

Abstract
Background and purpose:
We have previously shown that β-adrenoceptors continuously stimulated with noradrenaline induces an increase in β3-adrenoceptors (GαiPCRs) and a decrease in β1-adrenoceptors (GαsPCRs) at functional, genomic and protein levels. This compensatory modification induced by noradrenaline is probably one of the consequences of cardiac depression observed in heart disease. Therefore, we investigated further the interaction between β1- and β3-adrenoceptors in neonatal rat cardiomyocytes.
Experimental approach:
Functional studies were performed by cyclic adenosine monophosphate (cAMP) accumulation assays in cells untreated or treated with dobutamine and ICI 118551 (β1-adrenoceptor) or CL-3162436243 (β3-adrenoceptor) for 24 h in the presence or absence of protein kinase inhibitors. β-adrenoceptor and protein kinase expression was monitored by quantitative reverse transcription-polymerase chain reaction (RT-PCR) and by Western blotting, respectively.
Key results:
Chronic β1- or β3-adrenoceptor stimulation reduced β1-adrenoceptor-mediated cAMP accumulation in association with a decrease in β1-adrenoceptor mRNA and protein levels through protein kinase C (PKC), phosphoinositide 3-kinase (PI3K) and p38 mitogen-activated protein kinase (p38MAPK) activation. In contrast, both treatments induced an increase in β3-adrenoceptor expression and β3-adrenoceptor-inhibited forskolin response through PKC, extracellular-signal-regulated kinases 1 and 2 (ERK1/2) and p38MAPK phosphorylation, although no β3-adrenoceptor response was observed in untreated cells. ERK1/2 and p38MAPK were activated by both treatments. The modulation of β1- or β3-adrenoceptor function did not require stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) although chronic β1-adrenoceptor stimulation activated SAPK/JNK. β3-adrenoceptor treatment activated Akt although PI3K was not involved in β3-adrenoceptor up-regulation.
Conclusion and implications:
We show for the first time that chronic β1- or β3-adrenoceptor stimulation leads to the modulation of β1- and β3-adrenoceptors by a cross-regulation involving PKC, PI3K p38MAPK and MEK/ERK1/2 pathway, and through protein kinase A when β1-adrenoceptors are chronically activated.
Keywords: cardiomyocytes, heart, β1-adrenoceptor, β2-adrenoceptor, β3-adrenoceptors, cAMP accumulation, up-regulation, down-regulation, MAP kinases, Akt
Introduction
Three β-adrenoceptors (β1, β2 and β3-adrenoceptors; nomenclature follows Alexander et al., 2008) are expressed in heart (Dzimiri, 1999; Rozec and Gauthier, 2006). These receptor subtypes belong to the G-protein coupled receptor (GPCR) superfamily and modulate cardiac function after stimulation by the catecholamines, noradrenaline and adrenaline (Dzimiri, 1999). Increases in heart rate and force of contraction are mediated mainly by β1-adrenoceptors and to a lesser extent by β2-adrenoceptors through a coupling to the Gs protein pathway (Dzimiri, 1999). In contrast, the β3-adrenoceptor subtype coupled to Gi protein decreases cardiac contractility (Dzimiri, 1999; Rozec and Gauthier, 2006). It is well known that heart failure induces an increase in circulating cathecholamines, which leads to an alteration of cardiac function through a decrease in β1-adrenoceptor density (Cohn et al., 1984; Dzimiri, 1999; Tilley and Rockman, 2006). In contrast, β3-adrenoceptors were found up-regulated in failing human and animal hearts (Cheng et al, 2001; Moniotte et al., 2001; Morimoto et al., 2004; Zhang et al., 2005). Similarly, β1-adrenoceptor expression was decreased and β3-adrenoceptor level increased both in hearts from diabetic and from physically trained rats (Dinçer et al., 2001; Barbier et al., 2007). It is noteworthy that catecholamines, particularly noradrenaline, increase during physical exercise, and likewise during heart failure (Pagliari and Peyrin, 1995; Dzimiri, 1999; Tilley and Rockman, 2006). We have shown previously that chronic stimulation of cardiomyocytes with noradrenaline decreases β1-adrenoceptor function and expression, and enhances β3-adrenoceptor function (Germack and Dickenson, 2006). Interestingly, Thomas et al (1992) have reported a similar opposite regulation in adipocytes chronically stimulated with isoprenaline. In addition, β1-adrenoceptor expression was lower in transgenic mice specifically overexpressing human β3-adrenoceptor in the heart (Kohout et al., 2001) whereas β3-adrenoceptor knockout mice exhibited an increase in β1-adrenoceptor mRNA in adipose tissues (Susulic et al., 1995).
It is well known that β1 and β2-adrenoceptors are subject to desensitization, internalization and down-regulation following chronic exposure to agonists leading to a decrease in β-adrenoceptor responsiveness, whereas β3-adrenoceptors are resistant to desensitization (Dzimiri, 1999; Rozec and Gauthier, 2006). Indeed, the β1 and β2-adrenoceptors contain consensus sites mainly in the C-terminal region that can be phosphorylated by G-protein receptor kinases (GRKs), mainly GRK2, and cAMP protein kinase A (PKA) that are involved in desensitization of the receptors. In contrast, the β3-adrenoceptor subtype lacks phosphorylation sites preventing its desensitization by this mechanism (Dzimiri, 1999; Tilley and Rockman, 2006). β-adrenoceptor internalization requires the recruitment of β-arrestins and phosphoinositide 3-kinase (PI3K), which bind to phosphorylated β-adrenoceptors (Dzimiri, 1999; Naga Prasad et al., 2001; Nienaber et al., 2003). The down-regulation of GPCRs following persistent stimulation also involves transcriptional and post-transcriptional mechanisms leading to a decrease in mRNA and protein synthesis (Dzimiri, 1999). Headley et al. (2004) demonstrated that extracellular signal-regulated kinase (ERK1/2), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and p38 mitogen-activated protein kinase (p38MAPK) pathways can modulate β1-adrenoceptor mRNA stability. Finally, Li et al. (1998) showed that protein kinase C (PKC) induced the down-regulation of β1-adrenoceptors at protein and mRNA levels in rat C6 glioma cells. Similarly, isoproterenol-induced adenylyl cyclase activity was impaired in human embryonic kidney (HEK) 293 cells overexpressing constitutively active PKC isoforms (α, βII, ε and ζ) and transfected with β1- or β2-adrenoceptors (Guimond et al., 2005).
Overall, these observations suggest that compensatory interactions between β1- and β3-adrenoceptors take place during chronic stimulation with the nonselective agonists, noradrenaline or isoprenaline. In addition, β1-adrenoceptor desensitization and down-regulation involve PI3K and protein kinases whereas the implication of these kinases in β3-adrenoceptor up-regulation is unknown. Therefore, we investigated the possible interaction between β3-adrenoceptor up-regulation and β1-adrenoceptor down-regulation at the functional and expression levels, and identified kinases implicated in the functional receptor modulation following chronic β1- or β3-adrenoceptor selective stimulation in neonatal rat cardiomyocytes.
In this study, we demonstrate for the first time that a cross-regulation at the expression and functional level between β1- and β3-adrenoceptors occurs following chronic receptor stimulation via the activation of PKC, PI3K, p38 MAPK and MEK/ERK1/2 pathways.
Methods
Cell culture
All animal care and experimental procedures complied with the UK Home Office Policy and approved by the Nottingham Trent University Ethical Committee. Neonatal ventricular myocytes were prepared from 1–4 day-old Wistar rats using the Neonatal Cardiomyocyte Isolation System (Worthington Biochemical Corporation, Lornes laboratories, Reading, UK). As described previously (Germack and Dickenson, 2006), the cells were preplated three times for 30 min in a humidified incubator (95% air/5% CO2 at 37°C) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM L-glutamine, 10% (v/v) foetal calf serum and penicillin/streptomycin (100 U·mL−1) in order to minimize fibroblast contamination. Cardiomyocyte-rich cultures (>90%) were plated onto fibronectin-coated plates at a final density of 1.25 × 105 cells·cm−2 in supplemented DMEM. For cAMP accumulation assay, the cells were plated onto 24-well plates and for RT-PCR and Western blotting analysis onto 6-well plates. After 3 days, confluent and spontaneously beating cells were serum-starved and untreated or treated with 10 µM dobutamine (a β1-adrenoceptor selective agonist) and 1 µM ICI118551 (ICI, β2-adrenoceptor selective antagonist) or 10 µM procaterol (a β2-adrenoceptor selective agonist) and 1 µM CGP 20712A (CGP, β1-adrenoceptor selective antagonist) or 2 µM CL-316243 (a β3-adrenoceptor selective agonist) for 24 h. Because we did not observe any difference in the functional β1-adrenoceptor down-regulation induced by chronic β3-adrenoceptor stimulation in the presence and absence of propranolol at the concentration, which inhibits β1 and β2-adrenoceptors (data not shown), we did not combine CL-316243 treatment with an antagonist such as nadolol or propranolol to block β1- and β2-adrenoceptors. Similarly, Yoshida et al. (1994) showed that the treatment of obese diabetic mice with CL-316243 induces anti-obesity and anti-diabetic effects through specifically β3-adrenoceptors without involving β1- and β2-adrenoceptors (potency β1 : β2 : β3= 0:1:100 000). Finally, the CL-316243-inhibited forskolin response was only observed in noradrenaline-treated neonatal rat cardiomyocytes and not modified by the presence of propranolol (Germack and Dickenson, 2006). Therefore, these studies indicate that the treatment of cardiomyocytes with CL-316243 in the present investigation mediates its effects only through β3-adrenoceptor stimulation. The signalling pathway involved in the regulation of β1- and β3-adrenoceptor cAMP responses following dobutamine/ICI or CL-316243 treatments was investigated in the absence or presence of protein kinase inhibitors (50 µM PD 98059, MEK1 inhibitor; 10 µM SB 203580, p38MAPK inhibitor; 10 µM SP 600125, SAPK/JNK inhibitor; 100 nM wortmannin, PI3K inhibitor; 1 µM KT 57201, PKA inhibitor; or 10 µM GF 109203, PKC inhibitor).
cAMP accumulation assay
Following the treatment of cardiomyocytes with dobutamine/ICI or procaterol/CGP or CL-316243 for 24 h, assays were carried out in serum-free DMEM in a humidified incubator (95% air/5% CO2 at 37°C). Agonists as required by the experiments were added as described in the figure legends. The cells were incubated for 3 h in a humidified incubator (95% air/5% CO2 at 37°C) with 500 µL of serum-free DMEM containing [3H]adenine (2 µCi per well). [3H]adenine-labelled cells were washed twice with Hanks/Hepes buffer and then incubated in 500 µL per well serum-free DMEM containing the cAMP phosphodiesterase inhibitor, rolipram (10 µM) for 15 min at 37°C in a humidified incubator. Agonists were added 5 min prior to the incubation with 1.5 µM forskolin (10 min). Antagonist or inhibitors were added 30 min before agonist. Incubations were terminated by the addition of 500 µL 5% trichloroacetic acid after removing the medium. [3H]cAMP was isolated by sequential Dowex-alumina chromatography as previously described (Germack and Dickenson, 2006). After elution, the levels of [3H]cAMP were determined by liquid scintillation counting.
Quantitative RT-PCR
Total RNA was extracted from untreated or treated cardiomyocytes for 24 h with dobutamine/ICI or procaterol/CGP or CL-316243 combining RNA isolation reagent, RNABee (AMS Biotechnology, Oxon, UK) and RNeasy Mini Kit (Qiagen Ltd, Crawley, UK) according to the manufacturer's instructions. Reverse transcription was performed with 3 µg total RNA for the synthesis of cDNA using oligo d(T)15 (Promega, Southampton, UK) and Superscript II reverse transcriptase (Invitrogen Ltd, Praisley, UK) according to the manufacturer's instructions. Quantitative PCR was carried out with a Bio-Rad iCycler system using iQ SYBR green Supermix kit from Bio-Rad (Bio-Rad Laboratories Ltd, Hemel Hempstead, UK). The PCR protocol used was 3 min initial denaturation at 95°C followed by 45 cycles of denaturation (20 s at 95°C), annealing (30 s at 65°C) and elongation (30 s at 72°C). For exact quantification, standard curves were generated by serial dilutions (1 × 106–1 × 102 single stranded cDNA molecules) for each target gene used as external standards. Standard cDNAs were prepared as followed: initial PCR was conducted with 1 µg cDNA in the presence of deoxyribonucleotides triphosphate (dNTPS) (1.25 mM), 200 ng of respective primers (Table 1) and 1.5 U Taq DNA polymerase (Promega, Southampton, UK). Thirty five cycles of the amplification steps involved 1 min denaturation at 94°C and 1 min annealing at 65°C and 1 min extension at 72°C. The PCR products were analysed using 1.5% agarose gel electrophoresis and extracted from the gel using QIAquick gel extraction kit (Qiagen Ltd, Crawley, UK). The purified products were again subjected to PCR and purified using QIAquick PCR purification kit (Qiagen Ltd, Crawley, UK) followed by quantification by spectrophotometry (λ:260/280). β-actin mRNA was used as an internal standard to normalize β1-, β2- and β3-adrenoceptor mRNA.
Table 1.
Sequences of the specific primers for the three rat β-adrenoceptors (β1-AR, β2-AR, β3-AR) and for β-actin
| Primer | PCR product Base pairs | Reference | |
|---|---|---|---|
| β1-AR | 368 | ||
| Forward | 5′-TTCGGTAGACGTGCTATGTGTGAC-3′ | ||
| Reverse | 5′-TCTTCTTCACCTGTTTCTGGGCCT-3′ | ||
| β2-AR | 328 | ||
| Forward | 5′-TGCGTGATTGCAGTGGATCGCTAT-3′ | ||
| Reverse | 5′-CTATCTTCTGCAGCTGCCTTTTGG-3′ | ||
| β3-AR | 202 | Bensaid et al. (1993) | |
| Forward | 5′-AACTCTGCCTTCAACCCGCTCAT-3′ | ||
| Reverse | 5′-TTCATGTGGGAAATGGACGCTCAC-3′ | ||
| β-actin | 301 | Germack and Dickenson (2006) | |
| Forward | 5′-CGTAAAGACCTCTATGCCAA-3′ | ||
| Reverse | 5′-GGTGTAAAACGCAGCTCAGT-3′ |
Western blot analysis
Cardiomyocytes untreated or treated with dobutamine/ICI or procaterol/CGP or CL-316243 for 24 h were washed with ice-cold phosphate-buffered saline (PBS) and lysed in ice-cold hypotonic buffer [30 mM Tris-HCl, 5 mM MgCl2, 100 mM NaCl, 1 mM EGTA, 0.1 mM phenylmethylsulphonylfluoride (PMSF), 5 µg·mL−1 leupeptin] for G protein and β-adrenoceptor investigation. Cell membranes were collected by centrifugation for 30 min at 100 000 g and 4°C. The resulting plasma membrane pellets were dissolved in ice-cold detergent buffer [150 mM NaCl, 50 mM Tris.HCl, 5 mM ethylediaminetetraacetic acid (EDTA), 1% (v/v) IGEPAL CA-630, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulphate (SDS), 1 mM Na3VO4, 1 mM NaF, 1 mM benzamidine, 0.1 mM PMSF, 5 µg·mL−1 leupeptin]. For the study of the cell signalling, cardiomyocytes untreated or treated with dobutamine/ICI or CL-316243 for 24 h in the presence or absence of appropriate kinase inhibitors were washed with ice-cold PBS and lysed in ice-cold detergent buffer. Cell membrane homogenates or cell lysates were clarified by centrifugation (5 min; 9500×g) in an Eppendorf microcentrifuge. 100 µL of the cell lysate removed and stored at −20°C until required. Protein concentration was determined using Bio-Rad DC Protein assay (Bio-Rad laboratories, Hertfordshire, UK) with bovine serum albumin as the standard. Samples in lysis buffer were heated at 95°C in SDS-polyacrylamide gel electrophoresis (SDS/PAGE) sample buffer (v/v). Aliquots of the cell lysate (20–30 µg protein) were separated by SDS/PAGE (12% acrylamide gel), using a Bio-Rad Mini-Protean II system (1 h at 200 V). Proteins were transferred to nitrocellulose membranes using a Bio-Rad Trans-Blot system (1 h at 100 V in 25 mM Tris, 192 mM glycine and 20% MeOH). Following transfer, the membranes were washed with Tris-buffered saline (TBS) and blocked for 1 h at room temperature in blocking buffer (TBS, 5% (w/v) skimmed milk powder, 0.1% Tween-20). Blots were then incubated for 24 h at 4°C with primary antibody in blocking buffer against Gαi-protein, Gαs-protein, phosphorylated ERK1/2, P38 MAPK, JNK or PKB at 1:1000 dilution or β1-adrenoceptor at 1/200 dilution or β2- or β3-adrenoceptors at 1/100. The primary antibody was removed and the blot was extensively washed three times for 5 min in TBS/Tween 20. Blots were then incubated for 1 h at room temperature with the goat anti-rabbit secondary antibody coupled to horseradish peroxidase (DAKO Ltd, Cambridge, UK) at 1:1000 dilution in blocking buffer. Following removal of the secondary antibody, blots were extensively washed as previously described and developed using the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Little Chalfont, UK) and quantified by densitometry using GeneGenius BioImaging System (Syngene, Synoptics Ltd, UK). The uniform transfer of proteins to the nitrocellulose membrane was routinely monitored by transiently staining the membranes with Ponceau S stain (Sigma Chemical Co.) prior to application of the primary antibody. In addition, replicate samples from each experiment were analysed on separate blots using an antibody (1:1000) that recognizes β-actin or unphosphorylated (total) ERK1/2, P38 MAPK, JNK and PKB.
Statistical analysis
Results are expressed as means ± SEM. Concentration-response curves were analysed by computer-assisted iteration using the GraphPad Prism (GraphPad software, San Diego, CA). Statistical significance was determined by analysis of variance (ANOVA) followed by Dunnett's test, and P < 0.05 was considered as the limit of statistical significance.
Materials
Bovine serum albumin, DMEM, foetal calf serum, fibronectin, forskolin, CL-316243, deoxynucleotide mix, IGEPAL CA-630 and leupeptin were all obtained from Sigma Chemical Co. (Poole, Dorset, UK). Trichloroacetic acid was purchased from Calbiochem (Nottingham, UK). Dobutamine, procaterol hydrochloride, ICI 118551, CGP 20712A, SB 203580, PD 98059, SP 600125, wortmannin, KT 5720 and GF 109203X were all from Tocris (Bristol, UK). β-actin, phospho-ERK1/2 (Thr202/Tyr204), ERK1/2, phospho-SAPK/JNK (Thr183/Tyr185), SAPK/JNK, phospho-p38 MAPK (Thr180/Tyr182), p38 MAPK, phospho-PKB (Ser473) and PKB antibodies were purchased from New England Biolabs (Hitchin, UK). Mouse β1- (sc-568) and β2- (sc-570) adrenoceptor antibodies were provided by Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Mouse β3-adrenoceptor antibody was purchased from Alpha Diagnostic international (San Antonio, TX). G protein αi-2antibody was provided by Abcam (Cambridge, UK) and G protein αs was from Chemicon (Eastleigh, UK). ([8-3H]adenine was obtained from Amersham (Bucks, UK).
Results
Effect of chronic stimulation of β1, β2 or β3-adrenoceptors on the functional response induced by the β-adrenoceptor subtypes in neonatal rat cardiomyocytes
We have shown previously that β1-, β2- and β3-adrenoceptors are expressed in neonatal rat cardiomyocytes, and following chronic stimulation with noradrenaline, β1- and β2- are down-regulated whereas β3-adrenoceptors are up-regulated (Germack and Dickenson, 2006). Therefore, we investigated the functional response induced by β1-, β2- and β3-adrenoceptor stimulation following chronic stimulation of each subtype for 24 h (Figure 1, Table 2). Chronic stimulation of β1-adrenoceptors was performed using 10 µM dobutamine (a β1-adrenoceptor selective agonist) in the presence of 1 µM ICI118551 (ICI, a β2-adrenoceptor selective antagonist). Continuous activation of β2-adrenoceptors was carried out with 10 µM procaterol (a β2-adrenoceptor selective agonist) and 1 µM CGP 20712A (CGP, a β1-adrenoceptor selective antagonist). Finally, the pretreatment of cardiomyocytes with 2 µM CL-316243 (a β3-adrenoceptor selective agonist) stimulated chronically β3-adrenoceptors. The concentrations of agonists and antagonists were chosen according to their potency and maximal responses previously published (Germack and Dickenson, 2006). Following dobutamine/ICI (β1) or CL-316243 (β3) treatments, dobutamine-induced cAMP accumulation was decreased by 67% and 52%, respectively (Figure 1A) indicating an interaction between both β-adrenoceptor subtypes. In addition, the decrease in β1-adrenoceptor response was significantly higher when the cells were treated with dobutamine/ICI. Chronic stimulation of β2-adrenoceptors did not modify β1-adrenoceptor maximal response although the sensitivity to dobutamine was shifted to the right and decreased by around 10 times suggesting an interaction also between β1- and β2-adrenoceptors. Indeed, pretreatment with dobutamine/ICI resulted in a significant decrease in β2-adrenoceptor response by 69% and a decrease in sensitivity by 35 times whereas β3-adrenoceptors pretreatment had no effect on procaterol-induced cAMP accumulation (Figure 1B). Chronic stimulation of β2-adrenoceptors abolished procaterol response. As shown previously and in Figure 1C, β3-adrenoceptors are not functional in normal physiological conditions in neonatal rat cardiomyocytes (Germack and Dickenson, 2006). Chronic stimulation of β2-adrenoceptors did not induce functional β3-adrenoceptor expression. In pretreated cardiomyocytes with dobutamine/ICI or CL-316243, CL-316243 induced a significant inhibition of forskolin-mediated cAMP accumulation without changing the potency between both treatments strengthening the assumption of a compensatory regulation between β1- and β3-adrenoceptors (Table 2). In addition, CL-316243 treatment induced a higher β3-adrenoceptor response than β1-adrenoceptor treatment.
Figure 1.

Activation of cAMP accumulation induced by dobutamine, β1-selective agonist (A) and procaterol, β2-selective agonist (B), and effect of the selective β3-adrenoceptor agonist CL-316243 on forskolin (Forsk)-induced cAMP accumulation (C) in neonatal rat cardiomyocytes untreated, treated with 10 µM dobutamine in the presence of 1 µM ICI 118551, 10 µM procaterol in the presence of 1 µM CGP 20712A or 2 µM CL-316243 for 24 h. cAMP accumulation was measured as described under Methods. Data are expressed as percentage of the basal level of cAMP accumulation (100%, A, B) or as the percentage of 1.5 µM forskolin response in the absence of agonist (100%, C). Each point represents the mean ± SEM of four to six experiments performed in duplicate.
Table 2.
Effect of dobutamine/ICI118551, procaterol/CGP 20712A and CL-316243 treatments on dobutamine and procaterol-induced cAMP accumulation, and CL-316243-mediated inhibition of forskolin-induced cAMP response
| Agonist | Untreated |
Treatment |
|||
|---|---|---|---|---|---|
| β1 treatment | β2 treatment | β3 treatment | |||
| Dobutamine | Emax | 234 ± 11 | 77 ± 4*** | 219 ± 28 | 112 ± 6***, # |
| β1 response | pD2 | 6.46 ± 0.18 | 6.15 ± 0.30 | 5.05 ± 0.32*** | 6.65 ± 0.30 |
| Procaterol | Emax | 156 ± 10 | 49 ± 2*** | NR | 162 ± 12### |
| β2 response | pD2 | 7.67 ± 0.27 | 6.16 ± 0.29*** | – | 7.71 ± 0.31 |
| CL-316243 | Imax | NR | 23 ± 6 | NR | 30 ± 5# |
| β3 response | pIC50 | – | 8.04 ± 0.45 | – | 8.45 ± 0.23 |
Values are means ± SEM of four to six experiments performed in duplicate. The potencies of the agonists were evaluated by their EC50, concentration of agonist inducing 50% of the maximal cAMP accumulation expressed as –log10 EC50 (pD2) or their IC50, concentration of agonist inducing 50% of inhibition expressed as –log10 IC50 (pIC50). Emax is the maximal response expressed in % over basal response. Imax is the maximal percentage of inhibition.
P < 0.05 and
P < 0.001 versus untreated cells and
P < 0.05 and
P < 0.001 versus β1 treatment. NR means no response.
Effect of chronic stimulation of β1, β2 or β3-adrenoceptors on mRNA and protein expression of the β-adrenoceptor subtypes in neonatal rat cardiomyocytes
The functional studies suggest that a potential interaction exists between β1- and β2-adrenoceptors as well as β1- and β3-adrenoceptors. Therefore, we investigated whether this cross-regulation involves the modulation of the receptor expression by measuring the mRNA and protein level of the three receptors following the different treatments ( Figures 2 and 3). The expression of the β-adrenoceptors in untreated cells by real time PCR (Figure 2A) displayed a β1/β2/β3 ratio of 0.39/0.61/0.0008 indicating that β2-adrenoceptors are predominantly expressed in neonatal rat cardiomyocytes as recently reported by Morisco et al. (2008) using binding studies. The β1/β2/β3 mRNA ratio was 0.25/0.745/0.0017 or 0.24/0.75/0.0017 following β1- or β3-adrenoceptor treatments respectively. Although an increase in β3-adrenoceptors was observed, β3-adrenoceptor level remained very low, which may explain the absence of functional response in physiological conditions and a low functional activity (30%) following chronic stimulation. Chronic β1- or β3-adrenoceptor stimulation induced a decrease in β1-adrenoceptor mRNA level by 47% and 50% respectively (Figure 2B). At protein level, β1-adrenoceptor was reduced by 58% following β1-treatment and 42% subsequent to β3-adrenoceptor treatment (Figure 3A). β2-adrenoceptor treatment had no effect on β1-adrenoceptor expression (Figures 2B and 3A). A decrease in β2-adrenoceptor expression by around 40% occurred at mRNA and protein level when cardiomyocytes were treated with procaterol/CGP whereas β1- or β3-adrenoceptor treatments had no effect (Figures 2C and 3B). No effect of chronic β2-adrenoceptor stimulation on β3-adrenoceptor mRNA and protein level was observed (Figures 2D and 3C). The amount of β3-adrenoceptor transcripts was increased by 65% and 72%, and β3-adrenoceptor protein level by 43% and 68% following chronic stimulation of β1- or β3-adrenoceptors respectively (Figures 2D and 3C). The observed bands corresponded to the expected molecular weight (Diebold et al., 2001) and were modulated similarly to β-adrenoceptor mRNAs suggesting specific antibody binding. Overall, these data show that the modulation of receptor expression correlated well with the changes in β1- and β3-adrenoceptor functions.
Figure 2.
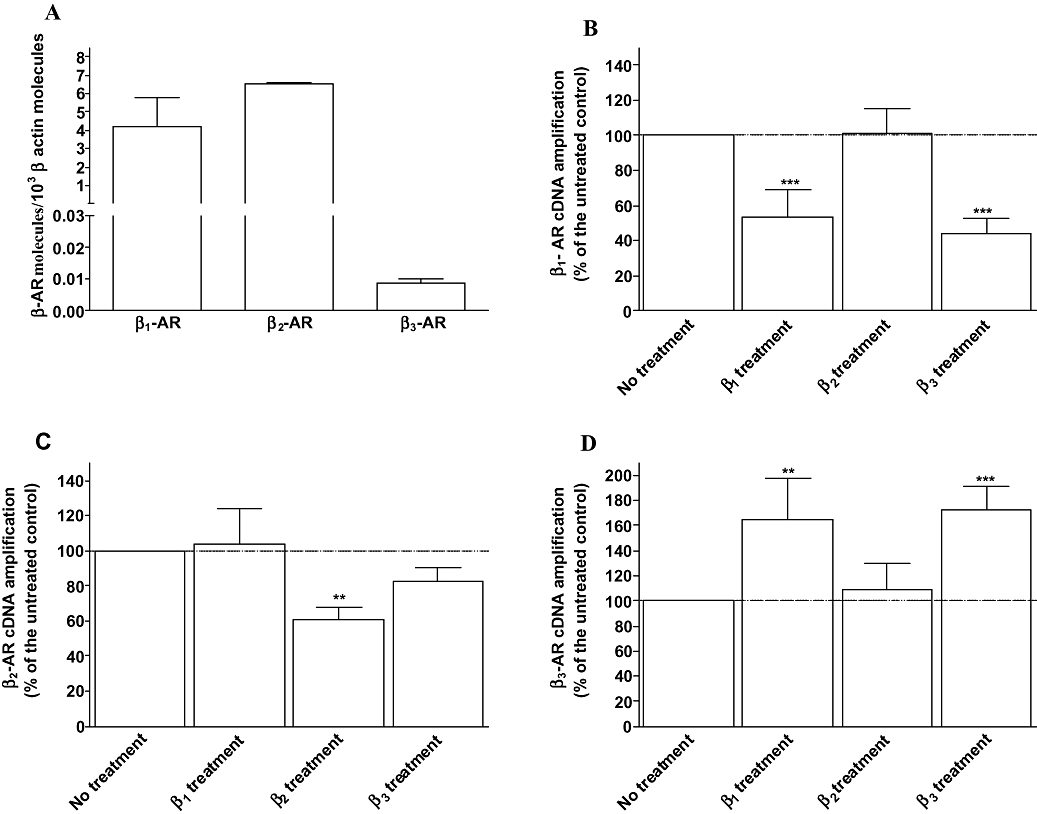
Quantitative expression of β-adrenoceptors in untreated neonatal rat cardiomyocytes (A) and expression of β1- (B), β2- (C) and β3-adrenoceptors (D) mRNA obtained from untreated (control), treated with 10 µM dobutamine in the presence of 1 µM ICI 118551 (β1 treatment), 10 µM procaterol in the presence of 1 µM CGP 20712A (β2 treatment) or 2 µM CL-316243 (β3 treatment) for 24 h. Total RNA was prepared, reverse transcription and real time PCR were carried out as described under Methods. Values were expressed as β-adrenoceptor molecules × 10−3β actin molecules in A and normalized to β-actin molecules and expressed as the percentage of untreated cardiomyocytes (100%) in B, C and D. Each point represents the mean ± SEM of five to six independent experiments. **P < 0.01 and ***P < 0.001 versus untreated control.
Figure 3.
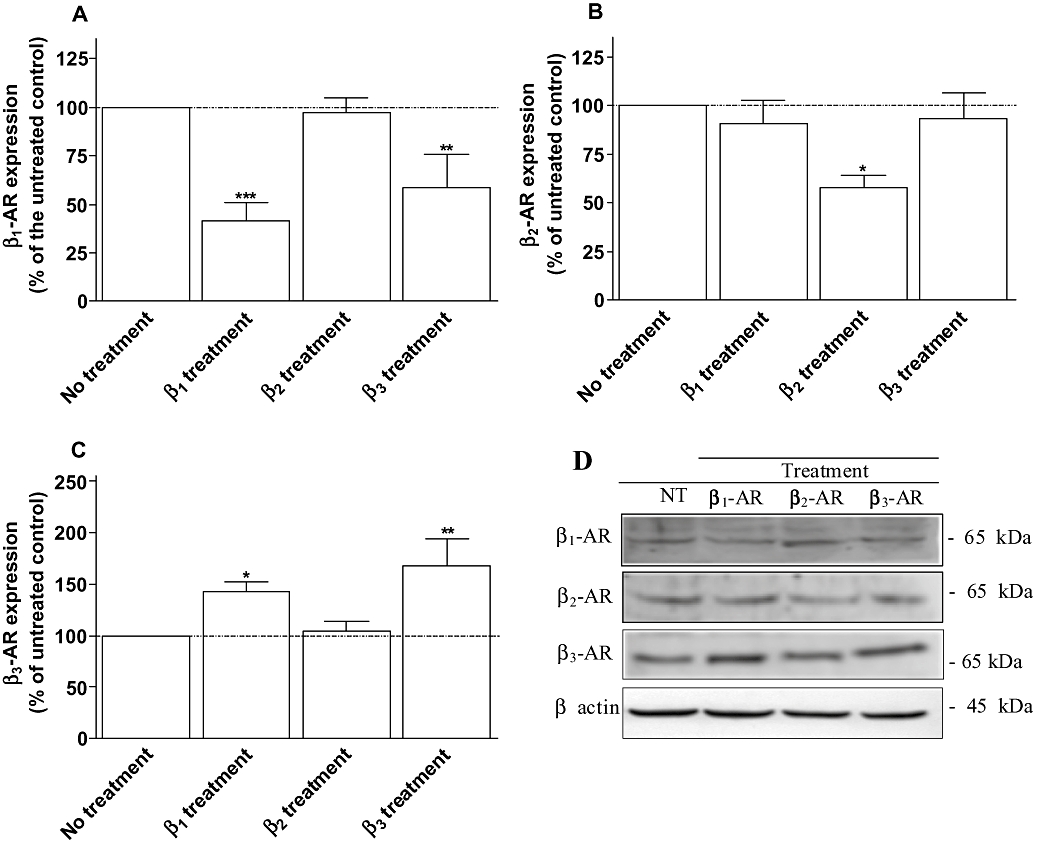
Expression of β1- (A), β2- (B) and β3-adrenoceptors (C) obtained from neonatal rat cardiomyocytes untreated (control), treated with 10 µM dobutamine in the presence of 1 µM ICI 118551 (β1 treatment), 10 µM procaterol in the presence of 1 µM CGP 20712A (β2 treatment) or 2 µM CL-316243 (β3 treatment) for 24 h. Cell membrane homogenates were monitored by Western blotting for β1-, β2- and β3-adrenoceptors as described under Methods. The same samples were also analysed on separate blots using an antibody that recognizes β actin to confirm equal loading on each lane. Representative immunoblots for each proteins are shown in panel C. Data are expressed as the percentage of untreated cardiomyocytes (100%) following the calculation of β-adrenoceptor/β-actin ratio for each lane. The combined results (panel A, B and C) obtained from densitometric analysis of blots represent the mean ± SEM of five to seven independent experiments. *P < 0.05 and **P < 0.01 versus untreated control.
Effect of chronic stimulation of β1, β2 or β3-adrenoceptors on Gαs and Gαi-2 protein in neonatal rat cardiomyocytes
Both, β1- and β3-adrenoceptor treatments induced a similar modulation of β1-adrenoceptor or β3-adrenoceptor expression, whereas the decrease in β1-adrenoceptor and increase in β3-adrenoceptor functional responses were higher in cardiomyocytes treated with dobutamine/ICI and CL-316243 respectively. These results indicate that the modulation of β1- and β3-adrenoceptor function may also involve modifications in receptor and G protein coupling.
Therefore, we investigated the expression of Gαs and Gαi-2 proteins in neonatal rat cardiomyocytes following β1-, β2- or β3-adrenoceptor treatment using Western blotting (Figure 4). Gαs protein level was not significantly modified in cardiomyocytes following the three treatments (Figure 4A). Inversely, Gαi-2 protein was increased by 44% when β1-adrenoceptors were chronically stimulated and by 94% and 112% following β2- or β3-adrenoceptor treatments respectively (Figure 4B). These results suggest that the higher inhibition of the response to forskolin induced by β3-adrenoceptor stimulation following CL-316243 treatment, compared with dobutamine/ICI treatment involves a better coupling to Gαi-2 protein.
Figure 4.
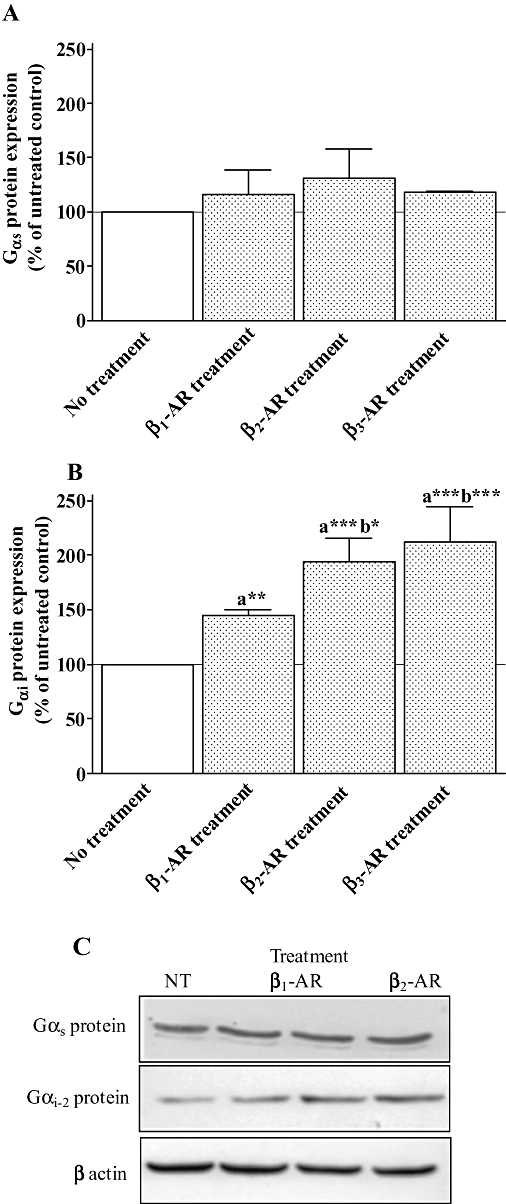
Expression of Gαs and Gαi proteins obtained from neonatal rat cardiomyocytes untreated (control), treated with 10 µM dobutamine in the presence of 1 µM ICI 118551 (β1 treatment), 10 µM procaterol in the presence of 1 µM CGP 20712A (β2 treatment) or 2 µM CL-316243 (β3 treatment) for 24 h. Cell membrane homogenates were monitored by Western blotting for Gαs (A) and Gαi (B) proteins as described under Methods. The same samples were also analysed on separate blots using an antibody that recognizes β actin to confirm equal loading on each lane. Representative immunoblots for each proteins are shown in panel C. Data are expressed as the percentage of untreated cardiomyocytes (100%) following the calculation of β-adrenoceptor/β-actin ratio for each lane. The combined results (panels A and B) obtained from densitometric analysis of blots represent the mean ± SEM of four to six independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, (a) versus untreated control and (b) versus β1-adrenoceptor treatment.
Effect of kinase inhibitors on the down-regulation of β1-adrenoceptor functional response induced by chronic stimulation of β1 or β3-adrenoceptors in neonatal rat cardiomyocytes
PI3K, PKA, PKC have been shown to be involved in the process of β1-adrenoceptor down-regulation (Li et al., 1998; Dzimiri, 1999; Naga Prasad et al., 2001; Nienaber et al., 2003). In addition, ERK1/2, SAPK/JNK and p38 seem to be also involved in the regulation of β-adrenoceptor expression following chronic stimulation (Headley et al., 2004). As β2-adrenoceptor treatment did not show any interaction at both expression and functional levels between β2- and β1-adrenoceptors, we investigated the signalling pathway involved in the down-regulation of the β1-adrenoceptor following chronic stimulation of β1- and β3-adrenoceptors. In order to determine the mechanisms implicated in β1-adrenoceptor down-regulation, cardiomyocytes were treated for 24 h with dobutamine/ICI or CL-316243 in the presence or absence of inhibitors of MAP kinase (MEK1, 50 µM, PD 98059), PI3K (100 nM, wortmannin), p38 MAPK (10 µM, SB 203580), SAPK/JNK (10 µM, SP 600125), PKA (1 µM, KT 57201) and PKC (10 µM, GF 109203). As shown in Figure 5, wortmannin, SB 203580 and GF 10920 produced a decrease in dobutamine-induced cAMP accumulation by 25%, 30% and 24% in untreated cells respectively. The inhibition of β1-adrenoceptor response in untreated cardiomyocytes was likely to occur following dobutamine/ICI and CL-316243 treatments. Therefore, to consider only the effect of both treatments on the modulation of β1-induced cAMP accumulation, the functional values obtained in the treated groups were corrected from the inhibitory effect of wortmannin, SB203580 or GF 10920 observed in untreated cells (decrease in untreated cells + value of treated cardiomyocytes for each individual experiments), as explained in the legend of Figure 5. PD 98059 and SP 600125 had no effect on the functional down-regulation induced by both β1- or β3-adrenoceptor treatments indicating that MEK and SAPK/JNK are not implicated in this process. Wortmannin, SB 203580 and GF 109203 reversed the inhibition of dobutamine response following β1-adrenoceptor treatment by 64%, 32% and 58%, and by 44%, 20% and 34% when cardiomyocytes were treated with CL-316243 (β3-adrenoceptor treatment) respectively. The PKA inhibitor, KT 57201 counteracted the decrease in β1-adrenoceptor function by 26% in the cells treated with dobutamine/ICI and did not modify β1-induced cAMP accumulation in CL-316243 treated cardiomyocytes.
Figure 5.
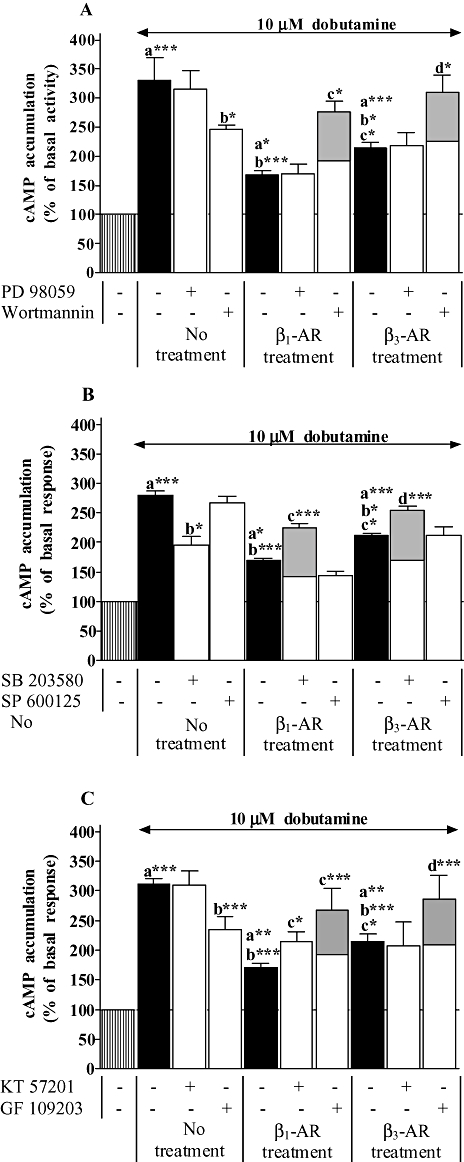
Effect of kinase inhibitors on dobutamine-induced cAMP accumulation in neonatal rat cardiomyocytes untreated, treated with 10 µM dobutamine in the presence of 1 µM ICI 118551 (β1 treatment), or 2 µM CL-316243 (β3 treatment) for 24 h. In addition, untreated and treated cells were incubated in panel A with 50 µM PD 98059 (MEK inhibitor) or 100 nM wortmannin (phosphoinositide 3-kinase inhibitor), in panel B with 10 µM SB 203580 (p38 MAPK inhibitor) or 10 µM SP 600125 (JNK inhibtor) and in panel C with 1 µM KT 57201 (PKA inhibitor) or 10 µM GF 109203 (PKC inhibitor). cAMP accumulation was measured as described under Methods. Data are expressed as the percentage of the basal level of cAMP accumulation (set at 100%, striped bars). Black and open bars represent cAMP accumulation induced by dobutamine in the absence and presence of protein kinase inhibitors in all groups respectively. Wortmanin (A), SB 203580 (B) and GF 109203 (C) elicited a significant decrease in cAMP accumulation induced by dobutamine in untreated cardiomyocytes. Grey filled bars represent the data corrected from the effect of these inhibitors on dobutamine response in untreated cardiomyocytes in order to consider only the effect of β1- or β3-adrenoceptor treatments on the modulation of β1-adrenoceptor response (decrease in untreated cells + value of treated cardiomyocytes for each individual experiments). Each bar represents the mean ± SEM of six to seven independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, (a) versus basal level, (b) versus dobutamine response in untreated cells, (c) versus dobutamine response in β1-adrenoceptor-treated cells and (d) versus dobutamine response in β3-adrenoceptor-treated cells.
Effect of kinase inhibitors on the up-regulation of β3-adrenoceptor functional response induced by chronic stimulation of β1 or β3-adrenoceptors in neonatal rat cardiomyocytes
β3-Adrenoceptor agonist-induced inhibition of responses to forskolin was investigated when cardiomyocytes were treated for 24 h with dobutamine/ICI or CL-316243 in the presence or absence of inhibitors of MEK1 (50 µM, PD 98059), PI3K (100 nM, wortmannin), p38 (10 µM, SB 203580), SAPK/JNK (10 µM, SP 600125), PKA (1 µM, KT 57201) and PKC (10 µM, GF 10920). As shown in Figure 6, PD 98059, SB 203580 and GF 10920 inhibited β1- or β3-adrenoceptor treatment-induced up-regulation of β3-adrenoceptor functional response indicating the involvement of MEK, p38 and PKC in the regulation of the β3-adrenoceptor subtype. KT 57201 also abolished the increase in β3-adrenoceptor response when the cells were treated with dobutamine/ICI but not with CL-316243. No effect of wortmannin and SP 600125 was observed indicating that PI3K and SAPK/JNK do not regulate β3-adrenoceptor up-regulation.
Figure 6.
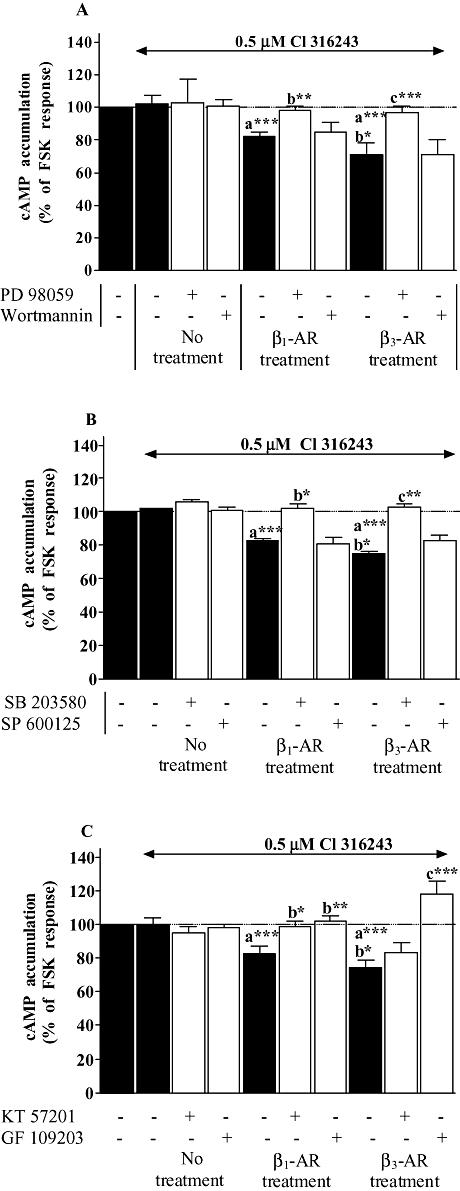
Effect of kinase inhibitors on CL-3162436243-inhibited forskolin response in neonatal rat cardiomyocytes untreated, treated with 10 µM dobutamine in the presence of 1 µM ICI 118551 (β1 treatment), or 2 µM CL-316243 (β3 treatment) for 24 h. In addition, untreated and treated cells were incubated in panel A with 50 µM PD 98059 (MEK inhibitor) or 100 nM wortmannin (phosphoinositide 3-kinase inhibitor), in panel B with 10 µM SB 203580 (p38 MAPK inhibitor) or 10 µM SP 600125 (JNK inhibtor) and in panel C with 1 µM KT 57201 (PKA inhibitor) or 10 µM GF 109203 (PKC inhibitor). cAMP accumulation was measured as described under Methods. Data are expressed as the percentage of 1.5 µM forskolin (FSK) response in the absence of agonist (100%, striped bars). Black and open bars represent the inhibition of FSK-mediated cAMP accumulation induced by CL-316243 in the absence and presence of protein kinase inhibitors in all groups respectively. Each bar represents the mean ± SEM of six to seven independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001, (a) versus CL-316243 response in untreated cells, (b) versus CL-316243 response in β1-adrenoceptor treated cells and (c) versus CL-316243 response in β3-adrenoceptor treated cells.
Effect of chronic stimulation of β1 or β3-adrenoceptors on ERK1/2, p38, SAPK/JNK and Akt activation in neonatal rat cardiomyocytes
The functional study indicates that PI3K, MEK, p38 but not SAPK/JNK are involved in the modulation of the receptors following the different treatments. Consequently, chronic stimulation of β1- and β3-adrenoceptors should lead to the activation of p38, extracellular signal regulated-kinase (ERK1/2, down-stream MEK target) and Akt (down-stream PI3K target). Therefore, we investigated the activation of ERK1/2, p38, SAPK/JNK and Akt by either dobutamine/ICI or CL-316243 treatments. Chronic stimulation of β1- or β3-adrenoceptor subtypes induced ERK1/2 phosphorylation by 17% and 49% respectively (Figure 7A). Significantly, lower ERK1/2 activation was obtained when the cells were treated with dobutamine/ICI compared with chronic β3-adrenoceptor stimulation. Both treatments activated p38 by around 60%, which correlated well with the involvement of this kinase in the regulation of β1- and β3-adrenoceptor functional response following dobutamine/ICI or CL-316243 treatments (Figure 7B). Chronic β1-adrenoceptor stimulation produced an increase in SAPK/JNK activation by 241% whereas β3-adrenoceptor treatment had no effect (Figure 7C). Interestingly, Akt was phosphorylated by 149% when cardiomyocytes were treated with CL-316243 and not with dobutamine/ICI (Figure 7D) indicating a possible involvement of the β3-adrenoceptor subtype in cell survival.
Figure 7.
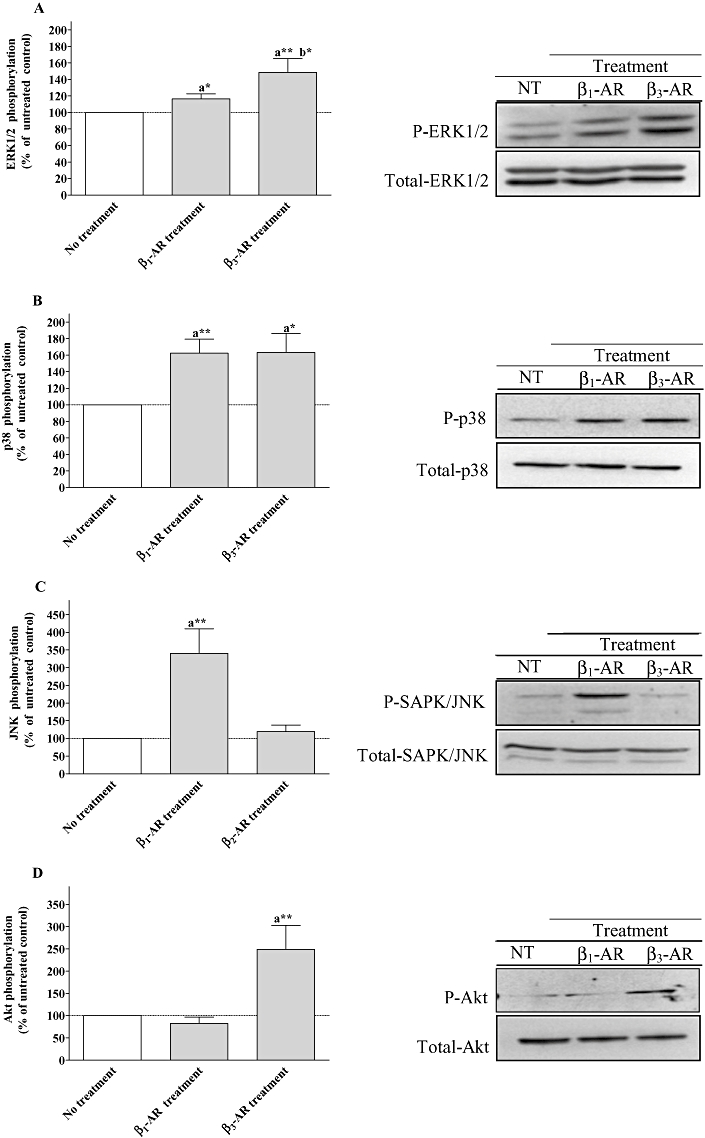
Effect of β1-adrenoceptor and β3-adrenoceptor treatments on ERK1/2, p38 MAPK, stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and Akt activation in neonatal rat cardiomyocytes. The cells were untreated or treated with 10 µM dobutamine in the presence of 1 µM ICI 118551 (β1-AR treatment), or 2 µM CL-316243 (β3-AR treatment) for 24 h. Following the treatments, cell lysates were analysed by Western blotting as described under Methods using phospho-specific antibodies. The same samples were also analysed on separate blot using an antibody that recognizes both unphosphorylated (total) and phosphorylated kinases to confirm equal loading on each lane. Data are expressed as percentage of the untreated control following the calculation of the phosphorylated/total ratio for each lane. The combined results (panel A for ERK1/2, panel B for p38 mitogen-activated protein kinase, panel C for SAPK/JNK and panel D for Akt) obtained from densitometric analysis of blots, represent the mean ± SEM of for to six independent experiments. *P < 0.05 and **P < 0.01, (a) versus untreated control, (b) versus β1-adrenoceptor treated cells.
Discussion and conclusions
We have previously shown that chronic β-adrenoceptor stimulation with noradrenaline induces a decrease in β1-adrenoceptors and an increase in β3-adrenoceptors at the functional, genomic and protein levels (Germack and Dickenson, 2006). For the first time, the present study provides evidence of a cross-regulation at the expression and functional level between β1- and β3-adrenoceptors following chronic receptor stimulation via PI3K, PKC, p38 MAPK and MEK/ERK1/2 pathway in neonatal rat cardiomyocytes.
Changes in cardiac rate and force are triggered by β1- and β2-adrenoceptors acting via the Gs/cAMP/PKA signalling pathway whereas β3-adrenoceptors mediate a decrease in contractility through a coupling to Gi protein (Dzimiri, 1999; Rozec and Gauthier, 2006). It is well established that β1- and β2-adrenoceptors are down-regulated following chronic stimulation (Dzimiri, 1999). As expected, in this study, chronic β1-adrenoceptor treatment led to a decrease in β1-adrenoceptor-induced cAMP accumulation and continuous β2-adrenoceptor stimulation abolished β2-adrenoceptor response, which was well correlated to the receptor expression. Interestingly, β2-adrenoceptor treatment decreased sensitivity to dobutamine without changing β1-adrenoceptor expression. Similarly, chronic β1-adrenoceptor stimulation reduced β2-adrenoceptor maximal response and efficiency with no alteration in β2-adrenoceptor level. These results indicate that the modification of β1- and β2-adrenoceptor functional response without modification of receptor expression may reflect an interaction between both subtypes at the membrane level. Indeed, Zhu et al. (2005) have demonstrated that adult cardiomyocytes elicit not only a colocalization and physical association between β1- and β2-adrenoceptors, but also a functional synergy in cAMP and contractile responses. The disruption of β1- and β2-adrenoceptor heterodimerization may also contribute to the decrease in cardiac function in addition to receptor down-regulation in heart diseases where catecholamines are elevated (Dzimiri, 1999; Tilley and Rockman, 2006). Chronic β3-adrenoceptor stimulation also produced a decrease in β1-adrenoceptor response and expression. In contrast, this treatment induced an increase in β3-adrenoceptor function and expression as observed in cardiomyocytes continuously exposed to β1-adrenoceptor treatment. It is noteworthy that the relative abundance of the β3-adrenoceptor in untreated neonatal rat cardiomyocytes was similar to the relative β3-adrenoceptor level observed in healthy human hearts using also real-time PCR (β1/β2/β3 ratio: neonatal rat 0.39/0.61/0.0008 vs. human 0.71/0.28/0.0012; Moniotte et al., 2007). Sepsis is also associated with impairment in β-adrenoceptor response and myocardial dysfunction (Silverman et al., 1993; Muller-Werdan et al., 2006). Interestingly, the relative proportion of the β3-adrenoceptor increased in human hearts from septic patients, from 0.0012 to 0.0024 (Moniotte et al., 2007) as observed in our study in neonatal rat cardiomyocytes following β1- or β3-adrenoceptor treatments (0.0008 to 0.0017). In addition, Moniotte et al. (2007) when using cytokines in murine cardiomyocytes as a model of sepsis showed a decrease in contractility by around 45% following β3-adrenoceptor stimulation. All together, these data indicate that even when it is up-regulated but expressed at low level, the β3-adrenoceptor may regulate cardiac function in pathological situation. Overall, our results clearly indicate that changes in β1- and β3-adrenoceptor regulation following chronic stimulation involves a cross-talk between both subtypes as observed in failing human and animal hearts (Cheng et al, 2001; Moniotte et al., 2001; Morimoto et al., 2004; Zhang et al., 2005).
The up-regulation of β3-adrenoceptor function following β3-adrenoceptor treatment was higher by 30% compared with chronic β1-adrenoceptor activation. In addition, both treatments did not produce a significant difference in the increase in β3-adrenoceptor expression suggesting an enhancement in the coupling between β3-adrenoceptors and Gαi protein. Indeed, the overexpression of Gαi protein in neonatal cardiomyocytes reduced isoprenaline-activated adenylyl cyclase, which was restored in the presence of Pertussis toxin (PTX) (Rau et al, 2003). Furthermore, the increase in Gαi protein level in addition to β-adrenoceptor down-regulation led to the desensitization of functional response, which was improved with PTX in failing rat heart (Kompa et al., 1999; Xiao et al., 2003). These works indicate that the function mediated by β-adrenoceptor is also regulated by coupling to Gαi protein when this protein is highly expressed. Although three isoforms Gαi-1, 2 and 3 have been identified, Gαi-2 is mainly expressed in the heart and up-regulated during heart failure (El-Armouche et al., 2003). In addition, the inhibition of cardiac Gαi-2 increased infarct size and apoptosis in transgenic mice expressing a Gαi-2 inhibitor peptide, when the mice were subjected to ischemia/reperfusion indicating an important role of this isoform in cardioprotection. In our study, Gαi-2 expression was increased by both, β1- or β3-adrenoceptor treatments as observed in neonatal cardiomyocytes chronically treated with noradrenaline and in failing rat heart (Reithmann et al., 1990; Kompa et al., 1999; Xiao et al., 2003). Interestingly, cardiomyocytes chronically stimulated with CL-316243 displayed a further increase in Gαi-2 protein level compared with β1-adrenoceptor treated cells by 32%. We can assume that an enhancement in β3-adrenoceptor coupling to Gαi protein may explain the difference in β3-adrenoceptor functional response between both, β1- and β3-adrenoceptor treatments. It is noteworthy that β2-adrenoceptor treatment also increased Gαi-2 protein as much as β3-adrenoceptor treatment without inducing β3-adrenoceptor functional response. This implies that the up-regulation of β3-adrenoceptor function requires an enhancement of the receptor expression taking place at the genomic level and an increase in Gαi protein as observed with chronic β1-adrenoceptor stimulation. In contrast, chronic β1-adrenoceptor stimulation led to a lower decrease in β1-adrenoceptor response by 31% compared with chronic β3-adrenoceptor activation suggesting that the increase in Gαi protein did not affect β1-adrenoceptor response as previously reported in cardiomyocytes from failing heart (Xiao et al., 2003).
β1-Adrenoceptor desensitization requires receptor phosphorylation by GRK2 (agonist dependent, homologous regulation) and PKA (agonist independent, heterologous regulation), which alters the coupling between the receptor and Gαs protein, leading to the reduction of β1-adrenoceptor functional response (Dzimiri, 1999; Tilley and Rockman, 2006). In the present study, the PKA inhibitor, KT 57 201 partially blocked the functional β1-adrenoceptor down-regulation following chronic β1-adrenoceptor stimulation, whereas this inhibitor had no effect on β3-adrenoceptor treatment-induced decrease in β1-adrenoceptor response. β1-adrenoceptors desensitization by PKA may explain the difference in β1-adrenoceptor down-regulation observed between chronic β1- and β3-adrenoceptor stimulation. Interestingly, the up-regulation of β3-adrenoceptor response was counteracted by KT 57201 only following chronic β1-AR activation. These data confirm previous reports that β3-adrenoceptors do not activate the Gαs/PKA signalling pathway in cardiomyocytes (Germack and Dickenson, 2006; Rozec and Gauthier, 2006). In our study, the PKC inhibitor, GF 109203, also reversed β1-adrenoceptor responsiveness following both, β1- or β3-adrenoceptor treatments. As previously shown, PKC mediates heterologous desensitization of β-adrenoceptors. Indeed, pretreatement of rat C6 glioma cells with phorbol 12-myristate 13-acetate (PMA, a PKC activator) produced β1-adrenoceptor down-regulation at protein and mRNA levels (Li et al., 1998). Guimond et al. (2005) have reported that overexpression of PKC isoforms (α, βII, ε and ζ) in HEK 293 cells transfected with β1-adrenoceptors resulted in the decrease in β1-adrenoceptor-induced adenylyl cyclase activity. Finally, PKC produced by the activation of angiotensin II receptors and overexpression α1B-ARs reduced heart rate and inotropic response induced by β1-ARs respectively (Schwartz and Naff, 1997; Lemire et al., 1998). The PKC inhibitor also impaired the functional β3-adrenoceptor up-regulation in chronic β1- or β3-adrenoceptor-treated cardiomyocytes. It is noteworthy that Gβγ subunits from GPCRs activate PI3K (Salazar et al., 2007) that in turn targets PKC ε, η, ζ and λ (Toker and Cantley, 1997). These observations may explain PKC activation mediated by chronic β1- or β3-adrenoceptor stimulation observed in our investigation.
We found in the present study that β1- or β3-adrenoceptor treatments in the presence of the PI3K inhibitor wortmannin restored β1-adrenoceptor function in cardiomyocytes indicating the recruitment of PI3K by either chronic β1- or β3-adrenoceptor stimulation in the regulation of β1-adrenoceptors. Indeed, β-adrenoceptor desensitization and down-regulation require PI3Kγ recruitment through the interaction with GRK2 (Naga Prasad et al., 2001; Ribas et al., 2007). In addition, cardiac overexpression of inactive PI3Kγ in mice prevented β-adrenoceptor down-regulation and desensitization to chronic isoprenaline administration, and preserved β-adrenoceptor-induced left ventricle contractility and adenylyl cyclase activity (Nienaber et al., 2003). Although the heart expressed two PI3K isoforms, PI3Kα and PI3Kγ the former form is mainly involved in the regulation of GPCRs (Salazar et al., 2007). In contrast to β1-adrenoceptor down-regulation, wortmannin had no effect on β3-adrenoceptor up-regulation induced by both, β1- or β3-adrenoceptor treatments. As shown previously, short term β3-adrenoceptor stimulation induced ERK1/2 activation through Gi/o/PI3K pathway in β3-adrenoceptor transfected CHO/K1 cells and human lung epithelial-derived cell line A549 (Gerhardt et al., 1998; Robay et al., 2005). In addition, Akt was also activated by the Gi/o/PI3K pathway in β3-adrenoceptor transfected cells (Gerhardt et al., 1998) as observed in our study where chronic β3-adrenoceptor but not β1-adrenoceptor treatments induced Akt phosphorylation. Liu et al. (2005) showed that Akt binds to GRK2 and this association inhibits Akt activity. This regulation may explain why chronic β1-adrenoceptor stimulation did not induce Akt phosphorylation while PI3K was activated. Overall, these data indicate that the chronic stimulation of both, β1- or β3-adrenoceptors, activates PI3K leading to β1-adrenoceptor down-regulation. In addition, continuous β3-adrenoceptor treatment triggered Akt activation suggesting the involvement of this subtype in cardioprotection. Indeed, the PI3K/Akt pathway is well known to induce cell viability through an antiapoptotic effect (Matsui and Rosenzweig, 2005).
Chronic β-adrenoceptor stimulation activates several members of the MAPK family including p38 MAPKs, SAPK/JNK and ERK1/2, which may be involved in the regulation of the β-adrenoceptor receptors (Palfi et al., 2005; Kim et al., 2006). The p38 MAPK inhibitor, SB 203580 inhibited the functional β1- and β3-adrenoceptor changes induced by β1- or β3-adrenoceptor treatments. In addition, both treatments activated p38 MAPK as previously suggested by Peter et al. (2007) regarding β1-adrenoceptors. Indeed, chronic β1-adrenoceptor stimulation enhances p38 MAPK activation in left ventricle from transgenic mice overexpressing β1-adrenoceptors. In contrast to SB 203580, the SAPK/JNK inhibitor, SP 6000125 did not modify the functional β1- and β3-adrenoceptor response modulated by both β1- or β3-adrenoceptor treatments. However, chronic β1-adrenoceptor stimulation activated SAPK/JNK as observed in rat heart following continuous isoprenaline administration (Palfi et al., 2005). Chronic β1- or β3-adrenoceptor stimulation also mediated ERK1/2 activation. Soeder et al. (1999) have shown that continuous β3-adrenoceptor stimulation leads to ERK1/2 activation through the Gαi pathway in 3T3F442A adipocytes as observed in the present study in neonatal cardiomyocytes. However, Robidoux et al. (2006) showed that β3-adrenoceptors activated ERK1/2 through a PKA-dependent and PKA-independent pathway in 3T3-L1 cells, the former involving src and epidermal growth factor receptor kinase. This seems to indicate that different pathways inducing ERK1/2 activation may be cell type dependent. However, only β3-adrenoceptor up-regulation was sensitive to PD 98059 (MEK inhibitor). It is well known that MAPKs are involved in the regulation of transcription factors and gene expression (Edmunds and Mahadevan, 2004). The modulation of β1- and β3-adrenoceptor mRNA levels suggests the involvement of a transcriptional regulation in the change of receptor expression following chronic β-adrenoceptor stimulation. Therefore, β1-adrenoceptor down-regulation and β3-adrenoceptor up-regulation should involve the activation of transcription factors like ICER or cAMP response element binding protein via MAPK stimulation. Further studies are required to investigate possible transcriptional mechanism involved in the cross-regulation between β1- and β3-adrenoceptors.
In conclusion, we have presented novel data showing that chronic stimulation of β1- or β3-adrenoceptors leads to an opposite regulation of the receptors at the expression and functional levels. The cross-talk between β1- and β3-adrenoceptors occurs via the activation of PKC, PI3K, p38 MAPK and MEK/ERK1/2 pathway, and through PKA when β1-adrenoceptors are continuously stimulated.
Acknowledgments
This work was supported by the British Heart Foundation Grant FS/03/095/16317.
Glossary
Abbreviations:
- DMEM
Dulbecco's modified Eagle's medium
- ERK1/2
extracellular responsive kinase
- GPCR
G-protein coupled receptor
- PKA
protein kinase A
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- p38MAPK
p38 mitogen-activated protein kinase
- PTX
Pertussis toxin
- SAPK/JNK
stress-activated protein kinase/c-Jun N-terminal kinase
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier J, Rannou-Bekono F, Marchais J, Tanguy S, Carre F. Alteration of beta(3)-adrenoceptors expression and their myocardial functional effects in physiological model of chronic exercise-induced cardiac hypertrophy. Mol Cell Biochem. 2007;300:69–75. doi: 10.1007/s11010-006-9370-9. [DOI] [PubMed] [Google Scholar]
- Bensaid M, Kaghad M, Rodriguez M, Le Fur G, Caput D. The rat beta 3-adrenergic receptor gene contains an intron. FEBS Lett. 1993;318:223–226. doi: 10.1016/0014-5793(93)80516-w. [DOI] [PubMed] [Google Scholar]
- Cheng H-J, Zhang Z-H, Onishi K, Ukai T, Sane DC, Cheng C-P. Upregulation of functional β3-adrenergic receptor in the failing canine myocardium. Circ Res. 2001;89:599–606. doi: 10.1161/hh1901.098042. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- Diebold Y, Ríos JD, Hodges RR, Rawe I, Dartt DA. Presence of nerves and their receptors in mouse and human conjunctival goblet cells. Invest Ophthalmol Vis Sci. 2001;42:2270–2282. [PubMed] [Google Scholar]
- Dinçer ÜD, Bidasee KR, Güner S, Tay A, Özçelikay AT, Altan VM. The effect of diabetes on expression of β1-, β2- and β3-adrenoceptors in rat hearts. Diabetes. 2001;50:455–461. doi: 10.2337/diabetes.50.2.455. [DOI] [PubMed] [Google Scholar]
- Dzimiri N. Regulation of β-adrenoceptor signalling in cardiac function and disease. Pharmacol Rev. 1999;51:465–501. [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J Cell Sci. 2004;117:3715–3723. doi: 10.1242/jcs.01346. [DOI] [PubMed] [Google Scholar]
- El-Armouche A, Zolk O, Rau T, Eschenhagen T. Inhibitory G-proteins and their role in desensitization of the adenylyl cyclase pathway in heart failure. Cardiovasc Res. 2003;60:478–487. doi: 10.1016/j.cardiores.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Gerhardt CC, Gros J, Strosberg AD, Issad T. Stimulation of the extracellular signal-regulated kinase ½ patway by human beta-3 adrenergic receptor: new [pharmacological profile and mechanism of action. Mol Pharmacol. 1998;55:255–262. doi: 10.1124/mol.55.2.255. [DOI] [PubMed] [Google Scholar]
- Germack R, Dickenson JM. Induction of β3-adrenergic receptor functional expression following chronic stimulation with noradrenaline in neonatal rat cardiomyocytes. J Pharmacol Exp Ther. 2006;316:392–402. doi: 10.1124/jpet.105.090597. [DOI] [PubMed] [Google Scholar]
- Guimond J, Mamarbachi AM, Allen BG, Rindt H, Hebert TE. Role of specific protein kinase C isoforms in modulation of β1- and β2-adrenergic receptors. Cellular Signalling. 2005;17:49–58. doi: 10.1016/j.cellsig.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Headley VV, Tanveer R, Greene SM, Zweifach A, Port JD. Reciprocal regulation of beta-adrenergic receptor mRNA stability by mitogen activated protein kinase activation and inhibition. Mol Cell Biochem. 2004;258:109–119. doi: 10.1023/b:mcbi.0000012841.03400.42. [DOI] [PubMed] [Google Scholar]
- Kim N, Kim H, Youm JB, Park WS, Warda M, Ko JH, et al. Site specific activation of ras/raf/ERK signalling in rabbit isoproterenol-induced left ventricular hypertrophy. BBA. 2006;1763:1067–1075. doi: 10.1016/j.bbamcr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Takaoka H, McDonald PH, Perry SJ, Mao L, Lefkowitz RJ, et al. Augmentation of cardiac contractility mediated by human β3-adrenergic receptor overexpressed in the hearts of transgenic mice. Circulation. 2001;104:2485–2491. doi: 10.1161/hc4501.098933. [DOI] [PubMed] [Google Scholar]
- Kompa AR, Gu XH, Evans BA, Summers RJ. Desensitization of cardiac β-adrenoceptor signalling with heart failure produced by myocardial infarction in the rat. Evidence for the role of Gi but not Gs or phosphorylating proteins. J Mol Cell Cardiol. 1999;31:1185–1201. doi: 10.1006/jmcc.1999.0951. [DOI] [PubMed] [Google Scholar]
- Lemire I, Allen BG, Rindt H, Hebert TE. Cardiac-specific overexpression of alpha1BAR regulates betaAR activity via molecular crosstalk. J Mol Cell Cardiol. 1998;30:1827–1839. doi: 10.1006/jmcc.1998.0746. [DOI] [PubMed] [Google Scholar]
- Li Z, Vaidya VA, Alvaro JD, Iredale PA, HS UR, Hoffman G, et al. Protein kinase C-mediated down-regulation of β1-adrenergic receptor gene expression in rat C6 glioma cells. Mol Pharmacol. 1998;54:14–21. doi: 10.1124/mol.54.1.14. [DOI] [PubMed] [Google Scholar]
- Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005;11:952–958. doi: 10.1038/nm1289. [DOI] [PubMed] [Google Scholar]
- Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling survival and function: the role of PI3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Moniotte S, Kobzik L, Feron O, Trochu J-N, Gauthier C, Balligand J-L. Up-regulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649–1655. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- Moniotte S, Belge C, Sekkali B, Massion PB, Rozec B, Dessy C, et al. Sepsis is associated with an upregulation of functional beta3 adrenoceptors in the myocardium. Eur J Heart Fail. 2007;9:1163–1171. doi: 10.1016/j.ejheart.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Morimoto A, Hasegawa H, Cheng HJ, Little W, Cheng CP. Endogenous β3-adrenoceptor activation contributes to left ventricular and cardiomyocytes dysfunction in heart failure. Am J Physiol. 2004;286:H2425–H2433. doi: 10.1152/ajpheart.01045.2003. [DOI] [PubMed] [Google Scholar]
- Morisco C, Marrone C, Galeotti J, Shao D, Vatner DE, Vatner SF, et al. Endocytosis machinery is required for beta1-adrenergic receptor-induced hypertrophy in neonatal rat cardiac myocytes. Cardiovasc Res. 2008;78:36–44. doi: 10.1093/cvr/cvn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Werdan U, Buerke M, Ebelt H, Heinroth KM, Herklotz A, Loppnow H, et al. Septic cardiomyopathy – a not yet discovered cardiomyopathy? Exp Clin Cardiol. 2006;11:226–236. [PMC free article] [PubMed] [Google Scholar]
- Naga Prasad SV, Barak LS, Rapacciuolo A, Caron MG, Rockman HA. Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by beta-adrenergic receptor kinase 1: a role in receptor sequestration. J Biol Chem. 2001;276:18953–18959. doi: 10.1074/jbc.M102376200. [DOI] [PubMed] [Google Scholar]
- Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, et al. Inhibition of receptor-localized PI3K preserves cardiac β-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 2003;112:1067–1079. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliari R, Peyrin L. Physical conditioning in rat's influences the central and peripheral catecholamine responses to sustained exercise. Eur J Appl Physiol Occup Physiol. 1995;71:41–52. doi: 10.1007/BF00511231. [DOI] [PubMed] [Google Scholar]
- Palfi A, Toth A, Kulcsar G, Hanto K, Deres P, Bartha E, Halmosi R, et al. The role of Akt and mitogen-activated protein kinase systems in protective effect of Poly(ADP-ribose) polymerase inhibition in Langendorff perfused and in isoproterenol-damaged rat hearts. J Pharmacol Exp Ther. 2005;315:273–282. doi: 10.1124/jpet.105.088336. [DOI] [PubMed] [Google Scholar]
- Peter PS, Brady JE, Yan L, Chen W, Engelhardt S, Wang Y, et al. Inhibition of p38a MAPK rescues cardiomyopathy induced by overexpressed β2-adrenergic receptor, but not β1-adrenergic receptor. J Clin Invest. 2007;117:1335–1343. doi: 10.1172/JCI29576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau T, Nose M, Remmers U, Weil J, Weissmüller A, Davia K. Overexpression of wild-type Galpha(i)-2 suppresses beta-adrenergic signaling in cardiac myocytes. FASEB J. 2003;17:523–525. doi: 10.1096/fj.02-0660fje. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, García-Hoz C, Jurado-Pueyo M, et al. The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Reithmann C, Gierschik P, Müller U, Werdan K, Jakobs KH. Pseudomonas exotoxin A prevents beta-adrenoceptor-induced up-regulation of Gi protein alpha-subunits and adenylyl cyclase desensitization in rat heart muscle cells. Mol Pharmacol. 1990;37:631–638. [PubMed] [Google Scholar]
- Robay A, Toumaniantz G, Leblais V, Gauthier C. Transfected β3- but β2-adrenergic receptors regulate cystic fibrosis transmembrane conductance regulator activity via a new pathway involving the mitogen-activated protein kinases extracellular signal-regulated kinases. Mol Pharmacol. 2005;67:648–654. doi: 10.1124/mol.104.002097. [DOI] [PubMed] [Google Scholar]
- Robidoux J, Kumar N, Daniel KW, Moukdar F, Cyr M, Medvedev AV, et al. Maximal beta3-adrenergic regulation of lipolysis involves Src and epidermal growth factor receptor-dependent ERK1/2 activation. J Biol Chem. 2006;281:37794–37802. doi: 10.1074/jbc.M605572200. [DOI] [PubMed] [Google Scholar]
- Rozec B, Gauthier C. β3-adrenoceptors in the cardiovascular system: putative roles in human pathologies. Pharmacol Ther. 2006;111:652–673. doi: 10.1016/j.pharmthera.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Salazar NC, Chen J, Rockman HA. Cardiac GPCRs: GPCR signalling in healthy and failing hearts. Biochim Biophys Acta. 2007;1768:1006–1018. doi: 10.1016/j.bbamem.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DD, Naff BP. Activation of protein kinase C by angiotensin II decreases beta 1-adrenergic receptor responsiveness in the rat heart. J Cardiovasc Pharmacol. 1997;29:257–264. doi: 10.1097/00005344-199702000-00015. [DOI] [PubMed] [Google Scholar]
- Silverman HJ, Penaranda R, Orens JB, Lee NH. Impaired beta-adrenergic receptor stimulation of cyclic adenosine monophosphate in human septic shock: association with myocardial hyporesponsiveness to catecholamines. Crit Care Med. 1993;21:31–39. doi: 10.1097/00003246-199301000-00010. [DOI] [PubMed] [Google Scholar]
- Soeder KJ, Snedden SK, Cao W, Della Rocca GJ, Daniel KW, Luttrell LM, et al. The β3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. J Biol Chem. 1999;17:12017–12022. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- Susulic VS, Frederich RC, Lawitts J, Tozzi E, Khan BB, Harper M-E, et al. Targeted disruption of the β3-adrenergic receptor gene. J Biol Chem. 1995;270:29483–29492. doi: 10.1074/jbc.270.49.29483. [DOI] [PubMed] [Google Scholar]
- Thomas RF, Holt BD, Schwinn DA, Liggett SB. Long-term agonist exposure induces up-regulation of β3-adrenergic receptor expression via multiple cAMP response elements. Proc Natl Acad Sci USA. 1992;89:4490–4494. doi: 10.1073/pnas.89.10.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley DG, Rockman HA. Role of β-adrenergic receptor signalling and desensitization in heart failure: new concepts and prospects for treatment. Expert Rev Cardiovasc Ther. 2006;4:417–432. doi: 10.1586/14779072.4.3.417. [DOI] [PubMed] [Google Scholar]
- Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Zhang SJ, Chakir K, Avdonin P, Zhu W, Bond RA, et al. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR) – but not beta1-AR–mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–1639. doi: 10.1161/01.CIR.0000087595.17277.73. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Sakane N, Wakabayashi Y, Umekawa T, Kondo M. Anti-obesity and anti-diabetic effects of CL 316,243, a highly specific beta 3-adrenoceptor agonist, in yellow KK mice. Life Sci. 1994;54:491–498. doi: 10.1016/0024-3205(94)00408-0. [DOI] [PubMed] [Google Scholar]
- Zhang ZS, Cheng HJ, Onishi K, Ohte N, Wannenburg T, Cheng CP. Enhanced inhibition of L-type Ca2+ current by β3-adrenergic stimulation in failing rat heart. J Pharmacol Exp Ther. 2005;315:1203–1211. doi: 10.1124/jpet.105.089672. [DOI] [PubMed] [Google Scholar]
- Zhu WZ, Chakir K, Zhang S, Yang D, Lavoie C, Bouvier M, et al. Heterodimerization of β1- and β2-adrenergic receptor subtypes optimises β-adrenergic modulation of cardiac contractility. Circ Res. 2005;97:244–251. doi: 10.1161/01.RES.0000176764.38934.86. [DOI] [PubMed] [Google Scholar]