

Abstract
Background and purpose:
Cannabinoid-2 (CB2) receptor-selective agonists have shown anti-nociceptive activity in models of neuropathic and inflammatory pain, and the two agonists most widely used, (+/−)AM1241 [(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone] and L768242 [(2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone] (GW405833), have been suggested to be protean agonists. Here we investigated the role of the constitutive activity of CB2 receptors in (+)AM1241 and L768242 protean agonism.
Experimental approach:
Pharmacological profiles of CB2 receptor ligands were evaluated in Chinese hamster ovary cells expressing recombinant human (hCB2) or rat (rCB2) receptors, by measuring modulation of cAMP. To assess the influence of constitutive activity on pharmacological profile, constitutive activity was abolished by pretreatment with AM630 [(6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone)], followed by extensive washing.
Key results:
In cell lines expressing either hCB2 or rCB2 receptors, (+)AM1241 did not reverse forskolin stimulation of cAMP levels. Conversely, L768242 was an inverse agonist at both hCB2 and rCB2 receptors. Abolition of constitutive activity disclosed (+)AM1241 and L768242 agonist activity, while activity of CP55940 [5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol] was unaffected and AM630 became a neutral antagonist. In presence of constitutively active CB2 receptors, (+)AM1241 antagonized CP55940, but when constitutive activity was abolished, it acted as a partial agonist with additive or antagonistic behaviour, depending on concentration.
Conclusions and implications:
These results show that (+)AM1241 and L768242 are protean agonists at both hCB2 and rCB2 receptors. Abolition of constitutive activity reveals the agonist activity of these compounds. Thus, differences between in vivo and in vitro profiles of CB2 receptor agonists could be due to different levels of constitutive activity in recombinant versus native CB2 receptors.
Keywords: AM1241, L768242, GW405833, protean agonist, constitutive activity, cannabinoid, CB2 receptor
Introduction
Cannabinoid effects are mediated by two distinct receptor subtypes: cannabinoid-1 (CB1) and cannabinoid-2 (CB2) receptors (Matsuda et al., 1990; Munro et al., 1993; nomenclature follows Alexander et al., 2008). Both CB1 and CB2 receptors belong to the superfamily of G protein-coupled receptors (GPCRs) and act mainly through inhibitory G proteins (Gi/o). The amino acid sequence identity between human CB1 and CB2 (hCB1, hCB2) receptors is unusually low for GPCRs subtypes that bind the same neurotransmitter/neuromodulator (Munro et al., 1993). Moreover, the sequence homology between orthologues, human versus rat and human versus mouse, is higher for CB1 receptors (95%) than for CB2 receptors (81%). The difference between mammalian CB2 receptors, more pronounced in the carboxy termini, may contribute to the variation in CB2 receptor pharmacology observed between species following receptor activation (Griffin et al., 2000; Mukherjee et al., 2004; Bingham et al., 2007; Yao et al., 2008).
Most of the pharmacological effects produced by cannabinoids have been attributed to the CB1 receptors that are expressed at high levels in the central nervous system (CNS), including regions involved in nociceptive perception and elaboration. However, activation of this receptor produces undesirable psychotropic side effects and therefore limits the development of a CB1 receptor agonist as an analgesic drug. In this scenario, in recent years the peripheral CB2 receptor has received increasing attention as a potential target for pain treatment.
This receptor is considered an innovative target for neuropathic pain therapies as its expression in the immune system and in distinct areas of the CNS (spinal cord and dorsal root ganglia) should avoid the unwanted centrally mediated side effects observed upon activation of CB1 receptors. Activation of CB2 receptors has been shown to have anti-nociceptive properties in various animal pain models, including chronic neuropathic and inflammatory type of pain (see Whiteside et al., 2007; Guindon and Hohmann, 2008; Beltramo, 2009). The two compounds most used to assess CB2 receptor agonist anti-nociceptive effects are (+/−)AM1241 [(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone] and L768242 [(2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone] (also named GW405833; Beltramo, 2009). Based on binding data, these compounds showed good affinity for CB2 receptors and selectivity over CB1 receptors (Yao et al., 2006; 2008; Bingham et al., 2007). Studies in rodent pain models showed that both compounds were anti-nociceptive and their action was either reversed by CB2 receptor-selective antagonists, such as SR144528 [N-[(1S)-endo-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl]5-(4-chloro-3-methylphenyl)-1-(4-methybenzyl)pyrazole-3-carboxiamide)] and AM630 [(6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone)], or absent in CB2 receptor -/- mice (Valenzano et al., 2005; Beltramo et al., 2006; Bingham et al., 2007). However, it should be noted that differences in the pharmacological profile of (+)AM1241 and (−)AM1241, that correspond to the R and S enantiomer respectively, have also been reported. In particular, (+)AM1241 has a higher affinity and selectivity for CB2 receptors and is also more potent in a cAMP-based functional assay compared with (−)AM1241 (Bingham et al., 2007). Surprisingly, however, (−)AM1241 showed a greater anti-nociceptive efficacy (Bingham et al., 2007), leading to the possibility that (−)AM1241 could activate other second messenger pathways with greater potency. This hypothesis would be in line with the theory of agonist-directed trafficking of response postulated by Kenakin (1995).
On the other hand, functional in vitro pharmacological characterization of both compounds is still incomplete, and conflicting data have recently been reported. For example, L768242 was shown to be a partial agonist (Valenzano et al., 2005) in a recombinant hCB2 receptor system, whereas another group reported this compound to be an inverse agonist on both hCB2 and rat CB2 (rCB2) receptor recombinant systems (Yao et al., 2008). In addition, it has been demonstrated that in recombinant systems, (+/−)AM1241 has inconsistent functional efficacies, behaving as an agonist or an antagonist depending on the assay conditions (Yao et al., 2006). This property has induced Yao et al. (2006) to suggest that (+/−)AM1241 may be a protean agonist. The phenomenon called protean agonism, after Proteus, the Greek god who could change shape, was first described by Kenakin on theoretical grounds, and is based on the observation that GPCR activation may occur spontaneously without any agonist binding (Kenakin, 2001). The receptor spontaneously binds G proteins and leads to the so-called constitutive activity of GPCRs (Kenakin, 2001). In this scenario, a protean ligand can act either as an agonist, an antagonist or an inverse agonist, depending on the level of constitutive activity of the receptor and on the intrinsic activity of the ligand. The existence of protean agonists has already been demonstrated for various ligands, such as proxyfan at the histamine H3 receptor (Gbahou et al., 2003); secretin at constitutively active mutants of secretin receptors (Ganguli et al., 1998); medetomidine and dexefaroxan analogue, RX831003 [2-(2-n-pentyl-2,3-dihydrobenzofuran-2-yl)-4,5-dihydro-1H-imidazole], at α2A-adrenoceptors (Jansson et al., 1998; Pauwels et al., 2002).
Considering the relevance that this phenomenon could have from a drug discovery perspective, the aim of this study was to provide essential experimental data to confirm the hypothesis that both (+/−)AM1241 and L768242 were indeed protean agonists. In order to obtain these data, we focused on testing the influence of constitutive activity on the pharmacological profile of R(+)AM1241, previously shown to be anti-nociceptive in rodent pain models (Beltramo et al., 2006), and L768242. We found that, in the presence of constitutively active receptors, these compounds were acting as either neutral or inverse agonists. By suppressing constitutive activity, we were able to unveil the agonist properties of both compounds. These experimental data confirm the protean agonist hypothesis for these two compounds.
Methods
Cell culture and transfection
Chinese hamster ovary (CHO) cells (ATCC, Manassas, VA, USA) were grown in F12 medium (Ham nutrient mixture) + 10% foetal bovine serum + 100 µg·mL−1 Penicillin/Steptomycin (PenStrep). They were stably transfected by using the Lipofectamine method with a pcDNA 3.1 expression vector (Invitrogen, Carlsbad, CA, USA) containing the coding sequence of either hCB2 (Accession # AY242132) or rCB2 receptor (Accession # NM_020543). After transfection, cells were maintained in the above complete medium containing G418 [3,5-dihydroxy-5-methyl-4-methylaminooxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-2-(1-hydroxyethyl)oxane-3,4-diol] (600 µg·mL−1) for clone selection. Clonal cell lines were grown at 37°C and 5% CO2 and were stable at least until passage no. 28. Splitting was performed by detaching the cells with 0.5% trypsin/EDTA.
Saturation binding
Membrane preparation from CHO stable cell lines (0.5 µg protein per well) expressing hCB2 or rCB2 receptors were used to perform radioligand-binding assays. Saturation experiments were performed in assay buffer [containing 50 mmol·L−1 Tris-HCl, pH 7.4, 2.5 mmol·L−1 EDTA, 5 mmol·L−1 MgCl2, 0.1% w/v fatty acid-free bovine serum albumin (BSA)], by using increasing concentrations from 0.01 to 10 nmol·L−1 of [3H]-CP55940 [5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol]. Non-specific binding was determined in presence of 1 µmol·L−1 unlabelled WIN55212-2. Binding reactions were conducted at room temperature (90 min incubation) and stopped by filtration through Multiscreen® HTS FB filter plates (#MSFBN6B50, Millipore, France) pre-soaked with 0.3% polyethyleneimine. After extensive washing the filters were dried, and radioactivity linked to filters was counted on a Microbeta Trilux counter (Perkin-Elmer Life Sciences, Waltham, MA, USA) by adding scintillation liquid in the plates.
Kd values were determined with ‘One site binding’ curve fitting of Prism software (GraphPad, San Diego, CA,USA).
cAMP assay
The Hit Hunter cAMP II assay enzyme fragment complementation chemiluminescent detection kit was used for the pharmacological characterization of clonal CB2 receptor-expressing cell lines. Cells (15 × 103 per well) in 50 µL complete medium were seeded onto 384-well white plates and incubated at 37°C and 5% CO2 for approximately 24 h before running the assay. In order to run the assay the medium was discarded, 10 µL of phosphate-buffered saline (PBS) containing IBMX (3-isobutyl-1-methylxanthine) (0.5 mmol·L−1) and 2.5 µL of vehicle were added to each well for basal level measurement. To measure functional activity, after adding the PBS containing IBMX, the agonist (2.5 µL) and forskolin (2.5 µL) were added to give a final concentration of 10 µmol·L−1. Cells were incubated for 30 min at 37°C. Finally, cAMP standard curve and the appropriate mixture of kit components were added (as described by the manufacturer, DiscoverX). Plates were incubated for 24 h at room temperature in the dark. Chemiluminescent signal was detected on Victor3 plate reader (Perkin-Elmer, Waltham, MA, USA) at 1 s·well−1.
In preliminary experiments, concentration–response curves (from 100 µmol·L−1 to 100 nmol·L−1) prepared by serial dilutions were used to establish the concentration of forskolin to be applied as stimulus. Based on these results, the experiments were performed by using a concentration of 10 µmol·L−1 of forskolin, unless otherwise specified. To perform ligand concentration–response curves, serial dilutions of the test compounds were prepared from a 10 mmol·L−1 stock in dimethyl sulphoxide (DMSO).
In some experiments, before performing the procedure described above, cells expressing rCB2 receptors were pretreated with 200 µg·mL−1Pertussis toxin (PTX) (from Sigma, St Louis, Mo, USA) for 24 h in order to block Gi protein activity.
To abolish constitutive activity of CB2 receptors, cells were resuspended in complete F12 medium containing 10 µmol·L−1 AM630, seeded onto 384-well plates and incubated for 24 h at 37°C and 5% CO2. At the end of the 24 h incubation the cells were extensively washed, six times for 10 min each, with F12 medium at 37°C and 5% CO2, and then stimulated with test compounds and processed for cAMP detection as described above. To assess the antagonist effect of (+)AM1241 cells were pre-incubated for 15 min at 37°C and 5% CO2.
GTPγS assay
Five micrograms of membranes from cells transfected with rCB2 receptors prepared in Tris-HCl 50 mmol·L−1 were used for each data point. AM630 was dissolved in Tris-HCl 50 mmol·L−1 containing 0.1% BSA and 0.5% DMSO. [35S] GTPγS (1250 Ci·mmol−1) was prepared in Tris-HCl 50 mmol·L−1 and used at the final concentration of 0.1 nmol·L−1. GDP concentration was 5 µmol·L−1. The assay was performed following standard procedure previously described in literature (Breivogel, 2006). Briefly, membranes were distributed in low binding 96-well plates (Corning, NY, USA) and incubated for 60 min at 30°C in buffer containing 50 mmol·L−1 Tris-HCl, 3 mmol·L−1 MgCl2, 0.2 mmol·L−1 EGTA, 100 mmol·L−1 NaCl, 0.1% BSA, 5 µmol·L−1 GDP, 0.5% DMSO, 0.1 nmol·L−1[35S] GTPγS (Perkin-Elmer) and AM630 at a concentration ranging: 10−12–10−5 mol·L−1. The assay was stopped by transferring the plate on ice; aliquots of assay mixture were transferred to filter plates and washed three times. Filter plates were dried for 1 h and radioactivity counted with a Microbeta Trilux counter.
Data analysis and statistical procedures
Data analysis was performed with GraphPad Prism 4 software (GraphPad, San Diego, CA, USA), using sigmoidal dose–response curve fitting to calculate EC50 values.
Data from cAMP assay were expressed as percentage of response to 10 µmol·L−1 forskolin. Counts per second (cps) relative to 10 µmol·L−1 forskolin were set to 100%, and the cps relative to the basal cAMP level were set to 0%. Data to evaluate cAMP concentration were calculated by interpolation of cAMP standard curve.
One-way anova followed by Bonferroni's post hoc test was used to perform statistical analysis, and P < 0.05 was considered statistically significant.
Materials
Synthetic cannabinoids were purchased from Tocris (Tocris Bioscience, Ellisville, MO, USA), except L768242 and (+)AM1241, which were synthesized by the Medicinal Chemistry Department of the Schering Plough Research Institute, Kenilworth, NJ, USA. Compounds were dissolved in 100% DMSO and stored as aliquots at −20°C. For cAMP detection the Hit Hunter cAMP II Assay enzyme fragment complementation chemiluminescent detection kit was used (DiscoverRx, Phremont, CA, USA).
For cell culture: medium F12 (Ham nutrient mixture), Penicillin/Steptomycin (PenStrep), foetal bovine serum, G418 (Geneticin), trypsin/EDTA (0.5% trypsin) were purchased from GIBCO (Invitrogen, Carlsbad, CA, USA). PBS, Hanks’ balanced salt solution (HBSS), HEPES buffer solution, Lipofectamine 2000 Kit were purchased from GIBCO (Invitrogen, Carlsbad, CA, USA). 384-well white plates, cell culture treated, for cAMP assay were purchased from Matrix (Thermo Fisher Scientific; Hudson, NH, USA). 96-well plates, white, non-binding-surface (NBS) for compound dilutions were purchased from Corning (New York, NY, USA).
DMSO, IBMX, forskolin and PTX were purchased from Sigma (St Louis, MO, USA) and [35S]GTPγS was from Perkin-Elmer.
Results
hCB2 and rCB2 receptors pharmacology in recombinant cell lines
The pharmacology of different cannabinoid compounds was evaluated on recombinant hCB2 and rCB2 receptors stably expressed in CHO cells. Saturation binding experiments using [3H]-CP55940 as radioligand indicated that the two cell clones expressed the receptors at similar levels, 11 and 14 pmol·mg−1 respectively (data not shown).
In cAMP assays, cannabinoid agonists and inverse agonists had no effect on forskolin-stimulated cAMP level in non-transfected CHO cells (data not shown). Basal cAMP level in both transfected and non-transfected cell lines was at the lower limit of the linear range of the standard curve (about 150 fmol·well−1). In this situation, it is difficult to draw any conclusion about basal receptor activity. Conversely, the cAMP levels after 10 µmol·L−1 forskolin stimulation were in the linear range and showed a significant difference between non-transfected and transfected cell lines (CHO 2225 ± 177 fmol·well−1; hCB2 receptor-CHO 1338 ± 344 fmol·well−1, P < 0.01 vs. CHO; rCB2 receptors-CHO 1055 ± 454 fmol·well−1, P < 0.01 vs. CHO). These data suggest the existence of constitutively active receptors in these recombinant cell lines. To further assess the existence of constitutive activity of CB2 receptors in absence of external stimulation such as forskolin, the effect of AM630 was evaluated on GTPγS binding to rCB2 receptors. Treatment of transfected cell membrane with AM630 reduces the basal level of GTPγS binding by 23.5 ± 9.0% further supporting the existence of constitutively active CB2 receptors in our experimental setting.
The reference agonists CP55940 and JWH133 [((6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran)] showed an order of potency, based on EC50 measurements, comparable at both receptors with CP55940 > JWH133 (Figure 1A and B). Considering that at both receptors CP55940 showed the highest efficacy of the ligands examined, it was taken as reference for the determination of intrinsic activity of other ligands. Maximal efficacy (Emax) of CP55940 was set to 100%, and Emax values of the ligands were calculated as the percentage of maximal CP55940 effect (Table 1).
Figure 1.

Concentration–response curves of cannabinoid compounds in cAMP functional assay. The curves show the effects of increasing concentration of the cannabinoid ligands on FSK-induced cAMP levels in stable CHO cells expressing the human (A) or the rat (B) CB2 receptor. Cells were stimulated in the presence of 10 µmol·L−1 FSK for 30 min. As expected CP55940 and JWH133 displayed an agonist profile and AM630 behaved as an inverse agonist. Conversely, (+)AM1241 was apparently inactive, whereas L768242 was an inverse agonists at both receptors. Data are expressed as percentage of FSK stimulation where 100% corresponds to 10 µmol·L−1 and 0% to basal intracellular cAMP levels. Each data point is the percentage mean values ± SEM of at least three independent experiments, each performed in quadruplicate. (+)AM1241, (+)(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone; AM630, (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone); CB2, cannabinoid-2; CHO, Chinese hamster ovary; CP55940, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol; FSK, forskolin; JWH133, ((6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran); L768242, (2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone.
Table 1.
Functional characterization of hCB2 and rCB2 receptors in cAMP assay
| Compound |
EC50 (nmol·L−1) (±SEM) |
Emaxa |
||
|---|---|---|---|---|
| hCB2 | rCB2 | hCB2 | rCB2 | |
| CP55940 | 9.7 (±2.5) | 4.0 (±1.7) | 100 | 100 |
| JWH133 | 43.0 (±12.5) | 54.2 (±3.2) | 102 | 69 |
| (+)AM1241 | ND | ND | −8 | −28 |
| L768242 | 11.8 (±1.5) | 7.2 (±2.1) | −42 | −99 |
| AM630 | 45.0 (±9.4) | 9.4 (±0.5) | −135 | −112 |
(+)AM1241, (+)(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone; AM630, (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone); CP55940, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol; hCB2, human CB2; JWH133, ((6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran); L768242, (2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone; ND, not determinable; rCB2, rat CB2.
% of CP55940 inhibition at 1 µmol·L−1.
The inverse agonist AM630, as expected, further increased the forskolin-stimulated cAMP levels, at both the hCB2 and rCB2 receptors (Figure 1A and B, Table 1). This effect was mediated by Gi protein, as demonstrated by its abolition in the rCB2 receptor cell line after treatment with PTX (205.9 ± 8.2% of forskolin stimulation before treatment vs. 94.0 ± 10.6% after treatment). Also the effect of CP55940 was blocked by PTX treatment (17.2 ± 1.0% of forskolin stimulation before treatment vs. 126.0 ± 4.0% after treatment) confirming that all observed effects are Gi-dependent.
When assessed for activity, (+)AM1241 and L768242, previously reported as selective CB2 receptor ligands, showed a peculiar pharmacological profile. At hCB2 and rCB2 receptors, (+)AM1241 behaved as a weak inverse agonist inducing a small increase in the forskolin-stimulated cAMP level (Figure 1A and B, Table 1). In the presence of such a weak effect, EC50 values could not be calculated and the compound was considered inactive. On the other hand, L768242, reported to be a partial agonist by Valenzano et al. (2005) and as an inverse agonist by Yao et al. (2008), was clearly an inverse agonist at hCB2 and rCB2 receptors (Figure 1A and B, Table 1).
It has been previously reported that the forskolin concentration might influence the intrinsic activity of protean agonists (Yao et al., 2006). In order to check for such effect, AM630, (+)AM1241 and CP55940 activity were analysed in the presence of different forskolin concentrations, from 2 to 32 µmol·L−1, in the cyclase assay. As shown in Figure 2, the increase in cAMP level induced by AM630 was stable at both hCB2 and rCB2 receptors, independent of the forskolin stimulus used. In addition, neither CP55940, nor (+)AM1241 modified their maximal efficacy at different levels of forskolin stimulation, showing that in this system forskolin did not influence compound efficacy. CP55940 remained a full agonist at the hCB2 and the rCB2 receptors, reducing the forskolin-induced cAMP level near to basal level at all forskolin concentrations. (+)AM1241 remained almost inactive or showed a modest inverse agonist activity (Figure 2A and B).
Figure 2.

AM630, CP55940 and (+)AM1241 maximal effects at human CB2 receptors (A) and at rat CB2 receptors (B) in functional cAMP assay after stimulation with various FSK concentrations. The graphs show the efficacy of the maximally active dose of CP55940 (1 µmol·L−1), (+)AM1241 (10 µmol·L−1) and AM630 (10 µmol·L−1) at different concentrations of FSK (µmol·L−1). Note that the first sets of data in (A) and (B) represent the effects of the standard dose of FSK (10 µmol·L−1). The ability of the cannabinoid compounds to modulate cAMP levels was not influenced by the concentration of FSK. Data are expressed as percentage of maximal FSK stimulation where 100% corresponds to the response obtained with FSK at the indicated concentration and 0% to basal intracellular cAMP levels. Data are mean values of two experiments performed in quadruplicate. (+)AM1241, (+)(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone; AM630, (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone); CB2, cannabinoid-2; CP55940, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol; FSK, forskolin.
Abolition of constitutive activity at hCB2 and rCB2 receptors
In order to study the pharmacological profile of compounds in the absence of constitutive activity, a protocol of pretreatment with the inverse agonist AM630 followed by extensive wash was established. It has been previously demonstrated that this type of protocol could block constitutive activation of receptors (Zhang et al., 2000; Seifert and Wenzel-Seifert, 2002; 2003; Milligan, 2003). Both hCB2 and rCB2 receptor cell lines were treated for 24 h with 10 µmol·L−1 AM630 and, after 1 h wash, they were challenged with compounds and cAMP level assessed after forskolin stimulation (see Methods).
Successful abolition of constitutive activity was assessed by testing increasing concentrations of AM630 after 24 h pretreatment. As shown in Figure 3A and B, in both hCB2 and rCB2 receptor cell lines, AM630 was no longer able to induce an increase in the cAMP level, as it did in non-pretreated cells. A second effect induced by block of constitutive activity was an increase of the level of cAMP induced by forskolin stimulation (Figure 3A and B).
Figure 3.
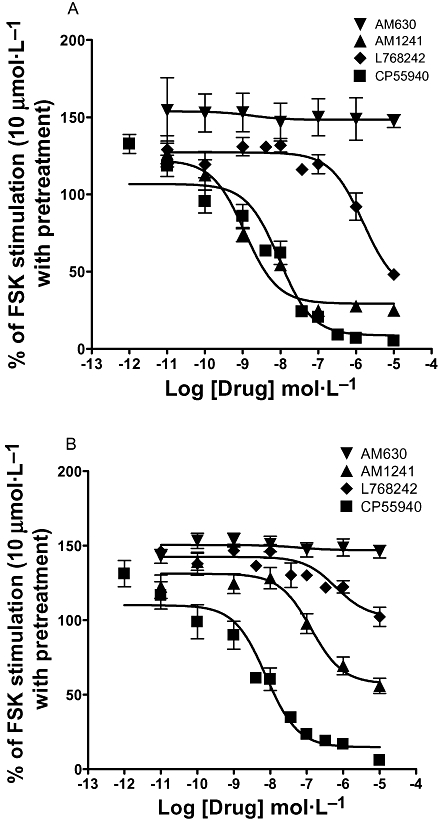
Concentration–response curves of cannabinoid compounds in cAMP functional assay at hCB2 receptors (A) and at rCB2 receptors (B) in absence of CB2 receptor constitutive activity. The curves show the effects of increasing concentration of cannabinoid ligands on FSK-induced cAMP levels in absence of constitutive activity. Constitutive activity was abolished by 24 h pretreatment with 10 µmol·L−1 AM630 followed by extensive washing. At both hCB2 receptors (A) and rCB2 receptors (B) AM630 leaves unaltered the FSK-stimulated cAMP level and CP55940 decreases it near to basal levels, acting as a full agonist. (+)AM1241 and L768242 also showed agonist activity as illustrated by the decrease of FSK-induced cAMP levels. Both compounds showed higher efficacy at hCB2 receptors compared with rCB2 receptors. Data are expressed as percentage of FSK stimulation where 100% corresponds to 10 µmol·L−1 FSK and 0% to basal intracellular cAMP levels in non-pretreated cells. Data are expressed as percentage mean values ± SEM of at least four independent experiments, each performed in quadruplicate. (+)AM1241, (+)(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone; AM630, (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone); CB2, cannabinoid-2; CP55940, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol; FSK, forskolin; hCB2, human CB2; L768242, (2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone; rCB2, rat CB2.
As expected, the activity of the full agonist CP55940 remained unaltered after pretreatment, and no significant differences were observed in its potency, neither at the hCB2 receptor (EC50 values were 9.7 nmol·L−1 before and 9.3 nmol·L−1 after pretreatment), nor at the rCB2 receptor (EC50 values were 4.0 nmol·L−1 before and 7.6 nmol·L−1 after pretreatment) (Figure 3A and B, Table 2). The statistical analysis confirmed that the concentration–response curves of CP55940 performed with or without AM630 pretreatment were not statistically different (P= 0.75 at hCB2 receptors and 0.33 at rCB2 receptors; one-way anova), proving that the inverse agonist was washed away and there was no residual antagonist effect.
Table 2.
Functional cAMP assay after abolition of receptor constitutive activity
| Compound |
Cyclase assay after AM630 treatment |
|||
|---|---|---|---|---|
|
EC50 (nmol·L−1) (95% confidence interval) |
Emaxa |
|||
| hCB2 | rCB2 | hCB2 | rCB2 | |
| CP55940 | 9.3 (5.5–15.6) | 7.6 (4.2–14.0) | 100 | 100 |
| (+)AM1241 | 1.4 (0.6–2.0) | 132.4 (61.4–285) | 90 | 63 |
| L768242 | >1000 (631–3815) | 628 (182–2169) | 69 | 24 |
| AM630 | ND | ND | ND | ND |
(+)AM1241, (+)(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone; AM630, (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone); CP55940, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol; hCB2, human CB2; L768242, (2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone; ND, not determinable; rCB2, rat CB2.
% of CP55940 inhibition at 1 µmol·L−1.
Pharmacological profile of (+)AM1241 and L768242 in absence of constitutively active CB2 receptors
After AM630 pretreatment, and thus in the absence of constitutive activity, (+)AM1241 revealed its agonistic activity at both the hCB2 receptor (EC50= 1.4 nmol·L−1 and Emax= 90%) and rCB2 receptor (EC50= 132.4 nmol·L−1 and Emax= 63%) (Figure 3A and B, Table 2). These data confirmed that (+)AM1241 can be considered a protean agonist, as its pharmacological profile was dependent on the constitutive activity of the receptor.
A similar profile was obtained with L768242. After block of constitutive activity, L768242 showed an agonist profile, although with lower potencies and efficacies at both hCB2 and rCB2 receptors (EC50 > 1000 nmol·L−1 and Emax= 69%; EC50= 628 nmol·L−1 and Emax= 24%, respectively) (Table 2). These data demonstrate that also L764282 is a protean agonist at the CB2 receptors, with a low intrinsic activity.
Based on the protean agonist theory, in the presence of constitutively active CB2 receptors (+)AM1241 should behave as an antagonist and in the absence of constitutive activity, it should behave as an agonist or, more precisely, as a partial agonist because its intrinsic activity is lower than the full agonist CP55940. To confirm these predictions, (+)AM1241 was tested for its ability to compete with CP55940 before and after pretreatment with AM630.
In normal conditions, (+)AM1241 dose-dependently blocked the agonist activity of CP55940 at both hCB2 and rCB2 receptors (Figure 4A and B). Rightward shifts of the CP55940 concentration–response curves were observed in the presence of increasing concentrations (from 10 nmol·L−1 to 10 µmol·L−1) of (+)AM1241. Indeed, the CP55940 EC50 values rose from 9.7 to 778 nmol·L−1 at the hCB2 receptor and from 4 to 2070 nmol·L−1 at the rCB2 receptor. In addition, due to the slight inverse agonist activity of (+)AM1241, at the lower CP55940 concentrations upward shifts of the curves were also observed at both CB2 receptors.
Figure 4.

Concentration–response curves of CP55940 in the presence of (+)AM1241 in cAMP functional assay with (A and B) and without (C and D) CB2 receptor constitutive activity. The assay was performed on hCB2 receptors (A and C) and rCB2 receptors (B and D). Cells were incubated with 1 µmol·L−1 (+)AM1241 for 15 min, then stimulated with CP55940 in the presence of 10 µmol·L−1 FSK for 30 min (A and B). The same procedure was followed in (C) and (D), after block of constitutive activity by treating cells for 24 h with 10 µmol·L−1 AM630 followed by 1 h extensive washing. In presence of constitutive activity, (+)AM1241 acted as an antagonist inducing a rightward shift of the concentration–response curve of CP55940 (A and B). In absence of constitutive activity (+)AM1241 had an additive effect at low CP55940 concentrations, whereas at high concentrations of both compounds, it behaved as an antagonist and shifted the CP55940 concentration–response curve to the right (C and D). Data are expressed as percentage of FSK stimulation where 100% corresponds to 10 µmol·L−1 FSK and 0% to basal intracellular cAMP levels. All experimental data of the curves are expressed as percentage mean values ± SEM of two independent experiments, each performed in quadruplicate. (+)AM1241, (+)(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone; AM630, (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone); CB2, cannabinoid-2; CP55940, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol; FSK, forskolin; hCB2, human CB2; rCB2, rat CB2.
After AM630 pretreatment, co-application of a fixed (+)AM1241 concentration had a double effect on CP55940 concentration response curve. At low CP55940 concentration, (+)AM1241 had an additive effect, but at high concentration of both compounds there was a rightward shift of the concentration–response curve revealing an antagonistic effect of (+)AM1241 (Figure 4C and D). Thus, in this experimental condition, (+)AM1241 behaves as a partial agonist with different efficacy at rCB2 and hCB2 receptors.
Discussion and conclusions
The CB2 receptor has received increasing attention in recent years, encouraged by data showing that CB2 receptor-selective agonists have anti-nociceptive properties in preclinical rodent models of neuropathic and inflammatory pain (Malan et al., 2001; 2002; Fox and Bevan, 2005; Whiteside et al., 2007; Guindon and Hohmann, 2008). As the CB2 receptor is mainly expressed in the periphery and only in some regions of the CNS (spinal cord and dorsal root ganglias), CB2 receptor-selective agonists are expected to elicit analgesic effects without displaying the unwanted psychotropic effects that have prevented the development of a CB1 receptor agonist drug.
The CB2 receptor-selective agonists most widely used to prove that activation of the CB2 receptors mediates analgesia have been (+/−)AM1241 and L768242 (GW405833) (Beltramo, 2009). While they showed efficacy in multiple pain models, at the same time they displayed inconsistent pharmacological profiles in vitro (Valenzano et al., 2005; Yao et al., 2006; Bingham et al., 2007; Yao et al., 2008).
To further explore the in vitro pharmacology of these agonists we have created CHO recombinant cell lines expressing hCB2 or rCB2 receptors. In these cell lines the pharmacology of reference agonists (CP55940 and JWH133) studied by functional assay (cAMP) was consistent with published data in terms of EC50 and Emax values (Griffin et al., 2000; Howlett et al., 2002; Mukherjee et al., 2004; Yao et al., 2008).
In these cell lines, (+)AM1241 appeared inactive or behaved as weak inverse agonist. On the other hand, L768242 showed a small inverse agonist activity at the hCB2 receptor and a full inverse agonist activity (Emax comparable to AM630) at the rCB2 receptor.
The phenomenon of diverse functional efficacy of one compound at a given receptor has already been described for other receptor/compound pairs: proxyfan at the histamine H3 receptor (Gbahou et al., 2003), secretin at constitutively active mutants of secretin receptors (Ganguli et al., 1998), medetomidine and the dexefaroxan analogue (RX831003) at α2A-adrenoceptors (Jansson et al., 1998; Pauwels et al., 2002), dichloroisoproterenol at β2-adrenoceptors (Chidiac et al., 1996). Ligands that behave in this way are called ‘protean’ agonists as these ligands change their apparent behaviour (Kenakin, 2001). By definition, a protean agonist is a ligand with functional efficacy dependent upon the relative level of constitutive activity exhibited by the system. It is well known that GPCRs can spontaneously form an active state and activate G proteins, triggering signal transduction cascades in absence of ligand binding. This condition of spontaneous receptor activity is known as constitutive activity, and the CB2 receptor is among the GPCRs that display constitutive activity (Milligan, 2003). On the other hand, a property of every compound is intrinsic activity, which reflects the ability of the ligand to interact with the receptor and to produce a response. If a ligand displays a positive high intrinsic activity, it will behave as a full agonist in systems with both high and low constitutive activity, showing always maximal efficacy. A ligand with a negative intrinsic activity will behave as a neutral antagonist in systems without constitutive activity and as an inverse agonist in systems with constitutive activity, showing either null or negative efficacy. A ligand without any intrinsic activity will behave as a neutral antagonist in systems both with and without constitutive activity, and such ligands are the real neutral antagonists. Instead, a protean agonist is a ligand with a low level of positive intrinsic activity. It will behave as a partial agonist in a system with a relatively low level of constitutive activity, but will behave as an inverse agonist when the receptor constitutive activity is high, thus showing positive, null or negative efficacy depending on the relative level of constitutive activity exhibited by the system.
Bingham et al. studying (+)AM1241 pharmacology found that in cell lines expressing mouse or rCB2 receptors, (+)AM1241 behaved as inverse agonist whereas at hCB2 receptors, it showed agonist activity. By using the inverse agonist SR144528, the assessment of constitutive activity showed similar levels in all three cell lines. The authors concluded that while it was tempting to speculate that (+)AM1241 could be a protean agonist, their data, based on the different pharmacological profile of (+)AM1241 at the human compared to rodent CB2 receptors, could not support this hypothesis (Bingham et al., 2007). However, no alternative explanation for the observed discrepancy was proposed. Conversely, (+/−)AM1241 was suggested to be a protean agonist (Yao et al., 2006) based on the different effect observed in various assays [calcium influx, extracellular signal-regulated kinase (ERK) phosphorylation and cAMP measurement] and on the switch from neutral antagonism to agonism in the cAMP assay when forskolin concentration was lowered (Yao et al., 2006). The reason for the discrepancy of this last result with our observation showing that forskolin had no effect on the activity of (+)AM1241 is unclear. However, it should be mentioned that Yao et al. used the AM1241 racemate in their experiments, while in our study we used the (+) enantiomer and this could have had an effect on the final outcome. In the ERK kinase assay, (+/−)AM1241 behaved as a partial agonist, whereas it displayed neutral antagonist activities in the cyclase and fluorometric imaging plate reader (FLIPR) assays. These differences were attributed to a different level of constitutive activity in the different assays: lower in the ERK assay and higher in the cAMP and FLIPR assays (Yao et al., 2006). However, the level of constitutive activity on the ERK assay was not tested, and inverse agonist activity is not detectable by the FLIPR assay. Therefore, even though the hypothesis was intriguing, crucial experimental evidence to confirm it, was missing. In addition, these results could also be explained by another phenomenon known as ‘agonist-directed trafficking of response’ (Kenakin, 1995). This theory postulates that certain ligands preferentially activate one effector system rather than another. As a consequence of this theory, a ligand acting on a receptor can trigger a certain signalling pathway and not another triggered by other ligands at the same receptor. Thus, ligands could have on the same receptor different intrinsic activity at different second messenger pathways, regardless of constitutive activity. As evidence has been published suggesting agonist-directed trafficking of response at CB2 receptors (Shoemaker et al., 2005), the results obtained with (+/−)AM1241 by Yao et al. (2006) could have an explanation other than protean agonism. The compound would activate the ERK kinase pathway, where it behaves as a partial agonist, while it would not modulate cAMP or intracellular Ca2+ pathway simply because it did not activate these effector systems.
In our experimental conditions, basal cAMP level was at the lower limit of the linear range of the standard curve and thus was not suitable to assess the presence of constitutive activity. However, the existence of constitutive activity of CB2 receptor in our cell lines is supported by different experimental data. First, the stimulation with forskolin in the parental cell line induces a higher level of cAMP than in the CB2 receptor transfected cell lines suggesting an inhibitory effect linked to constitutively active CB2 receptors. Second, administration of the inverse agonist AM630 induced a further increase of forskolin-stimulated cAMP level. This is in agreement with data showing that, after treatment with the inverse agonist SR144528, in the presence of constitutive activity there is an increase of cAMP levels above those stimulated by forskolin (Rhee and Kim, 2002; Bingham et al., 2007). The possibility that this increase of cAMP level could be elicited by Gs protein activation was ruled out by the result obtained with PTX that completely abolished this effect. Thirdly, the GTPγS assay application of AM630 reduced the basal level of GTPγS binding indicating the presence of constitutively active CB2 receptors. Thus, these cell lines represent a suitable tool for further investigating the pharmacology of CB2 receptor ligands in the presence of constitutive activity.
Therefore, we focused on the relevance of CB2 receptor constitutive activity in order to clarify if changes in constitutive active could actually be the cause of the puzzling pharmacological profiles of (+/−)AM1241 and L768242. Considering that application of inverse agonists stabilizes or enriches the inactive state of the receptor reducing signalling transduction (Milligan, 2003), in the case of CB2 receptors, inverse agonist pretreatment would reduce the inhibition of adenylate cyclase and consequently increase intracellular cAMP level.
In order to evaluate the effect of constitutive activity on the pharmacological profile of (+)AM1241 and L768242, this activity was abolished by using a protocol of inverse agonist pretreatment. Under this experimental condition, a full agonist (CP55940), an inverse agonist (AM630) and both (+)AM1241 and L768242 were tested at hCB2 and rCB2 receptors. The pretreatment indeed suppressed constitutive CB2 receptor activity as shown by the observation that AM630 became a neutral antagonist at both hCB2 and rCB2 receptors. In addition, unsurprisingly, the block of CB2 receptor constitutive activity resulted in a larger magnitude of forskolin-stimulated cAMP level in pretreated cells, as compared with non-pretreated cells.
As expected, CP55940 remained a full agonist in the presence or absence of constitutive activity at both hCB2 and rCB2 receptors. This confirms that after extensive washing, no residual AM630 was present to block receptor activity.
Conversely, upon AM630 pretreatment, (+)AM1241 and L768242 changed pharmacological profile at both hCB2 and rCB2 receptors. When the constitutive activity of the CB2 receptor was abolished, the agonist component of (+)AM1241 and L768242 was revealed at both receptors with a higher potency and efficacy at the hCB2 receptor. These results strongly support the hypothesis that both (+)AM1241 and L768242 are indeed protean agonists.
Having established that both compounds behave in the same way, in the second part of the study we decided to focus on AM1241, as it is the most widely used compound to study anti-nociceptive effects of CB2 receptor agonists. Having demonstrated that (+)AM1241 has a low intrinsic activity in our experimental setting, we inferred that in the presence of constitutive activity it should behave as an antagonist. Indeed, it antagonized the effect of the full agonist CP55940 at both hCB2 and rCB2 receptors. When CB2 receptor constitutive activity was abolished, (+)AM1241 induced a downward shift of the CP55940 concentration–response curve at both hCB2 and rCB2 receptors. However, in line with the low intrinsic activity of (+)AM1241, a residual antagonism could be observed at high (+)AM1241 concentrations, as described for partial agonists. These data further confirmed that (+)AM1241 is a protean agonist at the receptors and that this effect depends on the receptor constitutive activity.
The discrepancy between the null or negative efficacy of (+/−)AM1241, (+)AM1241 and L768242 in in vitro recombinant systems and their agonist efficacy in animal models of chronic pain could be explained by at least two hypotheses: (i) the absence of, or low CB2 receptor constitutive activity in vivo makes these compounds behave as agonists; and (ii) CB2 receptors are constitutively active in vivo but differences of cell environment between native and recombinant system, such as total receptor and G protein concentration, rate of G protein activation/deactivation, or different subtypes of Gαi or Gαo, system, make (+/−)AM121, (+)AM1241 and L768242 behave as agonists. Thus, an essential step forward to accept or reject these hypotheses would be the in vivo assessment of constitutive activity of CB2 receptors.
The physiological and clinical importance of receptor constitutive activity, and consequently of inverse and protean agonists, has become a important topic in recent years. In vivo, a protean agonist could behave differently depending on the level of receptor constitutive activity in different tissues or in diverse physiological or pathological conditions: as an agonist, causing receptor activation, or as an antagonist/inverse agonist, decreasing receptor activation. At present, it is not clear what therapeutic relevance a protean agonist would have. However, as it would set the level of receptor stimulation to a constant state without silencing it completely, as would happen with an inverse agonist, it could represent a valid treatment for pathologies caused by receptor constitutive activity that over-stimulate the system. This type of pathologies could be caused by mutations in the receptor or by overactivity of G protein or their regulators. Therefore, protean agonists could represent a new and promising class of drugs when constitutive receptor overactivity is the pathological element of a disease, but where the receptor tone also has a physiological role. Further studies will be essential to prove this hypothesis that could have important implications from the perspective of drug discovery.
Glossary
Abbreviations:
- AM1241
(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone
- AM630
(6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl) methanone)
- CHO
Chinese hamster ovary
- CP55940
5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol
- DMSO
dimethyl sulphoxide
- G418
3,5-dihydroxy-5-methyl-4-methylaminooxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-2-(1-hydroxyethyl)oxane-3,4-diol
- GPCR
G protein-coupled receptors
- IBMX
3-isobutyl-1-methylxanthine
- JWH133
((6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran)
- L768242
(2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone
- RX831003
2-(2-n-pentyl-2,3-dihydrobenzofuran-2-yl)-4,5-dihydro-1H-imidazole
- PTX
Pertussis toxin
Conflicts of interest
All authors were at the time of manuscript preparation, employees of Schering-Plough Research Institute, which has founded the research.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd) 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. edn (2008 revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006;23:1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- Beltramo M. Cannabinoid type 2 receptor as a target for chronic pain. MRMC. 2009;9:11–25. doi: 10.2174/138955709787001785. [DOI] [PubMed] [Google Scholar]
- Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA, et al. Species-specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol. 2007;151:1061–1070. doi: 10.1038/sj.bjp.0707303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel C. Cannabinoid receptor binding to membrane homogenates and cannabinoid-stimulated [35S]GTPgammaS binding to membrane homogenates or intact cultured cells. Methods Mol Med. 2006;123:149–162. doi: 10.1385/1-59259-999-0:149. [DOI] [PubMed] [Google Scholar]
- Chidiac P, Nouet S, Bouvier M. Agonist-induced modulation of inverse agonist efficacy at the beta 2-adrenergic receptor. Mol Pharmacol. 1996;50:662–669. [PubMed] [Google Scholar]
- Fox A, Bevan S. Therapeutic potential of cannabinoid receptor agonists as analgesic agents. Expert Opin Investig Drugs. 2005;14:695–703. doi: 10.1517/13543784.14.6.695. [DOI] [PubMed] [Google Scholar]
- Ganguli SC, Park CG, Holtmann MH, Hadac EM, Kenakin TP, Miller LJ. Protean effects of a natural peptide agonist of the G protein-coupled secretin receptor demonstrated by receptor mutagenesis. J Pharmacol Exp Ther. 1998;286:593–598. [PubMed] [Google Scholar]
- Gbahou F, Rouleau A, Morisset S, Parmentier R, Crochet S, Lin JS, et al. Protean agonism at histamine H3 receptors in vitro and in vivo. PNAS. 2003;19:11086–11091. doi: 10.1073/pnas.1932276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin G, Tao Q, Abood ME. Cloning and pharmacological characterization of the rat CB2 cannabinoid receptor. J Pharmacol Exp Ther. 2000;292:886–894. [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Jansson CC, Kukkonen JP, Nasman J, Huifang G, Wurster S, Virtanen R, et al. Protean agonism at alpha2A-adrenoceptors. Mol Pharmacol. 1998;53:963–968. [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Vanderah TW, Makriyannis A, Porreca F. Inhibition of pain responses by activation of CB(2) cannabinoid receptors. Chem Phys Lipids. 2002;121:191–200. doi: 10.1016/s0009-3084(02)00155-x. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol. 2003;64:1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Adams M, Whiteaker K, Daza A, Kage K, Cassar S, et al. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur J Pharmacol. 2004;505:1–9. doi: 10.1016/j.ejphar.2004.09.058. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Pauwels PJ, Rauly I, Wurch T, Colpaert FC. Evidence for protean agonism of RX 831003 at alpha 2A-adrenoceptors by co-expression with different G alpha protein subunits. Neuropharmacology. 2002;42:855–863. doi: 10.1016/s0028-3908(01)00201-5. [DOI] [PubMed] [Google Scholar]
- Rhee M-H, Kim S-K. SR144528 as inverse agonist of CB2 cannabinoid receptor. J Vet Sci. 2002;3:179–184. [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. The human formyl peptide receptor as model system for constitutively active G-protein-coupled receptors. Life Sci. 2003;73:2263–2280. doi: 10.1016/s0024-3205(03)00654-4. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Ruckle MB, Mayeux PR, Prather PL. Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J Pharmacol Exp Ther. 2005;315:828–838. doi: 10.1124/jpet.105.089474. [DOI] [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Lee GP, Valenzano KJ. The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr Med Chem. 2007;14:917–936. doi: 10.2174/092986707780363023. [DOI] [PubMed] [Google Scholar]
- Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB2 receptor? Br J Pharmacol. 2006;149:145–154. doi: 10.1038/sj.bjp.0706838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao BB, Hsieh GC, Frost JM, Fan Y, Garrison TR, Daza AV, et al. In vitro and in vivo characterization of A-796260: a selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in rodent pain models. Br J Pharmacol. 2008;153:390–401. doi: 10.1038/sj.bjp.0707568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Cheng H, Zhou YY, Wang DJ, Zhu W, Ziman B, et al. Inhibition of spontaneous beta 2-adrenergic activation rescues beta 1-adrenergic contractile response in cardiomyocytes overexpressing beta 2-adrenoceptors. J Biol Chem. 2000;275:21773–21779. doi: 10.1074/jbc.M909484199. [DOI] [PubMed] [Google Scholar]