

Abstract
The free fatty acid receptors FFA2 and FFA3 are recently de-orphanized G protein-coupled receptors that share a group of short-chain free fatty acids as endogenous ligands. The expression of FFA2 and FFA3 by immune cells, in parts of the gastro-intestinal tract and by white adipocytes has suggested their potential as therapeutic targets in conditions including inflammation and obesity. However, although FFA2 and FFA3 display distinct structure–activity relationships for stimulation by short-chain free fatty acids, the overlap between these endogenous agonists and the lack of synthetic small molecule ligands that display selectivity between these two receptors has, until recently, hindered efforts to resolve their individual functions. Recently, chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide has been described as an FFA2 selective ago-allosteric ligand, not only being a direct agonist but also acting as a positive allosteric modulator of the function of short-chain free fatty acids at FFA2. Mutation of a pair of key arginine residues near the top of transmembrane domains V and VII of both FFA2 and FFA3 eliminates the function of short-chain free fatty acids but is without effect on the direct agonist action of chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide at FFA2, confirming the distinct nature of the binding site of the ago-allosteric regulator from the orthosteric binding site for free fatty acids. An understanding of structure–activity relationships for ligands related to chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide is likely to provide greater insight into the mode of action and site of binding of this ligand, but further FFA2 and FFA3 selective ligands, preferably with higher potency/affinity, will be required to fully explore the physiological function of these receptors.
Keywords: G protein-coupled receptor, free fatty acid, ago-allosterism
Introduction
Although G protein-coupled receptors (GPCRs) are the molecular targets for a wide range of therapeutic small molecule medicines, only a small proportion of the GPCR family have been exploited in this way (Wise et al., 2004). In part, this reflects the importance of aminergic neurotransmitters and hormones in the regulation and homeostasis of key physiological functions that may become dysregulated in disease. However, a substantial number of GPCRs with expression patterns consistent with them playing important roles in tissues and functions that are known to be subverted in disease have yet to be exploited effectively. In a number of cases, this reflects a lack of knowledge of the endogenous modulators that regulate these receptors and such receptors are classified, therefore, as ‘orphans’ (Wigglesworth et al., 2006). A series of biomolecules, generally considered as metabolic intermediates, have recently been shown to act as agonists at certain, previously ‘orphan’, GPCRs. These include β-hydroxybutyrate (Taggart et al., 2005) [and/or butyrate (Thangaraju et al., 2009)] as an activator of GPR109A, aromatic D-amino acids as chemoattractant factors activating GPR109B (Irukayama-Tomobe et al., 2009), lactate as an agonist of GPR81 (Cai et al., 2008; Liu et al., 2009), kynurenic acid as an agonist of GPR35 (Wang et al., 2006a), succinate as an agonist of SUCNR1 (previously designated GPR91) (He et al., 2004) and oxaloacetate as an agonist of the OXGR1 receptor (previously designated GPR99) (He et al., 2004). Furthermore, free fatty acids of various chain length and degree of saturation have been shown to act as agonists at both GPR120 (Hirasawa et al., 2005) and GPR84 (Wang et al., 2006b) as well as the group of related GPCRs named free fatty acid receptors FFA1, FFA2 and FFA3 (previously designated GPR40, GPR43 and GPR41 respectively) (Briscoe et al., 2003; Brown et al., 2003; Itoh et al., 2003; Kotarsky et al., 2003; Le Poul et al., 2003; see Stoddart et al., 2008a for review). In a number of these cases, identification of endogenous ligands able to activate these GPCRs has not yet resulted in the identification (or at least the public disclosure) of synthetic, small molecule ligands able to act as agonists or antagonists of these receptors.
Phylogenetic analysis has clustered FFA1-3 into a branch of the rhodopsin-like, class A GPCRs that contains receptors for phospholipids, nucleotides, citric acid cycle intermediates and the protease-activated receptors, along with a number of orphan GPCRs (Surgand et al., 2006; Tikhonova et al., 2007). FFA1 has attracted considerable attention as a potential therapeutic target for the treatment of diabetes, based predominantly on its expression in pancreatic β-cells and the known role of medium- and longer-chain fatty acids, that are agonists at this GPCR, in modulating glucose-dependent release of insulin (Milligan et al., 2006; Stoddart et al., 2008a). This has resulted in the identification of a number of small molecule agonist and antagonist ligands at this receptor (Briscoe et al., 2006; Garrido et al., 2006; Bharate et al., 2008; Christiansen et al., 2008; Tan et al., 2008; Tikhonova et al., 2008; Humphries et al., 2009). A considerable number of recent reviews have centred on the function and pharmacology of this receptor and whether short-term stimulation or longer-term blockade might result in the most effective therapeutic treatment (Milligan et al., 2006; Brownlie et al., 2008; Costanzi et al., 2008; Hirasawa et al., 2008; Swaminath, 2008; Telvekar and Kundaikar, 2008). Significantly less discussion of the pharmacology and function of the related family members FFA2 and FFA3 is available and these receptors will, therefore, be the focus of the current review.
De-orphanization and modes of signal transduction of FFA2 and FFA3
Fatty acids with chain length of less than six carbons are endogenous agonists for FFA2 and FFA3 (Brown et al., 2003; Le Poul et al., 2003; Nilsson et al., 2003) but there are distinct chain length structure–activity relationships at the two receptors with FFA2 displaying potency C2 = C3 > C4 > C5 = C1 and FFA3 C3 = C4 = C5 > C2 = C1 (Figure 1). Brown et al. (2003) used recombinant cell systems to demonstrate that FFA2 was activated by the carboxylic acid, acetate and related molecules. This was also noted by Le Poul et al. (2003) and Nilsson et al. (2003). Brown et al. (2003) and Le Poul et al. (2003) also demonstrated that short-chain fatty acids were agonists at FFA3.
Figure 1.
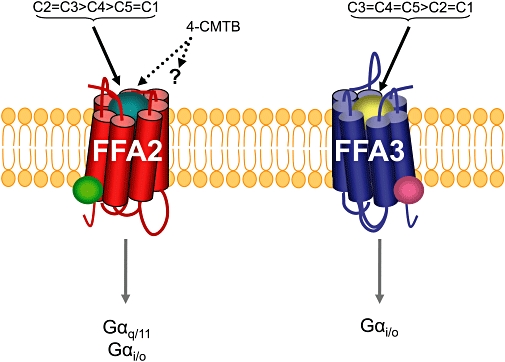
Ligand selectivity and G protein-coupling profiles of FFA2 and FFA3. Cartoons of the receptors FFA2 (red) and FFA3 (blue) are shown. The relationship of chain length (C) of free fatty acids as orthosteric agonists (implied by the teale or gold coloured balls) is shown for each receptor as is the potential for allosteric regulators of each receptor (green and pink coloured circles). chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide (4-CMTB) is the first described selective modulator of FFA2 and it is both a direct agonist and a positive allosteric modulator of the action of short-chain free fatty acids (Lee et al., 2008). The molecular basis of interaction of 4-CMTB with FFA2 and it's selectivity for FFA2 over FFA3 remains to be defined. FFA2/3, free fatty acid receptor 2/3.
Despite responding to the same group of endogenous agonists, FFA2 and FFA3 have distinct G protein-coupling specificities (Figure 1). FFA2 couples to both pertussis toxin-sensitive and -insensitive G proteins (Brown et al., 2003; Le Poul et al., 2003; Nilsson et al., 2003; Stoddart et al., 2008b) while FFA3 couples selectively to pertussis toxin-sensitive Gαi family G proteins (Brown et al., 2003; Le Poul et al., 2003; Stoddart et al., 2008b). However, as with many Gαi-coupled GPCRs (Kostenis et al., 2005), Le Poul et al., (2003) found that co-expression with the promiscuous G protein, Gα16 (Milligan and Kostenis, 2006), allowed activation of FFA3 to be coupled to the release of [Ca2+]i, despite an inability to couple directly to Gαq/11. Based on these observations, Stoddart et al. (2008b) employed a chimeric Gly66AspGαq-i5 variant (Heydorn et al., 2004) to link activation of FFA3 to the elevation of intracellular [Ca2+] and explore the structure–activity relationships of short-chain free fatty acids at this receptor. Furthermore, given the pleiotropic effects of short-chain free fatty acids, Stoddart et al. (2008b) also employed cells able to express either FFA2 or FFA3 in an entirely inducible manner (Figure 2) to ensure that effects of the ligands were actually mediated via the receptor.
Figure 2.
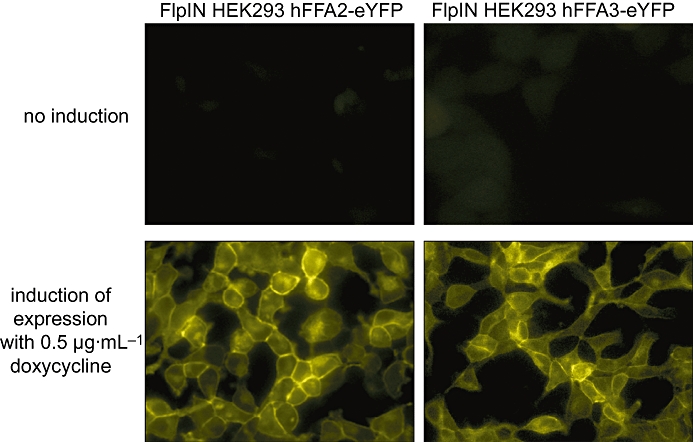
Cell lines able to express FFA2 or FFA3 in an inducible manner. Short-chain fatty acids such as acetate and propionate are pleiotropic in action and many effects in cells are likely to be mediated via non-receptor mechanisms. To define FFA2- and FFA3-specific effects of such ligands, Stoddart et al. (2008b) established Flp-In TREx HEK293 cell lines able to express C-terminally eYFP tagged forms of human FFA2 (left hand panels) or FFA3 (right hand panels) in an antibiotic dependent fashion. In the absence of doxycycline (top panels) the receptors cannot be detected but after treatment with doxycycline (bottom panels) the characteristic pattern of cell surface location is observed. Actions of short-chain fatty acids produced without treatment of the cells with doxycycline were considered unspecific. eYFP, enhanced yellow fluorescent protein; FFA2/3, free fatty acid receptor 2/3.
Identification, tissue distribution and physiological function of FFA2 and FFA3
The genes encoding FFA2 and FFA3 in man were first identified by Sawzdargo et al. (1997) within a tandemly encoded group of intronless genes, located at chromosome 19q13.1. This region also encompasses the genes encoding FFA1 and GPR42. The proteins predicted from these sequences contained seven putative transmembrane domains (TMDs) and a variety of other features common to class A GPCRs. Although having 98% homology with FFA3 (Sawzdargo et al., 1997), GPR42 has been suggested to be a pseudogene in man (Brown et al., 2003). However, very recent studies involving complete gene sequencing and analysis of human genome public databases have suggested that the six amino acid differences between FFA3 and GPR42 are actually polymorphisms rather than gene-specific differences and that a substantial number of individuals are likely to express functional GPR42 (Liaw and Connolly, 2009). FFA2 mRNA can be detected in a variety of tissues but highest expression is found in immune cells such as neutrophils, monocytes, peripheral blood mononuclear cells, B-lymphocytes and polymorphonuclear cells (Brown et al., 2003; Le Poul et al., 2003; Nilsson et al., 2003). Considerable levels of FFA2 mRNA were also detected in bone marrow and spleen but this probably reflects expression of the receptor by immune cells (Le Poul et al., 2003). FFA2 mRNA has also been detected in eosinophils (Nakajima et al., 2004). Nilsson et al. (2003) found expression of the receptor in skeletal muscle and heart, while FFA2 mRNA has also been reported in adipose tissue (Hong et al. et al., 2005; Ge et al., 2008) and rat distal ileum and colon (Karaki et al., 2006). FFA3 has a more widespread expression pattern than FFA2. Initial studies on FFA3 detected high levels of receptor mRNA in a range of human tissues with highest levels of expression in adipose tissue (Brown et al., 2003). Substantial expression was also detected in pancreas, spleen, lymph nodes, bone marrow and peripheral blood mononuclear cells (Brown et al., 2003; Le Poul et al., 2003). The expression of FFA3 in adipose cells has been confirmed (Xiong et al., 2004). Potential roles for FFA2 and FFA3 have recently been reviewed extensively (Stoddart et al., 2008a) and will not be covered here in detail. FFA2 may mediate the known effects of short-chain free fatty acids on leukocyte chemotaxis and movement towards sites of bacterial infection. Furthermore, using an FFA2 knockout mouse model, Ge et al. (2008) found that neither acetate nor propionate was able to inhibit lipolysis in primary adipocytes isolated from the FFA2−/− mouse, suggesting that FFA2 is able to inhibit lipolysis and, therefore, regulate plasma free fatty acid levels. However, based on the known effects of sodium propionate to increase circulating leptin levels in mice (Xiong et al., 2004) and that FFA3 knock-down has been reported to almost completely abolish the ability of propionate to induce leptin reporter expression and consequently leptin production (Xiong et al., 2004), FFA3 may also play an important role in signalling adipocyte mass and lipid stores. Short-chain free fatty acids such as acetate, propionate and butyrate are both a primary source of energy and also regulate development, insulin and glucagon secretion, and other physiological processes in ruminants such as cattle and sheep. It is also of note, therefore, that bovine forms of FFA2 and FFA3 have recently been cloned and shown to respond to these ligands (Wang et al., 2009). As both receptors have similar pharmacological profiles, the availability of selective agonists and antagonists would be invaluable in dissecting the relative contribution of each receptor in the normal physiology of adipocytes and in providing further support for conclusions reached from knockout and knock-down studies.
Structural features of FFA2 and FFA3 and the binding of free fatty acids
Both FFA2 and FFA3 are more closely related to each other than to any other GPCR. Despite this, they exhibit only 43% overall similarity (Stoddart et al., 2008a). As with the vast majority of class A GPCRs, both FFA2 and FFA3 contain cysteine residues at the top of the third TMD and in the second extracellular loops that are predicted to contribute to structure via the formation of intra-molecular disulphide bonds, although this remains to be demonstrated experimentally. They also contain an arginine at the bottom of TMD III within the highly conserved Asp-(Glu)-Arg-Tyr (D(E)RY) domain of class A GPCRs. FFA2 contains a Glu-Arg-Tyr motif in this location, whereas in FFA3 it is a Glu-Arg-Phe motif.
The orthosteric binding pocket
Both FFA2 and FFA3 respond to short-chain free fatty acids with carbon chain length C1 (formate) to C5 (valerate) (Brown et al., 2003; Stoddart et al., 2008b). However, the potency of these ligands is low, within the high µM to low mM range. Because of the lack of high affinity ligands that interact with FFA2 and/or FFA3, identification of residues that contribute to the binding of orthosteric ligands in these two GPCRs has so far been inferred simply from loss of function or impairment of signalling of point mutants of these receptors (Stoddart et al., 2008b) or from modelling studies (Lee et al., 2008). In initial studies designed to explore the contributions of the six amino acids predicted to differ between human FFA3 and the non-functional GPR42 sequence, Brown et al. (2003) systematically inter-converted each variant residue. Alteration of Arg174 of FFA3, that is predicted to be located in extracellular loop 2, to the corresponding Trp residue of GPR42 generated an FFA3 mutant that was no longer able to respond to the C3 fatty acid propionate. Conversely, introduction of Arg174 into GPR42 rendered the previously unresponsive protein able to generate signals to propionate (Brown et al., 2003). Alteration of the residue equivalent to Leu227 (residue 6.37 in the nomenclature of Ballesteros and Weinstein, 1995) of human FFA2 to Val at the bottom of TMD VI in the rat orthologue of FFA3 resulted in constitutive signalling (Brown et al., 2003). This region is often implicated in GPCR activation of G proteins.
Based on both the lack of function of fatty acid amides at each of FFA1-3 (Briscoe et al., 2003; Brown et al., 2003) and the loss of function of longer-chain fatty acids at forms of FFA1 in which the Arg residues at 5.39 and 7.35 were mutated (Sum et al., 2007), Stoddart et al., (2008b) aligned human FFA1, FFA2 and FFA3 and noted the conservation of these two residues. Mutation of Arg 5.39 in FFA2 to a range of amino acids, including Lys, resulted in loss of capacity of the C2 and C3 fatty acids acetate and propionate to cause elevation of intracellular Ca2+ in HEK293 cells transfected to express these variants transiently (Stoddart et al., 2008b). Because it was unclear in such studies if the mutated forms of FFA2 were able to reach the cell surface efficiently, Stoddart et al. (2008b) established stable HEK293 cell lines able to express C-terminally enhanced yellow fluorescent protein (eYFP)-tagged forms of either wild-type FFA2 or Arg 5.39 Ala FFA2, only upon addition of the antibiotic doxycycline (Figure 2). Imaging of such induced cells demonstrated similar sub-cellular distribution of the two forms and confirmed that Arg 5.39 Ala FFA2 was unable to respond to either acetate or propionate (Stoddart et al., 2008b). These cells had the additional benefit of providing proof that induction of expression of FFA2 was required for the short-chain free fatty acids to elevate intracellular Ca2+ and that this was not a non-specific effect of the ligands (Stoddart et al., 2008b). Construction of HEK293 cells able to express Arg 7.35 Ala FFA2-eYFP in an inducible fashion also demonstrated the importance of this residue for the function of acetate and propionate (Stoddart et al., 2008b). As noted earlier, FFA3 couples predominantly to pertussis toxin-sensitive G proteins of the Gi/Go subfamily (Brown et al., 2003; Le Poul et al., 2003). Generation and inducible expression of Arg 5.39 Ala FFA3-eYFP and Arg 7.35 Ala FFA3-eYFP also demonstrated the importance of these residues in FFA3 as propionate was unable to increase binding of [35S]GTPγS in membranes from such cells whereas a robust, concentration-dependent increase was produced following induction of wild-type FFA3-eYFP (Stoddart et al., 2008b). Molecular modelling, based on the structure of bovine rhodopsin, indicates the likely proximity of Arg 5.39 and Arg 7.35, as well as His 6.55, in both FFA2 and FFA3 and their importance in binding and function of the short-chain free fatty acids (Figure 3).
Figure 3.
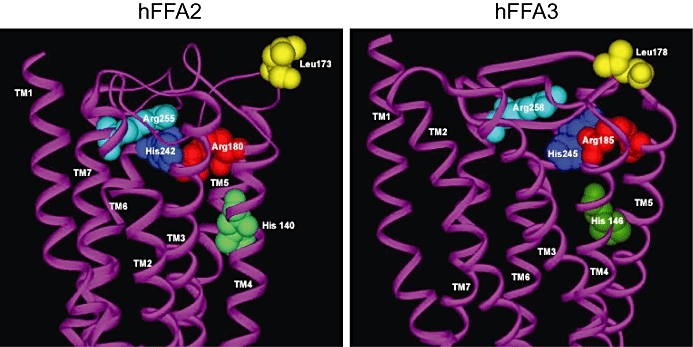
The orthosteric binding pocket of FFA2 and FFA3. Molecular models of human FFA2 (left) and FFA3 (right) were constructed as in Stoddart et al. (2008b) with the predicted carbon chain backbone of the TMDs shown in purple. Amino acids conserved between FFA2 and FFA3 (Arg 5.39, red, His 6.55, blue, Arg 7.35, cyan) that are important contributors to the binding and function of short-chain free fatty acids are shown filling the anticipated space. His 4.56 (green) plays a modulatory role that depends on the fatty acid chain length and the activity measured. Each amino acid is numbered relative to the start codon. Leu173 (yellow) in FFA2 is located in the predicted the second extracellular loop and has been suggested to be important in binding ago-allosteric phenylacetamides (Lee et al., 2008) that are selective for FFA2 over FFA3. The corresponding Leu178 (yellow) in FFA3 is also shown. FFA2/3, free fatty acid receptor 2/3; TMD, transmembrane domain.
As with many GPCRs (Crespo and Gutkind, 2004), both FFA2 and FFA3 are able to cause phosphorylation and activation of the ERK1/2 MAP kinases (Stoddart et al., 2008b). Phosphorylation of these kinases in response to both acetate and propionate in HEK293 cells induced to express variants of FFA3 was again lacking for Arg 5.39 Ala FFA3-eYFP and Arg 7.35 Ala FFA3-eYFP and, in agreement with the concept that FFA3 couples selectively to Gi/Go G proteins, this effect was abolished by pretreatment of the cells with Pertussis toxin (Stoddart et al., 2008b), which blocks GPCR-mediated activation of such G proteins by catalysing ADP-ribosylation of the cysteine residue located four amino acids from the C-terminus of the α subunit of members of this group (Milligan, 1988). By contrast, FFA2-mediated phosphorylation of the ERK1/2 MAP kinases was shown to be transduced by a combination of Gi/Go and Gq/G11 G proteins, as pretreatment with a mixture of Pertussis toxin and the selective Gq/G11 inhibitor YM-254890 (Takasaki et al., 2004) was required to ablate agonist function. However, as in Ca2+ elevation assays, both Arg 5.39 Ala FFA2 and Arg 7.35 Ala FFA2 failed to induce phosphorylation of the ERK1/2 MAP kinases in response to either acetate or propionate (Stoddart et al., 2008b). It seems reasonable, therefore, to indicate that the carboxylate of short-chain fatty acids interacts with these receptors in a similar manner to that of the longer-chain free fatty acids with FFA1 (Sum et al., 2007; Smith et al., 2009a).
His (4.56) is conserved between FFA1-3 and has been shown to contribute to binding/function in FFA1 (Sum et al., 2007; Smith et al., 2009a). Furthermore, although not conserved in FFA2/3, the polarity of Asn 6.55 in FFA1 is retained in FFA2 and FFA3 as both have a His at this position. Alteration of His 6.55 to Ala in FFA2 essentially eliminated the ability of short-chain free fatty acids to elevate [Ca2+]i and greatly reduced the ability of the ligands to promote phosphorylation of the ERK1/2 MAP kinases (Stoddart et al., 2008b). For FFA3, a His 6.55 Ala mutant was unable to respond to short-chain free fatty acids in any end point assessed, including stimulation of [35S]GTPγS binding, ERK1/2 MAP kinase phosphorylation or when Ca2+ signals were evoked by the co-expression of a chimeric Gq-Gi G protein (Stoddart et al., 2008b). Effects of mutation of His 4.56 to Ala were less dramatic. For FFA2, this resulted in an approximately 10-fold reduction in the potency of both acetate and propionate in measures of [Ca2+]i. Interestingly, for FFA3 the effect of the His 4.56 Ala mutant was end point-dependent. In measures of Gi-activation recorded in [35S]GTPγS binding studies, propionate was entirely unable to generate a response. However, although an artificial assay, when co-expressed with a chimeric Gq-Gi G protein, propionate was able to cause elevation of [Ca2+]i in single cell imaging studies via His 4.56 Ala FFA3, although only a single concentration of the agonist was tested and thus no potency information was provided (Stoddart et al., 2008b). These studies indicate that a similar orientation of the carboxylate group of the endogenous ligands of FFA2 and FFA3 as suggested for FFA1 (Sum et al., 2007) is likely with salt bridge interactions between the carboxylate anion of the ligand and guanidinium cation groups of Arg 5.39 and Arg 7.35 providing co-ordination (Figure 3). Indeed, such an interaction has been modelled by Lee et al. (2008). This is in agreement with other studies in which methyl substitutes of long-chain free fatty acids have reduced or lack activity at FFA1 (Itoh et al., 2003; Salehi et al., 2005) and the lack of activity of acetamide and propionamide at FFA2 and FFA3 (Stoddart et al., 2008b). How chain length selectivity between FFA2 and FFA3 and, in a more general sense, between FFA2/3 and FFA1 is defined remains to be explored. In modelling studies, Lee et al. (2008) suggest that the lipophilic chain of propionate is in a gauche conformation and oriented towards the Ser residue at position 6.54. However, no experimental studies have been reported in which this amino acid has been modified and, as it is conserved between FFA2 and FFA3, this presumably cannot underlie the difference in structure–activity relationships for short-chain free fatty acids at the two receptors. Interestingly, in His 4.56 Ala FFA2, a gain of function to the 6 carbon free fatty acid caproate was observed in both [Ca2+]i signalling and ERK1/2 MAP kinase assays whereas this was not observed for His 4.56 Ala FFA3 (Stoddart et al., 2008b). Given that free fatty acids with chain length >3 are less potent at FFA2 while potency at FFA3 is essentially the same for chain length 3–5, this observation may also provide insight into differences in the orthosteric binding pocket of FFA2 and FFA3 (Figure 3). However, further work remains to be performed. Furthermore, although both GPR84 (Wang et al., 2006b) and GPR120 (Hirasawa et al., 2005) are reported as receptors responsive to free fatty acids, they have no obvious homology with the FFA1-3 group and, therefore, presumably must bind ligands in a different manner. No direct studies have explored this issue but following identification of GPR120 selective agonists containing a carboxylate moiety, Suzuki et al. (2008) have suggested Arg 99 at the top of TMDII as a potential residue that might co-ordinate the acid. There are currently few small molecule ligands reported with selectivity between FFA1 and GPR120 (Briscoe et al., 2006; Suzuki et al., 2008) and thiazolidinedione peroxisome proliferator-activated receptor γ agonists such as ciglitazone and troglitazone activate both receptors, despite the limited overall sequence similarly of these two receptors.
An allosteric binding site in FFA2
A major limitation in efforts to study the biology and function of FFA2 and FFA3 is that, to date, there are no reported synthetic, orthosteric small molecule ligands. Recently, however, Lee et al. (2008) reported a pair of phenylacetamides as selective ‘allosteric agonists’ for FFA2. In a range of assays, including inhibition of cAMP levels and elevation of [35S]GTPγS binding, these ligands acted as direct agonists at FFA2, with between 100- and 1000-fold higher potency than acetate or propionate. They were also able to inhibit lipolysis in differentiated 3T3L1 cells (Lee et al., 2008), a standard, well-characterized cell-based adipocyte model. Furthermore, the phenylacetamides also acted as positive allosteric modulators of the function of short-chain free fatty acids, able to enhance the functional potency of acetate and propionate, although the estimated allosteric co-operativity factor was modest (Lee et al., 2008). However, such observations are consistent with the phenylacetamides binding to a distinct allosteric site (Langmead and Christopoulos, 2006) or potentially acting as bitopic or dualsteric ligands, molecules that have the potential to bind to multiple distinct sites on a single protein (Valant et al., 2008; Antony et al., 2009). Although no mutagenesis studies were performed, modelling studies were employed to suggest the location of this allosteric site. The models predict a contribution from Leu173, located in the second extracellular loop (Figure 3), and in differing models, contributions from Arg 7.35 of the orthosteric binding site that, as noted earlier, is central to the binding/function of the short-chain fatty acids. A series of hydrophobic residues located in TMDs II, III, VI and VII were also suggested to contribute to the binding of the p-chlorophenyl element of the ligands. Recently, Smith et al. (unpublished observations) have begun to explore this issue experimentally, taking advantage of the orthosteric binding pocket mutants studied by Stoddart et al. (2008b). Although able to confirm both the direct agonist and positive allosteric effects of chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide, as well as demonstrating the selectivity of this ligand for FFA2 over FFA3 and FFA1, Smith et al. (unpublished observations) were unable to provide evidence to support a key role for Arg 7.35 in the recognition/function of this ligand because chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide was at least as potent and efficacious in causing inhibition of cAMP production at Arg 7.35 Ala FFA2 as at the wild-type receptor. The second extracellular loop differs in length and is relatively diverse between FFA2 and FFA3 and such loops present a substantial challenge to model. However, at least based on a simple alignment, Leu173 in FFA2 is conserved as Leu178 in FFA3 (Figure 3), although in each receptor charged amino acids located close to this residue are not well conserved. As Sum et al. (2009) have recently provided evidence that negatively charged amino acids in extracellular loop 2 play an important role in generating an inactive state of FFA1 by co-ordinating Arg 5.39 and Arg 7.35, it may be that similar interactions contribute to the ground state of FFA2 and FFA3, but this remains to be tested. Selective ligands for FFA3, whether orthosteric or ago-allosteric, would be of great value to understanding the biology and function of this receptor.
Conclusions
Although there is a distinct structure–activity relationship for activation of FFA2 and FFA3 by fatty acids of carbon chain length 2–5, there is insufficient selectivity to use these ligands to define clearly the function these two receptors, particularly in tissues in which they are co-expressed. Until recently, insights have been restricted to studies employing siRNA-mediated knockdown of expression and/or the generation of knockout lines of mice. Despite these approaches, the specific functions of FFA2 and FFA3 remain uncertain and pharmacological tools able to activate or block each receptor selectively are required. The recent identification of a pair of phenylacetamides as selective ligands at FFA2 that act as both second-site agonists and positive allosteric modulators of the endogenous free fatty acids at this receptor is a helpful first step, but much remains to be achieved.
Acknowledgments
The Biotechnology and Biosciences Research Council grant BB/E019455/1 supports a small section of the work described herein. L.A.S. was funded by a BBSRC CASE studentship and N.J.S. is an NHMRC/NHF of Australia C.J. Martin Overseas Fellow.
Glossary
Abbreviations:
- eYFP
enhanced yellow fluorescent protein
- FFA2/3
free fatty acid receptor 2/3
- GPCR
G protein-coupled receptor
- TMD
transmembrane domain
Conflict of interest
The authors state no conflict of interest.
References
- Antony J, Kellershohn K, Mohr-Andrä M, Kebig A, Prilla S, Muth M, et al. Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 2009;23:442–450. doi: 10.1096/fj.08-114751. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G Protein Coupled Receptors. Methods Neurosci. 1995:366–428. [Google Scholar]
- Bharate SB, Rodge A, Joshi RK, Kaur J, Srinivasan S, Kumar SS, et al. Discovery of diacylphloroglucinols as a new class of GPR40 (FFAR1) agonists. Bioorg Med Chem Lett. 2008;18:6357–6361. doi: 10.1016/j.bmcl.2008.10.085. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11329. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- Brownlie R, Mayers RM, Pierce JA, Marley AE, Smith DM. The long-chain fatty acid receptor, GPR40, and glucolipotoxicity: investigations using GPR40-knockout mice. Biochem Soc Trans. 2008;36:950–954. doi: 10.1042/BST0360950. [DOI] [PubMed] [Google Scholar]
- Cai TQ, Ren N, Jin L, Cheng K, Kash S, Chen R, et al. Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem Biophys Res Commun. 2008;377:987–991. doi: 10.1016/j.bbrc.2008.10.088. [DOI] [PubMed] [Google Scholar]
- Christiansen E, Urban C, Merten N, Liebscher K, Karlsen KK, Hamacher A, et al. Discovery of potent and selective agonists for the free fatty acid receptor 1 (FFA(1)/GPR40), a potential target for the treatment of type II diabetes. J Med Chem. 2008;51:7061–7064. doi: 10.1021/jm8010178. [DOI] [PubMed] [Google Scholar]
- Costanzi S, Neumann S, Gershengorn MC. Seven transmembrane- spanning receptors for Free Fatty Acids as therapeutic targets for Diabetes Mellitus: pharmacological, phylogenetic, and drug discovery aspects. J Biol Chem. 2008;283:16269–16273. doi: 10.1074/jbc.R800014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo P, Gutkind JS. Activation of MAPKs by G protein-coupled receptors. Methods Mol Biol. 2004;250:203–210. doi: 10.1385/1-59259-671-1:203. [DOI] [PubMed] [Google Scholar]
- Garrido DM, Corbett DF, Dwornik KA, Goetz AS, Littleton TR, McKeown SC, et al. Synthesis and activity of small molecule GPR40 agonists. Bioorg Med Chem Lett. 2006;16:1840–1845. doi: 10.1016/j.bmcl.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen JL, et al. Activation of GPR43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology. 2008;149:4519–4526. doi: 10.1210/en.2008-0059. [DOI] [PubMed] [Google Scholar]
- He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- Heydorn A, Ward RJ, Jorgensen R, Rosenkilde MM, Frimurer TM, Milligan G, et al. Identification of a novel site within G protein α subunits important for specificity of receptor-G protein interaction. Mol Pharmacol. 2004;66:250–259. doi: 10.1124/mol.66.2.250. [DOI] [PubMed] [Google Scholar]
- Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- Hirasawa A, Hara T, Katsuma S, Adachi T, Tsujimoto G. Free fatty acid receptors and drug discovery. Biol Pharm Bull. 2008;31:1847–1851. doi: 10.1248/bpb.31.1847. [DOI] [PubMed] [Google Scholar]
- Hong YH, Nishimura Y, Hishikawa D, Tsuzuki H, Miyahara H, Gotoh C, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146:5092–5099. doi: 10.1210/en.2005-0545. [DOI] [PubMed] [Google Scholar]
- Humphries PS, Benbow JW, Bonin PD, Boyer D, Doran SD, Frisbie RK, et al. Synthesis and SAR of 1,2,3,4-tetrahydroisoquinolin-1-ones as novel G-protein-coupled receptor 40 (GPR40) antagonists. Bioorg Med Chem Lett. 2009;19:2400–2403. doi: 10.1016/j.bmcl.2009.03.082. [DOI] [PubMed] [Google Scholar]
- Irukayama-Tomobe Y, Tanaka H, Yokomizo T, Hashidate-Yoshida T, Yanagisawa M, Sakurai T. Aromatic D-amino acids act as chemoattractant factors for human leukocytes through a G protein-coupled receptor, GPR109B. Proc Natl Acad Sci USA. 2009;106:3930–3934. doi: 10.1073/pnas.0811844106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Karaki S, Mitsui R, Hayashi H, Kato I, Sugiya H, Iwanaga T, et al. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res. 2006;324:353–360. doi: 10.1007/s00441-005-0140-x. [DOI] [PubMed] [Google Scholar]
- Kostenis E, Waelbroeck M, Milligan G. Promiscuous Gα proteins in basic research and drug discovery. Trends Pharmacol Sci. 2005;26:595–602. doi: 10.1016/j.tips.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun. 2003;301:406–410. doi: 10.1016/s0006-291x(02)03064-4. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. Allosteric agonists of 7TM receptors: expanding the pharmacological toolbox. Trends Pharmacol Sci. 2006;27:475–481. doi: 10.1016/j.tips.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Liaw CW, Connolly DT. Sequence polymorphisms provide a common consensus sequence for GPR41 and GPR42. DNA Cell Biol. 2009 doi: 10.1089/dna.2009.0916. in press. [DOI] [PubMed] [Google Scholar]
- Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278:25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, et al. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol. 2008;74:1599–1609. doi: 10.1124/mol.108.049536. [DOI] [PubMed] [Google Scholar]
- Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem. 2009;284:2811–2822. doi: 10.1074/jbc.M806409200. [DOI] [PubMed] [Google Scholar]
- Milligan G. Techniques used in the identification and analysis of function of pertussis toxin-sensitive guanine nucleotide binding proteins. Biochem J. 1988;255:1–13. doi: 10.1042/bj2550001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Kostenis E. Hetero-trimeric G proteins: a short history. Br J Pharmacol. 2006;147:S46–S55. doi: 10.1038/sj.bjp.0706405. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Stoddart LA, Brown AJ. G protein-coupled receptors for free fatty acids. Cell Signal. 2006;18:1360–1365. doi: 10.1016/j.cellsig.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Iikura M, Okayama Y, Matsumoto K, Uchiyama C, Shirakawa T, et al. Identification of granulocyte subtype-selective receptors and ion channels by using a high-density oligonucleotide probe array. J Allergy Clin Immunol. 2004;113:528–535. doi: 10.1016/j.jaci.2003.12.036. [DOI] [PubMed] [Google Scholar]
- Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a Free Fatty Acid Receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun. 2003;303:1047–1052. doi: 10.1016/s0006-291x(03)00488-1. [DOI] [PubMed] [Google Scholar]
- Salehi A, Flodgren E, Nilsson NE, Jimenez-Feltstrom J, Miyazaki J, Owman C, et al. Free Fatty Acid Receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res. 2005;322:207–215. doi: 10.1007/s00441-005-0017-z. [DOI] [PubMed] [Google Scholar]
- Sawzdargo M, George SR, Nguyen T, Xu S, Kolakowski LF, O'Dowd BF. A cluster of four novel human G Protein-Coupled Receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem Biophys Res Commun. 1997;239:543–547. doi: 10.1006/bbrc.1997.7513. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J Biol Chem. 2009a;284:17527–17539. doi: 10.1074/jbc.M109.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2 and -3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008a;60:405–417. doi: 10.1124/pr.108.00802. [DOI] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Jenkins L, Brown AJ, Milligan G. Conserved polar residues in transmembrane domains V, VI and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J Biol Chem. 2008b;283:32913–32924. doi: 10.1074/jbc.M805601200. [DOI] [PubMed] [Google Scholar]
- Sum CS, Tikhonova IG, Neumann S, Engel S, Raaka BM, Costanzi S, et al. Identification of residues important for agonist recognition and activation in GPR40. J Biol Chem. 2007;282:29248–29255. doi: 10.1074/jbc.M705077200. [DOI] [PubMed] [Google Scholar]
- Sum CS, Tikhonova IG, Costanzi S, Gershengorn MC. Two arginine-glutamate ionic locks near the extracellular surface of FFAR1 gate receptor activation. J Biol Chem. 2009;284:3529–3536. doi: 10.1074/jbc.M806987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surgand JS, Rodrigo J, Kellenberger E, Rognan D. A chemogenomic analysis of the transmembrane binding cavity of human G-Protein-Coupled Receptors. Proteins. 2006;62:509–538. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Igari S, Hirasawa A, Hata M, Ishiguro M, Fujieda H, et al. Identification of G protein-coupled receptor 120-selective agonists derived from PPARgamma agonists. J Med Chem. 2008;51:7640–7644. doi: 10.1021/jm800970b. [DOI] [PubMed] [Google Scholar]
- Swaminath G. Fatty acid binding receptors and their physiological role in type 2 diabetes. Arch Pharm (Weinheim) 2008;341:753–761. doi: 10.1002/ardp.200800096. [DOI] [PubMed] [Google Scholar]
- Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. A novel Galphaq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- Tan CP, Feng Y, Zhou YP, Eiermann GJ, Petrov A, Zhou C, et al. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes. 2008;57:2211–2219. doi: 10.2337/db08-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telvekar VN, Kundaikar HS. GPR40 carboxylic acid receptor family and diabetes: a new drug target. Curr Drug Targets. 2008;9:899–910. doi: 10.2174/138945008785909301. [DOI] [PubMed] [Google Scholar]
- Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A is a G-protein-Coupled Receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69:2826–3282. doi: 10.1158/0008-5472.CAN-08-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova IG, Sum CS, Neumann S, Thomas CJ, Raaka BM, Costanzi S, et al. Bidirectional, iterative approach to the structural delineation of the functional ‘chemoprint’ in GPR40 for agonist recognition. J Med Chem. 2007;50:2981–2989. doi: 10.1021/jm0614782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, et al. Discovery of novel agonists and antagonists of the Free Fatty Acid Receptor 1 (FFAR1) using virtual screening. J Med Chem. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, et al. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J Biol Chem. 2008;283:29312–29321. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Gu Z, Heid B, Akers RM, Jiang H. Identification and characterization of the bovine G protein-coupled receptor GPR41 and GPR43 genes. J Dairy Sci. 2009;92:2696–2705. doi: 10.3168/jds.2009-2037. [DOI] [PubMed] [Google Scholar]
- Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem. 2006a;281:22021–22028. doi: 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- Wang J, Wu X, Simonavicius N, Tian H, Ling L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J Biol Chem. 2006b;281:34457–34464. doi: 10.1074/jbc.M608019200. [DOI] [PubMed] [Google Scholar]
- Wigglesworth MJ, Wolfe LA, Wise A. Orphan seven transmembrane receptor screening. Ernst Schering Found Symp Proc. 2006;2:105–143. doi: 10.1007/2789_2006_006. [DOI] [PubMed] [Google Scholar]
- Wise A, Jupe SC, Rees S. The identification of ligands at orphan G-protein coupled receptors. Annu Rev Pharmacol Toxicol. 2004;44:43–66. doi: 10.1146/annurev.pharmtox.44.101802.121419. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Miyamoto N, Shibata K, Valasek MA, Motoike T, Kedzierski RM, et al. Short-chain fatty acids stimulate leptin production in adipocytes through the G Protein-Coupled Receptor GPR41. Proc Natl Acad Sci USA. 2004;101:1045–1050. doi: 10.1073/pnas.2637002100. [DOI] [PMC free article] [PubMed] [Google Scholar]