

Abstract
It is becoming accepted that G-protein-coupled receptors (GPCRs) arrange in the neuronal membrane into homo- and hetero-oligomers and, therefore, these complexes mediate neurotransmission. New models are then needed to understand GPCR operation and predict the consequences of GPCR homo- or hetero-oligomerization. Although there is not any unifying theory addressing how hetero-oligomerization occurs, recent models have been devised to understand the thermodynamics of binding of neurotransmitters to GPCRs and the allosteric protomer-protomer interactions involved in neurotransmitter-mediated activation of GPCRs. Although a model to predict how signalling is produced via homo- or hetero-oligomerization is lacking, functional data show that receptor oligomers exist to produce a variety of effects in neurons in response to a single neurotransmitter.
Keywords: model, GPCRs, neurotransmitter, dimers, GPCR dimers, GPCR oligomers, receptor oligomer, heteromer, receptor heteromer, neurotransmission
G-protein-coupled receptors (GPCRs) are expressed in neurons as dimers and/or higher-order structures
Metabotropic neurotransmitter receptors are heptahelical transmembrane proteins coupled to heterotrimeric G proteins. There are different classes of metabotropic, also known as GPCRs or heptaspanning transmembrane receptors. The most relevant for neurotransmission are members of class C – to which metabotropic glutamate receptors belong – and of class A, which include quite a number of neurotransmitter (serotonin, dopamine, etc.) rhodopsin-like receptors.
Based on electrophoretic mobility studies in the early eighties, it was suggested that the functional specie of a class A GPCR, the beta2-adrenergic receptor, was a homodimer constituted by two identical protomers (Fraser and Venter, 1982). About 10 years later, Maggio et al. (1993a,b;) showed that another class A receptor, the muscarinic receptor, behaved structurally in a fashion analogous to a two-subunit receptor. At mid-1990s, homodimerization was reported for other class A members: dopamine (Ng et al., 1996) and adenosine receptors (Ciruela et al., 1995). Notwithstanding, occurrence of GPCR dimers was not seriously considered by the scientific community (see below) until the beginning of the 21st century. In the past few years, a number of reviews have addressed GPCR dimer formation (see Bouvier, 2001; Rios et al., 2001; George et al., 2002; Agnati et al. 2003; Franco et al. 2003; Terrillon and Bouvier 2004; Prinster et al. 2005; Springael et al. 2005; Fotiadis et al. 2006; Maurel et al. 2008).
A need for consensus on defining GPCR dimers/oligomers
Despite valuable attempts by IUPHAR (International Union of Basic and Clinical Pharmacology), the current nomenclature for GPCR dimers (Pin et al., 2007) does not cover all aspects underlying receptor dimerization/oligomerization. First of all, at the present stage of the knowledge about neurotransmitter receptors, a clear difference should be made to distinguish receptors that are heteromeric/oligomeric, and GPCRs that form heteromers/oligomers (see Ferréet al., 2007). Ionotropic neurotransmitter receptors are formed by different subunits and the complete assembly is required to build up the functional complex. Accordingly, they are homo or heteromeric receptors. In contrast, GPCR dimers/oligomers consist of complexes of protein molecules that are already able to bind the neurotransmitter; that is, each GPCR protomer in the oligomeric complex is considered to be a full receptor. Therefore, as of today, it is reasonable to address to these complexes as GPCR heteromers; that is, they are receptor heteromers and not heteromeric receptors. In summary, whereas ionotropic neurotransmitter receptors are heteromeric receptors, metabotropic neurotransmitter receptors assemble into GPCR homo- and heteromers. In the case of GPCR heterodimers, the nomenclature (Pin et al., 2007) consists of putting together the name of the two receptors (ordered alphabetically) separated by a hyphen. The nomenclature for the protomers, that is, the individual GPCRs, may be found in the Alexander et al. (2008) guide. For instance, the specie(s) constituted by adenosine A2A receptors and dopamine D2 receptors (Hillion et al., 2002) may be denoted as the A2A-D2 receptor heterodimer or as A2AR-D2R heteromer; similarly, the complex formed by adenosine A1 and dopamine D1 receptors (Ginés et al., 2000) may be denoted as the A1R-D1R heteromer. As indicated below, GPCRs are able to form trimers, dimers of dimers and oligomers. For the trimer formed by cannabinoid CB1, dopamine D2 and adenosine A2A receptors (Carriba et al., 2008), the nomenclature would be: ‘A2A-CB1-D2 receptor heteromer’.
The occurrence of GPCR homomers and heteromers also requires specific nomenclature to describe how protomers ‘cooperate’ in both neurotransmitter binding and receptor heteromer activation and signalling. It is worth noting that the pharmacology of receptors in heteromers is often different to that of homomers. In homodimers, cooperativity is the word to describe that the binding of the first neurotransmitter molecule to a protomer modifies the affinity of the binding of the second neurotransmitter molecule to the second protomer. It would be advisable to look for a term to describe the same fact in a heterodimer, that is, that the binding of a neurotransmitter molecule to a protomer modifies the affinity of the binding of another neurotransmitter molecule to the second protomer. More complexity arises when taking into account synthetic molecules and/or drugs interacting with homo- and heteromers. There is not any specific term to describe that the binding of a drug to a protomer modifies the affinity of the binding of a neurotransmitter molecule to the second protomer in either a homodimer or a heterodimer.
An increasing number of physiological and synthetic molecules, which are structurally unrelated to the natural agonist, have been reported to affect the binding of a neurotransmitter to its receptor. For instance, homocysteine reduces dopamine D2 receptor output. Hyper-homocysteinemia produced by L-dopa treatment in Parkinson's disease may contribute to the loss of the therapeutic effects of L-dopa; consequently, a combined therapy of L-dopa plus homocysteine may be more effective than the classical L-dopa treatment (Agnati et al., 2006). Homocysteine interacts with dopamine D2 receptors at a site that is different from the orthosteric centre to which dopamine binds. Homocysteine is then an allosteric modulator of dopamine D2 receptors, which are able to form both homo- and heterodimers. This allosteric modulation is conceptually different from the modulation exerted in a heterodimer by neurotransmitter binding to each of the partner receptors. Although technically this modulation is allosteric because there is not competition in the binding of the two neurotransmitters (because each binds to its respective orthosteric centre), the use of the term allosteric may be used with caution in the case of modulation exerted by molecules binding to orthosteric sites. For example, in the case of A2A-D2 receptor heteromers, adenosine binding to its orthosteric site influences the binding of dopamine to its orthosteric site. As adenosine does not bind to the same centre that dopamine, adenosine can be considered an allosteric modulator. But in fact the allosterism caused by adenosine upon D2-receptor-mediated actions differs mechanistically from that exerted by homocysteine.
Those described above are just some examples demonstrating facets of the cross-talk within the heteromer that may be equivocal if not adequately addressed. Due to the lack of a specific nomenclature, an adequate description of the details of the each facet is needed. A further example of the myriad of aspects arising from the fact that GPCR heteromerize/oligomerize is the cross-antagonism in GPCR action, that is, the fact that the antagonist selective for one receptor is able to antagonize not only the action mediated by ‘its’ receptor but also the action mediated by the partner receptor in a heterodimer. In fact, an agonist/antagonist may not only trigger the activation/blockade of one receptor in the heteromer but an agonist may influence the activation of the partner receptor and an antagonist may prevent activation of the partner receptor in the heterodimer. Apart from nomenclature aspects, these findings (Ferrada et al., 2009) and data in preparation are important to define new therapeutic targets and to design new drugs acting on GPCR dimers or oligomers.
Structural features in GPCR dimer formation
The first GPCR dimer whose structure was solved was a metabotropic glutamate receptor. Kunishima et al. (2000) determined three different crystal structures of the extracellular ligand-binding region of mGluR1 in a complex with glutamate and in two unliganded forms. The authors showed disulphide-linked homodimers whose active and resting conformations are modulated through the dimeric interface by a packed alpha-helical structure. The crystal of the extracellular region of metabotropic glutamate receptors was feasible due to its relatively large size. Despite other receptors have much shorter N-terminal domains, crystals of GPCR have been possible after some biotechnological engineering of the GPCR molecule. This has allowed solving the structure (including the seven transmembrane helices) of two modified class A receptors: the beta2-adrenergic (Cherezov et al., 2007) and the A2A adenosine receptor (Jaakola et al., 2008). Unfortunately, the interacting surface for receptor homodimers cannot be deduced from these structures. Once crystals of class A GPCRs have been obtained, efforts are being made to try to obtain crystals of (modified) GPCR heteromers. While waiting for crystal structures of receptor heteromers, what it is becoming evident is that the dimerization surface in GPCR involves not only extracellular regions but transmembrane domains and cytoplasmic epitopes.
Computational studies performed by Gouldson et al. (2000) indicated that GPCR may form dimers by swapping domains or just by placing one besides the other (contact dimer). Domain swapping would result in one orthosteric binding site every two GPCRs, whereas two orthosteric binding sites would be present in a contact GPCR dimer. Subsequent data indicate that domain swapping may occur for some GPCRs but that the general trend is the formation of contact dimers. According to Gouldson et al. (2000), the two possibilities are equivalent in terms of signalling and transmembrane helices 5 and 6 likely participate in the interaction between the two GPCRs in a dimer. Docking studies performed by Canals et al. (2003) indicate that helixes 5 and 6 from the dopamine receptor and helix 4 in the adenosine receptor were participating in the D2-A2A receptor heterodimer interface. Studies by the group of Javitch using cysteine cross-linking show formation of homo-tetramers at physiological expression levels of dopamine D2 receptors and that depending on the interface within the tetramer, the transmembrane domain 4 or 1 plays a key role (Guo et al., 2003; Guo et al., 2008). Insight into the structure of the prototypic member of class A GPCR, rhodopsin, has been possible. The deduced structure of the rhodopsin oligomer is useful to understand how GPCR dimerization/oligomerization may take place. On the one hand, the interacting surface involves transmembrane domains 4 and 5 but, on the other hand, contacts mainly between transmembrane domains 1 and 2 and the cytoplasmic loop connecting helices 5 and 6 facilitates the formation of rows of rhodopsin dimers (Fotiadis et al., 2006). It is then probable that depending on the receptor and the disposition of protomers in the heteromer, every single transmembrane domain may participate in the multiple interactions required to form an oligomeric complex.
The involvement of different cytoplasmic domains in GPCR dimer formation has also been demonstrated. In fact, electrostatic interactions between a basic epitope containing adjacent arginine residues and an acidic epitope constituted by arrays of aspartic or glutamic residues or phosphorylated amino acids are involved in receptor heteromerization (Canals et al., 2003; Azdad et al., 2009). The strength of the epitope-epitope interaction is even higher than that of a covalent bond. Looking for these epitopes in databases has been instrumental in our laboratory to identify novel heteromers. Finally, it should be noted that phosphorylation events may modulate dimer formation and/or affect dimer interface(s). For instance, phosphorylation of the acidic epitope by caseine kinase makes it available to interact with the basic epitope. In contrast, phosphorylation of serine or threonine residues adjacent to the basic epitope by protein kinase A would reduce the electrostatic attraction between epitopes (Woods and Ferré, 2005).
The development of sophisticated biophysical techniques has allowed the unequivocal detection of dimers (see Bouvier, 2001; Milligan, 2001; Agnati et al., 2003), and more recently, that of trimers and dimers of dimers and oligomers in living cells (Carriba et al., 2007; 2008; Philip et al., 2007; Guo et al., 2008; Maurel et al., 2008). A unifying model able to predict how GPCR protomers arrange into oligomers is lacking. Furthermore, it is of interest to know how heterotrimeric G proteins arrange in the GPCR dimer complex. Whereas a stoichiometry 1:1 for G protein and GPCR protomer was suspected, Herrick-Davis et al. (2005) has put forward the idea that one G protein interacts with a GPCR dimer.
A model able to handle ligand binding to GPCR dimers and quantitate allosteric modulation
Until 2005, all existing models for GPCR considered that the receptors were monomeric. Colquhoun (1973) and Thron (1973) pioneered some studies that led to the subsequent development of models for neurotransmitter/hormone receptors (De Lean et al., 1980; Costa and Herz, 1989; Onaran et al. 1993; Samama et al. 1993; Proska and Tucek 1995; Franco et al. 1996; Leff 1996; Weiss et al., 1996a,b,c; Hall 2000; Lorenzen et al. 2002). Those models consider receptors as monomers and are modifications of del Castillo and Katz (1957) model of nicotinic acetylcholine receptor activation. The most used model to explain receptor operation is the so-called ternary complex model (Figure 1; De Lean et al., 1980). For the last 25 years, this model has been instrumental to explain receptor operation. This model is very straightforward to understand the functioning of full or partial agonists, of neutral antagonists or inverse agonists, and also to understand how GPCR constitutive activity occurs. What none of those ‘monomeric’ models are able to adequately address is the variety of complex data that arise upon binding of ligands to GPCRs.
Figure 1.
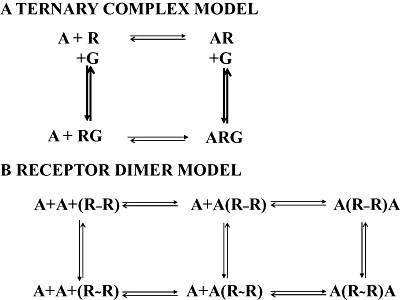
Analogies between the ternary-complex (A) and the two-state dimer (B) models. In the ternary-complex model, the ligand/neurotransmitter, A, may bind to non-active (R) or the active (RG) monomeric receptor. In a receptor dimer model, A may bind to the unoccupied dimer (RR) or to the semi occupied dimer (ARR) in the non-active (R-R) or active (R∼R) dimer. Equivalent ‘active’ forms of the receptor are denoted as RG in the ternary-complex model or R∼R in the dimer model; in the latter, there is no need to consider (in the scheme) G proteins, that, as other protein or non-protein molecule, may be considered to be allosteric modulators affecting the values of microscopic (kinetic) and equilibrium dissociation (macroscopic) constants and the value of the cooperativity index. Comparing the two schemes, the main difference is that the first model (top) relies on receptor monomers and the second (bottom) in receptor dimers and that in the receptor dimer model it is not necessary to include proteins other than receptors themselves.
Upon performing radioligand binding, complex data frequently arise. Non-linear Scatchard plots reflect complex binding in saturation experiments. In the case of competition assays, which are mandatory when the ligand does not exist in radiolabelled form, complex kinetics is detected by biphasic competition curves. Due to the inability of the above-described models to handle these situations, the so-called two-independent-site model was devised. The model is quite simple as it assumes the existence of two different conformations of the receptor: one having low affinity for the agonist (assumed to be the pool of non-G-protein-interacting receptor molecules) and another displaying high affinity for the agonist (assumed to correspond to the receptor–G protein complex). This model allows calculation of the equilibrium dissociation constants for the high- (KDH) and the low-affinity (KDL) binding, and provides a percentage of high- versus low-affinity sites. For instance, the A1 adenosine receptor would bind with a given proportion of low- versus high-affinity species the selective agonist, R-phenylisopropyladenosine, with a KDL of 1 nmol·L−1 and a KDH of 0.1 nmol·L−1 (Casadóet al., 1991). The model has been highly employed in past decades due to the lack of any alternative model and due to the fact that GPCRs were considered to be monomeric. As any other model, it relies on some assumptions such as the ‘independence’ of the sites, that is, no equilibrium may exist between G-uncoupled (displaying low affinity for the neurotransmitter) and G-coupled (displaying high affinity for the neurotransmitter) sites. Assuming this model to be correct, Casadóet al. (1991) reported an apparent conversion between these two independent sites. Therefore, if equilibrium between two GPRC sites happens, the ‘two-independent-site model’ has an intrinsic incongruence when facing real data. On the other hand, mathematical constraints of the ternary complex model lead to its inability to ‘explain’ complex binding data. Consequently, to rely on the ternary complex to explain the effects of agonists/antagonists but then use the ‘two-independent-site model’ for fitting data is also incongruent. A further limitation of these two models is that they cannot explain positive cooperativity that also occurs in GPCR pharmacology (Lazareno et al., 1998). In summary, the ternary-complex and the two-independent-site models have accomplished a relevant function for decades but it is now the time to look forward to other more suitable models, which have necessarily to take into account the occurrence of receptor dimers. Recently, devised ‘dimer’ models (see Figure 1 and below) have the real advantage of being similar conceptually to the ternary complex model but allowing to explain both negative and positive cooperativity and allowing to fit complex binding data without making any extra/aprioristic assumption.
To further illustrate that GPCR models have limitations, it is worth highlighting the work of Whorton et al. (2007), who have shown that detergent-extracted highly purified beta2-adrenergic receptors can be incorporated into a reconstituted high-density lipoprotein phospholipid bilayer particle, and that a single receptor efficiently activates Gs in the vesicle and displays GTP-sensitive ligand-binding properties. This is a beautiful example of experimental situation in which a two-independent-site approach can be used, because data indicate that complex kinetics on agonist binding are due to the presence of a mixture in the lipid bilayer of receptor monomers uncoupled and coupled to Gs. The claim, however, that these data refute the contribution of oligomers towards high-affinity agonist binding (Whorton et al., 2007) is not substantiated because in vivo or in vitro-detergent-free preparations the evidence points towards intra-membrane cross-talk, that is, allosteric interactions between protomers in dimers/oligomers (see below), as the cause of complex binding data. The report, therefore, exemplifies how the two-independent-site model may be used only in conditions at which two really independent GPCR sites happen.
When complex binding data occur, the easy choice was to assume (as discussed above) the occurrence of two independent populations of monomeric GPCRs. Another possibility now is the use dimer-based models, which naturally deals with the concept of cooperativity in the binding, that is, that matches complex binding data to cooperative binding. As negative cooperativity cannot be distinguished from binding site heterogeneity by equilibrium binding, Springael et al. (2006) used infinite tracer dilution conditions to demonstrate that negative cooperativity is established between chemokine CCR2/CCR5 receptor heteromers and that G proteins may modulate the inter-protomer heteromeric cross-talk. Once cooperativity is demonstrated for GPCRs, a possibility to explain it was to assume that there was communication between receptor monomers. Assuming a similar hypothesis, Franco et al. (1996) devised the ‘cluster-arranged cooperativity’ model, which also relied on monomers. But after assuming GPCRs as forming dimers, a new model to explain cooperativity, that is, allosteric interactions between protomers, was needed. Cooperative agonist binding likely results from conformational changes transmitted from one protomer to the second (in a dimer). Mesnier and Banères (2004) using a spectroscopy approach were able to show intra-molecular (i.e. inter-protomer) cross-talk in the leukotriene BLT1 receptor dimer. Vilardaga et al. (2003) has developed a fluorescence-based technique for real-time monitoring of the activation switch of GCPRs in living cells; by using this technique in cells expressing alpha2A-adrenergic and mu-opioid receptor heteromers, it has been possible to detect a conformational cross-talk between protomers (Vilardaga et al., 2008). In summary, it is becoming more and more evident that complex binding data come from cooperativity when ligands bind to a receptor dimer/oligomer molecule, that is, when, at least, there are two (orthosteric) binding sites (one per protomer) in the GPCR hetero-dimer/oligomer.
Two similar models based on receptor dimers have been recently devised (Franco et al., 2005; Albizu et al., 2006). The group of Durroux (Albizu et al., 2006) assumes equilibrium between receptor dimers able to establish a molecular cross-talk and also the possibility of equilibrium between monomeric and dimeric species. Franco et al. (2005; 2006;) propose the ‘two-state’ dimer model based only in dimeric species, one of its advantages being the possibility to obtain ‘dimer-specific’ parameters. GPCR dimer models are quite convenient as they are quite similar to the ternary complex model but just taking into account GPCR dimers instead of GPCR monomers. Then, two agonist molecules may bind instead of only one as in the monomeric ternary complex model. Compared with the ternary complex model, GPCR dimer models perform equally well in understanding the effects of full or partial agonists, of neutral antagonists or inverse agonists, and the occurrence of constitutive activity. But dimer models have the added value of being able to explain cooperativity (positive or negative) on ligand binding to GPCRs, which is something that the monomeric-based models are unable to adequately address. As discussed below, these models make possible a convenient management of both non-complex and complex binding data.
For non-complex binding, the choice of the model is quite irrelevant as only one affinity equilibrium constant can be calculated. The success of the two-independent-site model was due to the possibility of providing parameters for complex binding of ligands/drugs to GCPRs. As indicated above, those were KDH and KDL and the percentage of high- versus low-affinity sites. For complex binding data, dimer models provide two dissociation constants that are more meaningful that those provided by the two-independent-site model. A model does not allow per se fitting radioligand-binding data. For instance, the ternary complex model does not allow at all fitting of complex radioligand-binding data. Therefore, a specific development is required to convert concepts and equilibria between receptor forms into equations useful to fit binding data. In the case of GPCR dimer models, this has been possible (see Casadóet al., 2007). Some few equations are provided below that are useful to fit binding data and that demonstrate that fitting binding data is very easy as is straightforward the meaning of the different parameters obtained. First of all, to obtain the two equilibrium constants describing the affinity of the binding of a ligand to the two protomers in a dimer, complex binding data1 must be fitted to the following equation:
From data of binding (Abound) versus the concentration of the radioligand (A in the equation), the two equilibrium constants (KD1 and KD2) and the total binding (RT being the total number of dimers) can be automatically obtained using any commercially available fitting data package, which is available in every laboratory where radioligand binding is performed. As indicated, KDA1 and KDA2 are the macroscopic dissociation constants describing the binding of the first and the second radioligand molecule (A) to the dimeric receptor. On performing parallel analysis with real complex data, the two-state dimer model is simpler (three parameters: KD1, KD2 and RT) than the two-independent-site (four parameters: KDH, KDL, RH and RL) and performs better. The dimer model is of election even assuming mixtures of monomer and dimer receptor populations.
On making the development of the dimer model useful to handle binding data, Casadóet al. (2007) have defined a parameter to quantitate cooperativity. A dimer homotropic cooperativity index (DC) for the radioligand is defined as log (4KD1/KD2). The meaning of DC is similar to the Hill coefficient in the case of enzymes (Giraldo, 2008), that is, negative or positive values of DC indicate respectively negative or positive cooperativity, whereas a lack of cooperativity is deduced from DC values approaching zero.
Quite often, parameters for the binding of a molecule/drug, B, are obtained from competition assays. Usually in competition assays, the radioligand (A) binds to the GPCR following simple binding data. This leads to a significant simplification in the equation needed to fit data of binding of the radioligand (Abound) as a function of the concentration of the competing ligand (B in the equation):
where A represents the radioligand concentration, RT is the total amount of receptor dimers and KDA1 and KDA2 are the macroscopic dissociation constants describing the binding of the first and the second radioligand molecule (A) to the dimeric receptor. These two constants are already known after fitting data of (saturation) assays of binding as a function of the radioligand concentration (see above). B represents the assayed competing compound concentration and KDB1 and KDB2 are, respectively, the KD of the first and second binding of B. KDAB is a novel parameter that can be described as a hybrid KD, which is the dissociation constant of B binding to a receptor dimer semi-occupied by A.
In the case that both the radioligand A and the competitor B bind to the receptor in a non-cooperative manner, the equation is simplified to:
The DC for the competing ligand (DCB) is easily calculated using an equation similar to that described above [DCB= log (4KDB1/KDB2)].
A comparison between the ternary complex, the two-independent-site and a receptor dimer model, clearly points towards the overall advantages of using ‘dimer-based’ models (Table 1). In summary, dimer models are instrumental to understand GPCR functioning and the equations devised by Casadóet al. (2007) are of election to fit binding data to GPCR dimers and, importantly, even to a mixture of GPCR monomers, dimers and oligomers. A summary on how a dimer model provides useful parameters from both saturation and competition assays is provided in Figure 2.
Table 1.
Characteristics of three different GPCR models
| Model | Simple binding data | Complex binding data | Negative cooperativity | Positive cooperativity | Possibility to quantitate cooperativity | Possibility to quantitate allosteric modulation |
|---|---|---|---|---|---|---|
| Ternary1 (GPCRs as monomers) | Yes | No2 | No2 | No | No | No |
| Two-independent-site1 (GPCRs as monomers) | Yes | Yes2 | Yes2 | No | No | No |
| Dimer (GPCR dimers but with the possibility to include monomers) | Yes | Yes | Yes | Yes | Yes | Yes3 |
‘Yes’ indicates that the model is able to explain the feature or able to provide a convenient equation to fit radioligand-binding data.
The only difference between these two models is that in the two-independent-site model, the active (also known as the G-protein-coupled: RG) and the non-active (also known as uncoupled: R) forms of the GPCR monomer cannot be in equilibrium. In the ternary complex, these two forms interconnect (i.e. an equilibrium exists between R and RG) and, therefore, are not ‘independent’.
In cases of complex binding data, the ternary complex and the two-independent-site models are incompatible (unless under some particular conditions, which are seldom encountered in vivo).
See Gracia et al. (2008).
GPCR, G-protein-coupled receptor.
Figure 2.

Scheme of the GPCR-dimer-specific parameters that can be obtained from saturation curves and from competition assays. The equilibrium dissociation constants for a radioligand can be obtained from saturation assays (top) in which saturation isotherm data of bound ligand versus concentration are fitted to the suitable equation (see text). This provides the number of dimers in the preparation, the dissociation constant of the first and second binding of A to the receptor dimer (KD1A and KD2A) and a measure of cooperativity (dimer cooperativity index for A, DCA). When the ligand (B) does not exist in radiolabelled form, competition assays are performed (bottom). Data of degree of competition versus concentration of the competing ligand can be fitted to the suitable equation (see text) to obtain: the two equilibrium dissociation constants for the binding of B to the dimer (KD1B and KD2B), the cooperativity index for the binding of B (DCB) and also a ‘novel’ equilibrium constant for the binding of B to a dimer semi-occupied by A (KDAB). GPCR, G-protein-coupled receptor.
How neurons can display differently flavoured patterns in response to the same neurotransmitter or how different GPCR heteromers may ‘smell’ differently the same neurotransmitter
A key question in the GPCR field is why nature has allowed hetero-oligomer formation. There is increasing evidence that GPCR heteromers serve to provide signalling diversity. Signalling diversity is already achieved by means of the occurrence of different receptor subtypes that for a given neurotransmitter provide different signalling outputs. For instance, P2 receptors are represented by eight metabotropic (P2Y) and seven ionotropic (P2X) receptors. To ask why so many receptor subtypes are necessary for triggering biological properties and functions, Volontéet al. (2006) envision receptor subtypes as a combinatorial receptor web. This web would be a dynamic architecture of P2 proteins demonstrating economic efficiency and ensuring a ‘fine-tuning’ of the neural responses to the natural neuromodulators. It seems, however, that signalling diversity mediated by the existence of different subtypes of receptor for a given neurotransmitter is not enough for the higher neural functions performed by mammals, especially by humans. Then, it is reasonable to think that nature has provided GPCRs with the possibility to heteromerize/oligomerize to provide the required signalling diversity. A way to understand how this diversity may impact on neurotransmission is by means of a metaphor using the sense of smell.
Species rely on odorant compounds to locate food, predators or toxins. The sense of smell is also involved in animal communication, and revealing the underlying mechanisms will therefore facilitate a deeper understanding of animal behaviour (see Zarzo, 2007 for review). Due to the variety of substances in nature, mammals have required hundreds of odorant receptors to make possible to distinguish between danger, pleasure, etc. Each olfactory receptor cell possesses only one type of odorant receptor, and each receptor can detect a limited number of odorant substances. To process the high amount of events needed for neural transmission, one would think in a similar approach, that is, the occurrence of a large gene family giving rise to an equivalent number of neurotransmitter receptors. This would require hundreds of different neurotransmitters. What nature seems to have designed for carrying out neurotransmission is just the opposite; that is, instead of having several hundreds of neurotransmitters, whose production and handling would be energetically insurmountable, neurotransmission relies on a bunch of neurotransmitters/neuromodulators interacting with few different specific receptors. But, in this situation, the signalling diversity has to be provided at the receptor level and this seems to be achieved (elegantly) by forming receptor heteromers. The number of possible heteromers is quite high taking into account that neurotransmitter receptors may combine with each other to form dimers, trimers, tetramers and high-order oligomers. Therefore, GPCR heteromers may be considered devices that ‘smell’ differently a same neurotransmitter (Figure 3). A few examples on how different neurons expressing different GPCR heteromers may display differently flavoured patterns in response to the same neurotransmitter are given below.
Figure 3.
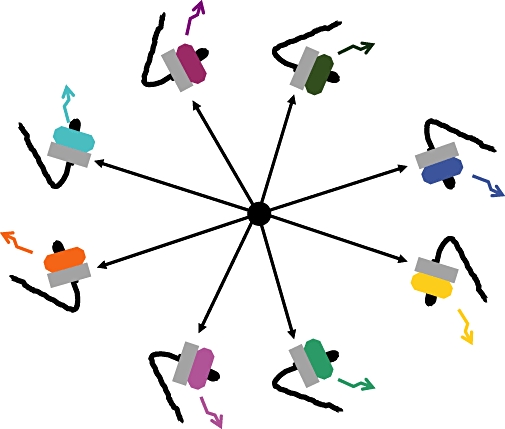
Variety of signalling by GPCR heteromers. Each neuron, represented by a nose, expressing a given heterodimer (grey protomer plus a coloured protomer) may display differently flavoured signalling patterns (coloured arrow) in response to the same neurotransmitter (black circle in the middle of the image), whose specific receptor is the grey protomer in the receptor heterodimer. GPCR, G-protein-coupled receptor.
The examples that follow are restricted to heteromers formed by two receptor subtypes of a given neurotransmitter. Heteromerization of the different subtypes of opioid receptors has been widely described. Hetero-oligomerization of mu- and delta-opioid receptors (George et al., 2000) leads to a different pharmacology and to different G protein coupling properties if compared with the properties of mu or delta opioid receptors expressed individually. More recently, Rozenfeld and Devi (2007) have shown that heterodimerization of mu with delta opioid receptors leads to a constitutive recruitment of beta-arrestin2 to the receptor complex, ultimately resulting in changes in the spatio-temporal regulation of extracellular signal-regulated kinase 1/2 signalling.
The reported occurrence of receptor subtypes for the same neurotransmitter, one coupled to Gs and the other coupled to Gi, has been difficult to understand. Recent data indicate that one (relevant) reason for co-expression of Gi- and Gs-coupled receptors is the formation of heteromers that ‘smell’ the neurotransmitter differently than homomers. One example is the adenosine A1-A2A receptor heteromer (Ciruela et al., 2006). Results obtained using samples from necropsies indicate that A1-A2A receptor heteromers exist in human brain striatum and therefore the A1R-A2AR-heteromer-mediated adenosinergic modulation of striatal neurotransmission is expected to occur in the human brain. At the presynaptic level, A1-A2A receptor heteromers exert a tight control of the glutamatergic neurotransmission in striatum. In fact, at low concentrations of adenosine, which has a higher affinity for the A1 than for the A2A receptor, the signalling through the heteromer goes mainly via the A1 receptor-mediated pathway and a negative modulation of glutamate release occurs. In contrast, higher adenosine concentration activates the A2A receptor in the heteromer and this shuts down (by antagonistic intra-membrane/inter-protomer cross-talk) the signal originating at A1 receptors; the final outcome is just the opposite, that is, an A2A receptor-mediated positive modulation of glutamate release (Ciruela et al., 2006). Therefore, the heteromer functions as a ‘concentration-dependent switch’ or, in other words, a sensor of the concentration of the neuromodulator.
There was controversy concerning co-expression in the same neuron of dopamine D1 receptors, which couple to Gi proteins, and dopamine D2 receptors, which couple to Gs. Lee et al. (2004) reported that dopamine D1 and D2 receptors may arrange into D1-D2 heteromers. Whereas activation of D1 leads to increases in cAMP levels and activation of D2 receptor does the opposite, the D1-D2 heteromer couples to a Gq protein and, therefore, dopamine may lead to increases in intracellular calcium levels. Criticisms that D1-D2 receptor heteromers may not occur in neurons due to lack of co-expression were overcame by in vivo assays showing that D1-D2 receptor heteromer-signalling is more readily detected in mice that are 8 months in age compared with animals that are 3 months old, and that activation of Gq through the heteromer increases levels of calcium/calmodulin-dependent protein kinase II in the nucleus accumbens, unlike activation of Gs/olf-coupled D1 receptors (Rashid et al., 2007).
Interestingly, D1 receptors may also form heteromers with D3 receptors, whose function in the striatum has remained elusive. Marcellino et al. (2008) have recently given evidence for D1-D3 receptor heteromer formation in striatum. This heteromer allows a synergistic D1-D3 intra-membrane receptor-receptor interaction, by which D3 receptor stimulation enhances D1 receptor agonist affinity. In agreement to the synergistic D1-D3 intra-membrane/inter-protomer cross-talk, experiments in reserpinized mice showed that D3 receptor stimulation potentiates D1 receptor-mediated behavioural effects by a different mechanism than that mediated by D2 receptor stimulation (Marcellino et al., 2008). These results suggest that a main significant functional feature of the D3 receptor is to obtain a stronger dopaminergic response in the striatal neurons that co-express the two receptors.
Many more examples may be provided to illustrate how heteromers may ‘smell’ differently a given neurotransmitter; a detailed review of these data is out of the scope of the present article. Still, more examples are likely to appear in the coming years that collectively will impact deeply in our view of how neurotransmission takes place.
Acknowledgments
This work was supported by the Spanish Ministerio de Ciencia y Tecnología (Grant SAF2006-00170) and by Fundació La Marató de TV3 (Grant 060110).
Glossary
Abbreviations:
- DC
dimer homotropic cooperativity index
- GPCR
G-protein-coupled receptor
- KD
equilibrium dissociation constant
- R
number of receptor molecules (monomer or dimer)
Footnotes
Conflict of interests
None.
Note added in proof
This article was written before the appearance of the Ferréet al. (2009) paper in which a new conceptual framework for receptor heteromers is given and a new nomenclature is proposed.
References
- 1.Agnati LF, et al. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol Rev. 2003;55:509–550. doi: 10.1124/pr.55.3.2. [DOI] [PubMed] [Google Scholar]
- Agnati LF, Ferré S, Genedani S, Leo G, Guidolin D, Filaferro M, et al. Allosteric modulation of dopamine D2 receptors by homocysteine. J Proteome Res. 2006;5:3077–3083. doi: 10.1021/pr0601382. [DOI] [PubMed] [Google Scholar]
- Albizu L, Balestre M-N, Breton C, Pin J-P, Manning M, Mouillac B, et al. Probing the existence of G-protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol. 2006;70:1783–1791. doi: 10.1124/mol.106.025684. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptor and channels. 3rd edition (2008 revision) Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azdad K, Gall D, Woods AS, Ledent C, Ferré S, Schiffmann SN. Dopamine D(2) and adenosine A(2A) receptors regulate NMDA-mediated excitation in accumbens neurons through A(2A)-D(2) receptor heteromerization. Neuropsychopharmacology. 2009;34:972–986. doi: 10.1038/npp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Canals M, Marcellino D, Fanelli F, Ciruela F, de Benedetti P, Goldberg SR, et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem. 2003;278:46741–46749. doi: 10.1074/jbc.M306451200. [DOI] [PubMed] [Google Scholar]
- Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology. 2007;32:2249–2259. doi: 10.1038/sj.npp.1301375. [DOI] [PubMed] [Google Scholar]
- Carriba P, Navarro G, Ciruela F, Ferré S, Casadó V, Agnati L, et al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods. 2008;5:727–733. doi: 10.1038/nmeth.1229. [DOI] [PubMed] [Google Scholar]
- Casadó V, Mallol J, Canela EI, Lluis C, Franco R. The binding of [3H]R-PIA to A1 adenosine receptors produces a conversion of the high-to the low-affinity state. FEBS Lett. 1991;286:221–224. doi: 10.1016/0014-5793(91)80978-c. [DOI] [PubMed] [Google Scholar]
- Casadó V, Cortés A, Ciruela F, Mallol J, Ferré S, Lluis C, et al. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol Ther. 2007;116:343–354. doi: 10.1016/j.pharmthera.2007.05.010. [DOI] [PubMed] [Google Scholar]
- del Castillo J, Katz B. Interaction at end-plate receptors between different choline derivatives. Proc R Soc Ser B. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Mallol J, Canela EI, Lluis C, Franco R. Immunological identification of A1 adenosine receptors in brain cortex. J Neurosci Res. 1995;42:818–828. doi: 10.1002/jnr.490420610. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. Rang HP. A Symposium on Drug Receptors. Baltimore, MD: University Park Press; 1973. The relationship between classical and cooperative models for drug action; pp. 149–182. [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Ferrada C, Moreno E, Casadó V, Bongers G, Cortés A, Malloll J, et al. Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol. 2009;157:64–75. doi: 10.1111/j.1476-5381.2009.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007;30:440–446. doi: 10.1016/j.tins.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pun JP, Volkow ND, Waldhoer M, Woods AS, Franco R. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis DD, Jastrzebska B, Philippsen A, Müller DJ, Palczewski K, Engel A. Structure of the rhodopsin dimer: a working model for G-protein-coupled receptors. Curr Opin Struct Biol. 2006;16:252–259. doi: 10.1016/j.sbi.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Ciruela F, Mallol J, Lluis C, Canela EI. The cluster-arranged cooperative model: a model that accounts for the kinetics of binding to adenosine receptors. Biochemistry. 1996;35:3007–3015. doi: 10.1021/bi952415g. [DOI] [PubMed] [Google Scholar]
- Franco R, Canals M, Marcellino D, Ferré S, Agnati L, Mallol J, et al. Regulation of heptaspanning-membrane-receptor function by dimerization and clustering. Trends Biochem Sci. 2003;28:238–243. doi: 10.1016/S0968-0004(03)00065-3. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferré S, Fuxe K, Cortés A, Ciruela F, et al. Dimer-based model for heptaspanning membrane receptors. Trends Biochem Sci. 2005;30:360–366. doi: 10.1016/j.tibs.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferrada C, Ferré S, Fuxe K, et al. The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol. 2006;69:1905–1912. doi: 10.1124/mol.105.020685. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Venter JC. The size of the mammalian lung beta2-adrenergic receptor as determined by target size analysis and immunoaffinity chromatography. Biochem Biophys Res Commun. 1982;109:21–29. doi: 10.1016/0006-291x(82)91560-1. [DOI] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, et al. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- George SR, O'Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- Ginés S, Hillion J, Torvinen M, Le Crom S, Casadó V, Canela EI, et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci USA. 2000;97:8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo J. On the fitting of binding data when receptor dimerization is suspected. Br J Pharmacol. 2008;155:17–23. doi: 10.1038/bjp.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouldson PR, Higgs C, Smith RE, Dean MK, Gkoutos GV, Reynolds CA. Dimerization and domain swapping in G-protein-coupled receptors: a computational study. Neuropsychopharmacol. 2000;23:S60–S77. doi: 10.1016/S0893-133X(00)00153-6. [DOI] [PubMed] [Google Scholar]
- Gracia E, Cortés A, Meana JJ, García-Sevilla J, Herhsfield MS, Canela EI, et al. Human adenosine deaminase as an allosteric modulator of human A1 adenosine receptor: abolishment of negative cooperativity for [3H](R)-PIA binding to the caudate nucleus. J Neurochem. 2008;107:161–170. doi: 10.1111/j.1471-4159.2008.05602.x. [DOI] [PubMed] [Google Scholar]
- Guo W, Shi L, Javitch JA. The fourth transmembrane segment forms the interface of the dopamine D2 receptor homodimer. J Biol Chem. 2003;278:4385–4388. doi: 10.1074/jbc.C200679200. [DOI] [PubMed] [Google Scholar]
- Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, et al. Dopmine D2 receptros form higher order oligomers at physiological expression levels. EMBO J. 2008;27:2293–2304. doi: 10.1038/emboj.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA. Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol Pharmacol. 2000;58:1412–1423. doi: 10.1124/mol.58.6.1412. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Harrigan TJ, Mazurkiewicz JE. Inhibition of Serotonin 5-Hydroxytryptamine2C Receptor Function through Heterodimerization: receptor dimers bind two molecules of ligand and one G protein. Biol Chem. 2005;280:40144–40151. doi: 10.1074/jbc.M507396200. [DOI] [PubMed] [Google Scholar]
- Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, et al. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem. 2002;277:18091–18097. doi: 10.1074/jbc.M107731200. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Gharagozloo P, Kuonen D, Popham A, Birdsall NJ. Subtype-selective positive cooperative interactions between brucine analogues and acetylcholine at muscarinic receptors: radioligand binding studies. Mol Pharmacol. 1998;53:573–589. doi: 10.1124/mol.53.3.573. Erratum in: Mol Pharmacol 1999 55: 194. [DOI] [PubMed] [Google Scholar]
- Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, et al. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–35678. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- Leff P. The two-state model of agonist action: challenges to pharmacological receptor theory. Proc West Pharmacol Soc. 1996;39:67–68. [PubMed] [Google Scholar]
- Lorenzen A, Beukers MW, van der Graaf PH, Lang H, van Muijlwijk-Koezen J, de Groote M. Modulation of agonist responses at the A1 adenosine receptor by an irreversible antagonist, receptor-G protein uncoupling and by the G protein activation state. Biochem Pharmacol. 2002;64:1251–1265. doi: 10.1016/s0006-2952(02)01293-5. [DOI] [PubMed] [Google Scholar]
- Maggio R, Vogel Z, Wess J. Coexpression studies with mutant muscarinic/adrenergic receptors provide evidence for intermolecular ‘cross-talk’ between G-protein-linked receptors. Proc Natl Acad Sci U S A. 1993a;90:3103–3107. doi: 10.1073/pnas.90.7.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio R, Vogel Z, Wess J. Reconstitution of functional muscarinic receptors by co-expression of amino- and carboxyl-terminal receptor fragments. FEBS Lett. 1993b;319:195–200. doi: 10.1016/0014-5793(93)80066-4. [DOI] [PubMed] [Google Scholar]
- Marcellino D, Ferre S, Casado V, Cortes A, Le Foll B, Mazzola C, et al. Identification of dopamine D1-D3 receptor heteromers: indications for a role of synergistic D1-D3 receptor interactions in the striatum. J Biol Chem. 2008;283:26016–26025. doi: 10.1074/jbc.M710349200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, et al. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5:561–567. doi: 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnier D, Banères JL. Cooperativity conformational changes in a G-protein-coupled receptor dimer, the lekotriene B(4) receptor BTL1. J Biol Chem. 2004;279:49464–49670. doi: 10.1074/jbc.M404941200. [DOI] [PubMed] [Google Scholar]
- Milligan G. Oligomerisation of G-protein-coupled receptors. J Cell Sci. 2001;114:1265–1271. doi: 10.1242/jcs.114.7.1265. [DOI] [PubMed] [Google Scholar]
- Ng GY, O'Dowd BF, Lee SP, Chung HT, Brann MR, Seeman P, et al. Dopamine D2 receptor dimers and receptor-blocking peptides. Biochem Biophys Res Commun. 1996;227:200–204. doi: 10.1006/bbrc.1996.1489. [DOI] [PubMed] [Google Scholar]
- Onaran HO, Costa T, Rodbard D. Betagamma subunits of guanine nucleotide-binding proteins and regulation of spontaneous receptor activity: thermodynamic model for the interaction between receptors and guanine nucleotide-binding protein subunits. Mol Pharmacol. 1993;43:245–256. [PubMed] [Google Scholar]
- Philip F, Sengupta P, Scarlata S. Signaling through a G protein-coupled receptor and its corresponding G protein follows a stoichiometrically limited model. J Biol Chem. 2007;282:19203–19216. doi: 10.1074/jbc.M701558200. [DOI] [PubMed] [Google Scholar]
- Pin J-P, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, et al. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev. 2007;59:5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- Prinster SC, Hague C, Hall RA. Heterodimerization of G protein-coupled receptors: specificity and functional significance. Pharmacol Rev. 2005;57:289–298. doi: 10.1124/pr.57.3.1. [DOI] [PubMed] [Google Scholar]
- Proska J, Tucek S. Competition between positive and negative allosteric effectors on muscarinic receptors. Mol Pharmacol. 1995;48:696–702. [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MMC, Furtak T, Ghundi E, Cheng M, et al. D1-D2 dopamine receptor heterooligomers with unique pharamacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natil Acad Sci USA. 2007;104:654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther. 2001;92:71–87. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007;21:2455–2465. doi: 10.1096/fj.06-7793com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Springael JY, Urizar E, Parmentier M. Dimerization of chemokine receptors and its functional consequences. Cytokine Growth Factor Rev. 2005;16:611–623. doi: 10.1016/j.cytogfr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Springael J-Y, Le Minh PN, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol. 2006;69:1652–1661. doi: 10.1124/mol.105.019414. [DOI] [PubMed] [Google Scholar]
- Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thron CD. On the analysis of pharmacological experiments in terms of an allosteric receptor model. Mol Pharmacol. 1973;9:1–9. [PubMed] [Google Scholar]
- Vilardaga JP, Bunemann M, Krasel C, Castro M, Lohse MJ. Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat Biotechnol. 2003;21:807–812. doi: 10.1038/nbt838. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol. 2008;4:126–131. doi: 10.1038/nchembio.64. [DOI] [PubMed] [Google Scholar]
- Volonté C, Amadio S, D'Ambrosi N, Colpi M, Burnstock G. P2 receptor web: complexity and fine-tuning. Pharmacol Ther. 2006;112:264–280. doi: 10.1016/j.pharmthera.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model I. Model description. J Theor Biol. 1996a;178:151–167. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model II. Understanding apparent affinity. J Theor Biol. 1996b;178:169–182. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor-occupancy model III. Resurrecting efficacy. J Theor Biol. 1996c;181:381–397. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SGF, Huang B, Zare RN, Kobilka B, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AS, Ferré S. Amazing stability of the arginine-phosphate electrostatic interaction. J Proteome Res. 2005;4:1397–1402. doi: 10.1021/pr050077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzo M. The sense of smell: molecular basis of odorant recognition. Biol Rev Camb Philos Soc. 2007;82:455–479. doi: 10.1111/j.1469-185X.2007.00019.x. [DOI] [PubMed] [Google Scholar]