705549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmoselhi AB, Samson SE, Grover AK. SR Ca2+ pump heterogeneity in coronary artery: Free radicals and IP3-sensitive and -insensitive pools. Am J Physiol. 1996;271:C1652–C1659. doi: 10.1152/ajpcell.1996.271.5.C1652. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- Gaubert ML, Sigaudo-Roussel D, Tartas M, Berrut G, Saumet JL, Fromy B. Endothelium-derived hyperpolarizing factor as an in vivo back-up mechanism in the cutaneous microcirculation in old mice. J Physiol. 2007;585:617–626. doi: 10.1113/jphysiol.2007.143750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard M, Roddy MA, Creager SJ, Creager MA. Aging progressively impairs endothelium-dependent vasodilation in forearm resistance vessels of humans. Hypertension. 1996;27:849–853. doi: 10.1161/01.hyp.27.4.849. [DOI] [PubMed] [Google Scholar]
- Giusti B, Porciani MC, Brunelli T, Evangelisti L, Fedi S, Gensini GF, et al. Phenotypic variability of cardiovascular manifestations in Marfan syndrome. Possible role of hyperhomocysteinemia and C677T MTHFR gene polymorphism. Eur Heart J. 2003;24:2038–2045. doi: 10.1016/j.ehj.2003.08.020. [DOI] [PubMed] [Google Scholar]
- Goffi L, Chan R, Boccoli G, Ghiselli R, Saba V. Aneurysm of a jejunal branch of the superior mesenteric artery in a patient with Marfan's syndrome. J. Cardiovasc Surg. 2000;41:321–323. [PubMed] [Google Scholar]
- González JM, Briones AM, Starcher B, Conde MV, Somoza B, Daly C, et al. Influence of elastin on rat small artery mechanical properties. Exp Physiol. 2005;90:463–468. doi: 10.1113/expphysiol.2005.030056. [DOI] [PubMed] [Google Scholar]
- Gray JR, Davies SJ. Marfan syndrome. J Med Genet. 1996;33:403–408. doi: 10.1136/jmg.33.5.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatrick AG, Malcolm PN, Burnand KG, Irvine AT. A superficial femoral artery aneurysm in a patient with Marfan's syndrome. Eur J Vasc Endovasc Surg. 1998;15:459–460. doi: 10.1016/s1078-5884(98)80212-8. [DOI] [PubMed] [Google Scholar]
- Jiang X, Yang F, Tan H, Liao D, Bryan RM, Jr, Randhawa JK, et al. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler Thromb Vasc Biol. 2005;25:2515–2521. doi: 10.1161/01.ATV.0000189559.87328.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jondeau G, Boutouyrie P, Lacolley P, Laloux B, Dubourg O, Bourdarias JP, et al. Central pulse pressure is a major determinant of ascending aorta dilation in Marfan syndrome. Circulation. 1999;99:2677–2681. doi: 10.1161/01.cir.99.20.2677. [DOI] [PubMed] [Google Scholar]
- Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge DP, Dietz HC. Marfan's syndrome. Lancet. 2005;366:1965–1976. doi: 10.1016/S0140-6736(05)67789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanie N, Kamata K. Contractile responses in spontaneously diabetic mice. I. involvement of superoxide anion in enhanced contractile response of aorta to norepinephrine in C57BL/KsJ(db/db) mice. Gen Pharmacol. 2000;35:311–318. doi: 10.1016/s0306-3623(02)00115-5. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Wilson DG, Stuart G, Musumeci F, Cones CJ, Davies S, et al. Fibrillin expression and deposition by vascular endothelial cells: implications for the Marfan syndrome. Circulation. 1996;94:I-350.. [Google Scholar]
- Kilpatrick EV, Cocks TM. Evidence for differential roles of nitric oxide (NO) and hyperpolarization in endothelium-dependent relaxation of pig isolated coronary artery. Br J Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurant P, Adrian M, Berthelot A. Effect of age on mechanical properties of rat mesenteric small arteries. Can J Physiol Pharmacol. 2004;82:269–275. doi: 10.1139/y04-026. [DOI] [PubMed] [Google Scholar]
- Lay CS, Yu CJ, Tyan YS. Abdominal aortic dissection with acute mesenteric ischemia in a patient with Marfan syndrome. J Chin Med Assoc. 2006;69:326–329. doi: 10.1016/S1726-4901(09)70267-9. [DOI] [PubMed] [Google Scholar]
- Lounsbury KM, Hu Q, Ziegelstein RC. Calcium signaling and oxidant stress in the vasculature. Free Radic Biol Med. 2000;28:1362–1369. doi: 10.1016/s0891-5849(00)00222-7. [DOI] [PubMed] [Google Scholar]
- Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- McCulloch AI, Bottrill FE, Randall MD, Hiley CR. Characterization and modulation of EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br J Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JJ, Hollenberg MD, Andrade-Gordon P, Triggle CR. Multiple mechanisms of vascular smooth muscle relaxation by the activation of proteinase-activated receptor 2 in mouse mesenteric arterioles. Br J Pharmacol. 2002;135:155–169. doi: 10.1038/sj.bjp.0704469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnor WR. Properties of the vascular wall. In: Collins N, editor. Hemodynamics. Baltimore: Williams & Wilkins; 1989. pp. 84–90. [Google Scholar]
- Mulvany MJ, Aalkjaer C. Structure and function of small arteries. Physiol Rev. 1990;70:921–961. doi: 10.1152/physrev.1990.70.4.921. [DOI] [PubMed] [Google Scholar]
- Murdoch JL, Walker BA, Halpern BL, Kuzma JW, McKusick VA. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med. 1972;286:804–808. doi: 10.1056/NEJM197204132861502. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Itoh S, Makita S, Ohira A, Arakawa N, Hiramori K. Peripheral resistance vessel dysfunction in Marfan syndrome. Am Heart J. 2000;139:661–666. doi: 10.1016/s0002-8703(00)90045-0. [DOI] [PubMed] [Google Scholar]
- Newaz MA, Yousefipour Z, Oyekan A. Oxidative stress-associated vascular aging is xanthine oxidase-dependent but not NAD(P)H oxidase-dependent. J Cardiovasc Pharmacol. 2006;48:88–94. doi: 10.1097/01.fjc.0000245402.62864.0a. [DOI] [PubMed] [Google Scholar]
- Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa Y, Stepp DW, Chilian WM. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am J Physiol Heart Circ Physiol. 2000;279:H459–H465. doi: 10.1152/ajpheart.2000.279.2.H459. [DOI] [PubMed] [Google Scholar]
- Ohba M, Shibanuma M, Kuroki T, Nose K. Production of hydrogen peroxide by transforming growth factor-beta1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J Cell Biol. 1994;126:1079–1088. doi: 10.1083/jcb.126.4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Haldar S, Deshpande S, Irani K, Kass DA. Wall stiffness suppresses Akt/eNOS and cytoprotection in pulse-perfused endothelium. Hypertension. 2003;41:378–381. doi: 10.1161/01.hyp.0000049624.99844.3d. [DOI] [PubMed] [Google Scholar]
- Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- Ramsey MW, Goodfellow J, Jones CJ, Luddington LA, Lewis MJ, Henderson AH. Endothelial control of arterial distensibility is impaired in chronic heart failure. Circulation. 1995;92:3212–3219. doi: 10.1161/01.cir.92.11.3212. [DOI] [PubMed] [Google Scholar]
- Reinhardt DP, Chalberg SC, Sakai LY. The structure and function of fibrillin. Ciba Found Symp. 1995;192:128–143. doi: 10.1002/9780470514771.ch7. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Murphy RA. Muscle length, shortening, myoplasmic [Ca2+], and activation of arterial smooth muscle. Circ Res. 1990;66:1354–1361. doi: 10.1161/01.res.66.5.1354. [DOI] [PubMed] [Google Scholar]
- Rosenbloom J. Extracellular matrix 4: the elastic fiber. FASEB J. 1993;7:1208–1218. [PubMed] [Google Scholar]
- Safar ME, London GM. Swales JD. Textbook of Hypertension. Blackwell Scientific Publications: London; 1994. The arterial system in human hypertension; pp. 85–102. [Google Scholar]
- Savolainen H, Savola J, Savolainen A. Aneurysm of the iliac artery in Marfan's syndrome. Ann Chir Gynaecol. 1993;82:203–205. [PubMed] [Google Scholar]
- Schulz E, Anter E, Keaney JF., Jr Oxidative stress, antioxidants, and endothelial function. Curr Med Chem. 2004;11:1093–1104. doi: 10.2174/0929867043365369. [DOI] [PubMed] [Google Scholar]
- Sener G, Paskaloglu K, Toklu H, Kapucu C, Ayanoglu-Dulger G, Kacmaz A, et al. Melatonin ameliorates chronic renal failure-induced oxidative organ damage in rats. J Pineal Res. 2004;36:232–241. doi: 10.1111/j.1600-079X.2004.00113.x. [DOI] [PubMed] [Google Scholar]
- Sheremet'eva GF, Ivanova AG, IuV B, Gens AP, Kocharian EZ. A comparative study of the aortic wall in patients with Marfan's syndrome and Erdheim's disease. Angiol Sosud Khir. 2004;10:22–29. [PubMed] [Google Scholar]
- Shimokawa H. Primary endothelial dysfunction: atherosclerosis. J Mol Cell Cardiol. 1999;31:23–37. doi: 10.1006/jmcc.1998.0841. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Tare M, Coleman HA, Parkington HC. Glycyrrhetinic derivatives inhibit hyperpolarization in endothelial cells of guinea pig and rat arteries. Am J Physiol Heart Circ Physiol. 2002;282:H335–H341. doi: 10.1152/ajpheart.2002.282.1.H335. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelium-dependent hyperpolarizations: the history. Pharmacol Res. 2004;49:503–508. doi: 10.1016/j.phrs.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Vitarelli A, Conde Y, Cimino E, D'Angeli I, D'Orazio S, Stellato S, et al. Aortic wall mechanics in the Marfan syndrome assessed by transesophageal tissue Doppler echocardiography. Am J Cardiol. 2006;97:571–577. doi: 10.1016/j.amjcard.2005.09.089. [DOI] [PubMed] [Google Scholar]
- Walia M, Sormaz L, Samson SE, Lee RM, Grover AK. Effects of hydrogen peroxide on pig coronary artery endothelium. Eur J Pharmacol. 2000;400:249–253. doi: 10.1016/s0014-2999(00)00393-9. [DOI] [PubMed] [Google Scholar]
- Wilkinson IB, Qasem A, McEniery CM, Webb DJ, Avolio AP, Cockcroft JR. Nitric oxide regulates local arterial distensibility in vivo. Circulation. 2002;105:213–217. doi: 10.1161/hc0202.101970. [DOI] [PubMed] [Google Scholar]
- Williams A, Davies S, Stuart AG, Wilson DG, Fraser AG. Medical treatment of marfan syndrome: a time for change. Heart. 2008;94:414–421. doi: 10.1136/hrt.2006.109454. [DOI] [PubMed] [Google Scholar]
- Wilson DG, Bellamy MF, Ramsey MW, Goodfellow J, Brownlee M, Davies S, et al. Endothelial function in Marfan syndrome: selective impairment of flow-mediated vasodilation. Circulation. 1999;99:909–915. doi: 10.1161/01.cir.99.7.909. [DOI] [PubMed] [Google Scholar]