

Abstract
Background:
β2-adrenoceptor agonists are effective bronchodilators. In vitro studies demonstrated long-lasting airway smooth muscle relaxation by salmeterol after washout, the quick disappearance of this effect in presence of antagonists and its recovery after antagonist removal. Current explanations invoke salmeterol accumulation in the membrane (‘diffusion microkinetic’ model) or the existence of salmeterol-binding ‘exosites’. An alternative model based on ‘rebinding’ of a dissociated ligand to the receptor molecules also produces an apparent decrease in the ligand's dissociation rate in the absence of competing ligands.
Purpose and approach:
Computer-assisted simulations were performed to follow the receptor-occupation by a salmeterol-like ligand and a competing ligand as a function of time. The aptness of the models to describe the above in vitro findings was evaluated.
Key results:
The ‘diffusion microkinetic’ model is sufficient to explain a long-lasting β2-adrenoceptor stimulation and reassertion as long as the membrane harbors a high concentration of the agonist. At lower concentration, ‘rebinding’ and, in second place, ‘exosite’ binding are likely to become operational.
Conclusions and implications:
The ‘rebinding’ and ‘exosite’ binding mechanisms take place at a sub-cellular/molecular scale. Pending their demonstration by experiments on appropriate, simple models such as intact cells or membranes thereof, these mechanisms remain hypothetical in the case of salmeterol. Airway smooth muscle contraction could also be governed by additional mechanisms that are particular to this macroscopic approach.
Keywords: salmeterol, β2-adrenoceptor, bronchodilation, reassertion, diffusion microkinetic model, exosites, rebinding, simulations
Introduction
β2-adrenoceptor agonists are widely used as bronchodilators. While the first generation of these drugs were only short acting and unable to provide extended protection from bronchoconstriction in patients with nocturnal asthma (Anderson, 1993), ensuing ones like salmeterol and formoterol caused significant bronchodilation for at least 12 h after a single administration (Maesen et al., 1990; Beach et al., 1993; Palmqvist et al., 1997).
While in vitro studies on isolated airway smooth muscle preparations such as guinea-pig trachea and human bronchi confirmed the aptness of these agonists to produce long-lasting relaxation, these studies also shed light on some remarkable differences between their kinetic properties (Jeppsson et al., 1989; Ball et al., 1991; Johnson et al., 1993; Nials et al., 1993a; 1994; Anderson et al., 1994). When the tissues were preincubated with these agonists and subsequently superfused with fresh medium to remove the free drug, the action of formoterol in the washout medium exhibited a concentration-dependent duration; that is, from relatively short for low initial concentrations to sustained for supramaximally effective concentrations. By contrast, the action of salmeterol was pseudo-irreversible (for up to 12 h under washout) regardless of its initial concentration (Johnson et al., 1993; Nials et al., 1994). Yet, the relaxating effect of these agonists could be rapidly reversed by washing of the tissues with high concentrations of sotalol and other antagonists provided that they display sufficiently high affinity for β2-adrenoceptors. Similar behavior of formoterol and salmeterol has also been observed when monitoring their ability to promote cAMP accumulation in isolated cell systems (McCrea and Hill, 1993; 1996; Clark et al., 1996; Green et al., 1996; Rong et al., 1999). This behaviour is difficult to reconcile with the simple traditional view that agonist- and antagonist- β2-adrenoceptor interactions are competitive bimolecular processes obeying the law of mass-action and that, when free, these ligands only reside in the extracellular aqueous space from where they have to move to and from the receptor. Indeed, while this view does not preclude long-lasting binding of agonists, it cannot explain why the termination of the agonist response is accelerated in the presence of antagonists. Even more surprising was that airway smooth muscle relaxation reappeared when the antagonist was washed away even though no agonist was added to the washout medium (Ball et al., 1991; Lindén et al., 1991; Voss, 1994). This latter phenomenon has been denoted as ‘reassertion’ of relaxation and it is quite peculiar to lipophilic β2-adrenoceptor agonists. Although this reassertion declines rapidly upon repeating the antagonist washout cycle in the case of formoterol, it is very persistent in the case of salmeterol as it was still perceived after 10 cycles (Anderson et al., 1994). Yet, the results from a subsequent study suggested that the persistent reassertion of the salmeterol-mediated relaxation requires its initial concentration to be high enough (Bergendal et al., 1996).
Formoterol and salmeterol possess adequate lipophilic properties to permit their incorporation in the plasma membrane (Rhodes et al., 1992; Bergendal et al., 1996). This phenomenon is reversible, resulting in constant partitioning of the drugs between the membrane and the surrounding aqueous phase. This implies that the plasma membrane can act as a depot/reservoir for the ligand rather than merely functioning as an inert substratum for the receptor. The diffusion ‘microkinetic’ model (Anderson, 1993; Anderson et al., 1994) was essentially based on this consideration and it is generally regarded to offer an adequate explanation for the (above-mentioned) in vitro properties of formoterol. According to this model, the membrane acts as a reservoir for formoterol from where it progressively leaches out into the aqueous medium to interact with the active site of the β2-adrenoceptor (Johnson and Coleman, 1995; Johnson, 2001). As the concentration of formoterol determines the initial ‘size’ of the depot, it will also determine the time lag during which a sufficient amount of this agonist can be released in the medium to cause effective airway smooth muscle dilation in in vitro washout experiments. By competing with this released formoterol, antagonists can rapidly terminate this response provided that already bound formoterol dissociates sufficiently swiftly from the receptor. After removal of the antagonist, constantly released formoterol can stimulate the β2-adrenoceptor again so that relaxation is reasserted. The microkinetic model has also been invoked to explain the in vitro properties of salmeterol, albeit with an important modification. Because of the very high partitioning of salmeterol in synthetic plasma membranes as well as its slow release from such membranes (t1/2 of 25 min) (Rhodes et al., 1992; Bergendal et al., 1996), this agonist was proposed to reach the receptor by lateral diffusion through the membrane instead of having to move into the aqueous phase first and even to gain (and leave) the central core of the receptor by lateral diffusion between the receptor's membrane-spanning alpha helices (Anderson, 1993; Johnson et al., 1993; Anderson et al., 1994; Coleman et al., 1996; Teschemacher and Lemoine, 1999). From the structural point of view, salmeterol is about 25 Å long and it was shown to preferentially accumulate in the outer monolayer of synthetic membranes wherein it assumes a highly specific orientation analogous to the phospholipids themselves (Rhodes et al., 1992; Johnson et al., 1993). In support of the microkinetic model, subsequent experiments revealed an even slower release of salmeterol from tracheal strips (t1/2 of 3 h) (Austin et al., 2003).
Yet, some of the in vitro observations with salmeterol were hard to explain by the microkinetic model alone. Among them, the concentration-independent duration of its relaxing effect on airway smooth muscle preparations (Johnson et al., 1993) as well as the apparent persistence of the salmeterol-receptor complexes seen in the early radioligand binding studies (Jack, 1991; Nials et al., 1993b) were considered to be at odds with the ability of the membrane-associated drug to re-equilibrate with the aqueous phase (Anderson et al., 1994; Naline et al., 1994). Moreover, salmeterol has also been reported to exert β2-adrenoceptor-independent in vitro effects at high concentrations. It was argued that, if all the actions of salmeterol resulted from a microkinetic mechanism, they should be of equally long duration (Coleman et al., 1996). Instead, these independent in vitro effects were found to be reversible on washing and, in contrast to the long-lasting β2-adrenoceptor-mediated smooth muscle relaxation, they displayed closely the same half-lives as the release of salmeterol from synthetic membranes (Swales and Paterson, 1990; Ball et al., 1991; Rhodes et al., 1992; Coleman et al., 1996; McCrea and Hill, 1996). Coleman et al. (1996) therefore concluded that there are two processes that contribute to the duration of action of salmeterol; with a minor role for the microkinetic mechanism and a more dramatic role for a process like ‘exosite’ binding that keeps the agonist in the vicinity of the active site of the β2-adrenoceptor.
The exosite model relies heavily on the structural characteristics of salmeterol and this is probably why it constitutes a favourite interpretation of the pharmacological properties of this molecule. Besides a saligenin head that is responsible for β2-adrenoceptor activation, salmeterol also possesses an extended lipophilic phenylalkoxyalkyl side chain (Figure 1– top). This is quite unlike most other β-adrenoceptor agonists. Structure-activity relationship studies also stressed that the position of the oxygen atom in the side chain does not influence the lipophilicity of this agonist but that it is critical for its long duration of action (Johnson and Coleman, 1995; Johnson, 2001). These findings were considered to provide firm support for a model according to which phenylalkoxyalkyl side chain of salmeterol is able to undergo a highly stable association with an accessory site (the ‘exosite’) located either in the immediate vicinity of the β2-adrenoceptor or even within the receptor molecule itself. Whatever the exact location, the exosite's ability to keep the phenylalkoxyalkyl side chain in place permits the active saligenin head of salmeterol to freely enter and leave the active site of the receptor. In this respect, it has been evoked that, while the position of the oxygen atom in the alkyloxalkyl side chain does not affect the average depth of penetration of the whole salmeterol molecule in the membrane, it could dictate the average depth of the ‘hinge’ and thereby the efficiency of the docking process (Chester et al., 1987; Mason et al., 1991; Herbette, 1994; Johnson, 2006). Departure of the saligenin head will temporarily free the active site, thereby allowing a competing antagonist molecule to sneak in and terminate the relaxing effect of salmeterol. Upon withdrawal of the antagonist, the saligenin head of salmeterol will flip back in the active site from its tethered position so that relaxation resumes (Johnson et al., 1993; Johnson, 2001).
Figure 1.
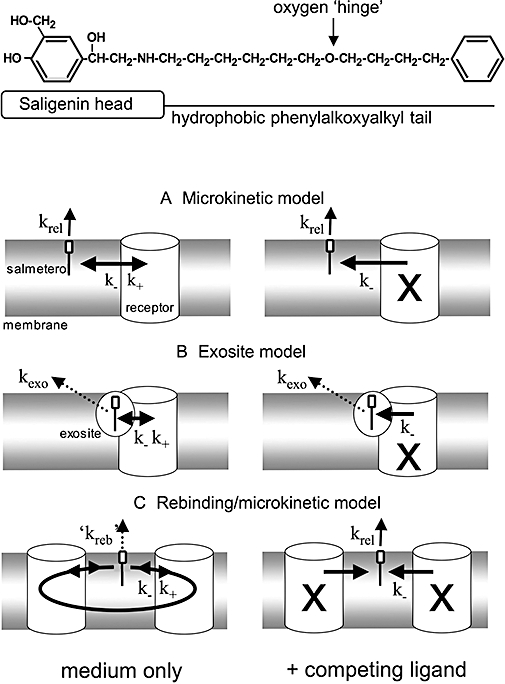
Chemical structure of salmeterol (top) and potential mechanisms for explaining the time-dependencies of its in vitro effects. The saligenin head of salmeterol will bind to the ‘active site’ of the β2-adrenoceptor while the hydrophobic phenylakoxyalkyl tail is important for membrane incorporation and for binding to a potential ‘exosite’. In all models, only membrane- or exosite-associated salmeterol molecules are able to undergo reversible bimolecular interaction with the active site of the receptor (with k+ and k- as association and dissociation rate constants, respectively). Association is prevented when the receptor active sites are occupied by a competing ligand such as a β2-adrenoceptor antagonist (occupied receptors denoted with an X). The following models were tested for their ability to explain the time-related β2-adrenoceptor stimulation by salmeterol under washout conditions either in medium only (left panels) or in medium containing an excess of competing ligand (right panels). (A) Microkinetic model: The membrane acts as a ‘sink’ from where salmeterol is continuously released with the first-order rate constant krel. (B) Exosite model: The side chain of salmeterol binds to an accessory ‘exosite’ located close to or within the β2-adrenoceptor. This allows the saligenin head of salmeterol to continuously sneak in- and out of the active site of the receptor. The membrane still acts as a ‘sink’ but, as the release of salmeterol from the membrane is much faster than its dissociation from the exosite (with first-order rate constant kexo), only the second process was taken into account to calculate the time-wise decline in the amount of salmeterol that can produce receptor stimulation. (C) Rebinding model: This model takes account of the capability of dissociated salmeterol to reassociate to the same or other receptor molecules. The constant shuffling of the involved salmeterol molecules between nearby receptors delays their escape from the membrane. Hence, krel from the microkinetic model is replaced by the much smaller ‘kreb’. Rebinding is prevented in the presence of high concentrations of competing ligand. Under this condition, dissociated salmeterol will escape from the membrane with a rate described by krel.
Provided that exosite binding is long lasting, it can adequately explain the persistent action of salmeterol, even at low concentrations. Along, if one assumes that the exosite is specifically linked to the β2-adrenoceptor, it can also explain the fast reversibility of action when other receptors are involved. Finally, the exosite model is also compatible with the most of the reassertion data. However, additional structural and kinetic arguments led some to cast doubt on the pertinence of this model (Bergendal et al., 1996; Teschemacher and Lemoine, 1999). Based on the trendiest variant of the exosite hypothesis (i.e. that it displays pharmacological specificity and high affinity for the phenylalkoxyalkyl side chain of salmeterol), other molecules should be able to attenuate the reassertion behaviour of salmeterol (by competing with salmeterol for binding to the exosite) if they possess the same phenylalkoxyalkyl group (Bergendal et al., 1996). Yet, CGP 54103 and D 2543, two structural mimetics of this phenylalkoxyalkyl group, failed to do so (Bergendal et al., 1996). Although these findings plead against the existence of ‘independent’ exosites, they do not exclude a model wherein receptor active site binding must precede exosite binding. Another puzzling observation was that when strips from guinea pig trachea were treated with a sub-maximally effective salmeterol concentration, reassertion of relaxation was found to fade away after a few antagonist challenges while the phenomenon persisted at higher salmeterol concentrations (Bergendal et al., 1996). This concentration dependency did not comply with a model wherein salmeterol endures long-lasting binding to an exosite that is part of, or closely associated to, the receptor. Finally, there is presently still no tangible proof for existence of such exosite. To be close to, but yet distinct from the receptor's active site, the exosite was proposed to be located deep into the central core domain of the β2-adrenoceptor (Jack, 1991; Johnson et al., 1993). In agreement with this proposal, site-directed receptor mutatagenesis studies as well as photoaffinity labelling experiments with [125I]iodoazido-salmeterol suggest that the phenylalkoxyalkyl side chain of a bound salmeterol molecule resides deeper within the β2-adrenoceptor than its saligenin head (Green et al., 1996; Isogaya et al., 1998; Rong et al., 1999). However, such binding pocket can only correspond to the exosite if it is able to retain the salmeterol molecule in the presence of a function-blocking concentration of antagonist (Rong et al., 1999). As the photo-reactive azide is positioned at the hydrophobic end of [125I]iodoazidosalmeterol, photoaffinity labelling experiments with this radioligand should be well-suited to test this hypothesis. Although such experiments were underway at the time of publication (Rong et al., 1999), no information has been disclosed since. Hence, the existence of an exosite at the β2-adrenoceptor still awaits a firm demonstration.
The above considerations prompted us to reexamine the aptness and limitations of the microkinetic and exosite models to describe the peculiar pharmacokinetic properties of salmeterol by performing computer-assisted simulations to follow, in the situation of an exponentially vanishing ‘depot’, the degree receptor occupancy by a drug as a function of time under washout and reassertion conditions. As none of the models explained the totality of the experimental observations, we also addressed the ability of dissociated ligands to undergo rebinding to the receptor.
Methods
Simulations
Ligand- and competing ligand-receptor interactions are defined to be bimolecular in nature and to be reversible. Computer-assisted simulations were performed to follow the receptor-occupation by the ligand and the competing ligand as a function of time as previously described (Vauquelin et al., 2001; Vauquelin and Van Liefde, 2006). Simulated data rely on the differential equations describing reversible bimolecular ligand (L)- and competing ligand (C)- receptor (R) interactions (reaction equations below) along with a first-order decline in free ligand concentration by release from the membrane, ‘exosite’ or rebinding ‘trap’.
Equation for ligand-receptor binding:
Equation for competing ligand-receptor binding:
The differential equations (below) yield changes in the Ligand concentration and the percentage of free and occupied receptors over very small time (t) scales.
The ligand and the competing ligand have the same association rate constant at all times (i.e. k+= kc+= 1 × 108 M−1·min−1). The receptor dissociation rate constant of the competing ligand is also constant at all times (i.e. kc−= 0.23 min−1 for t1/2= 3 min). The ligand first-order rate constants for receptor dissociation (k-), release from the membrane (krel), exosite dissociation (kexo) and release from the rebinding ‘trap’ (‘kreb’) correspond to the half-lives given in Results and Discussion section and the legend of each figure. At the initial concentration (i.e. the starting point of the simulations), free ligand is in equilibrium with ligand bound to the indicated percentages of receptor molecules. The major processes affecting ligand-receptor interaction under each condition are schematically represented in Figure 1. Release from the membrane is not taken into account when krel > kexo or ‘kreb’.
Curves in Figures 2–6 are obtained by integrating the outcomes of these equations for the desired periods of time (abscissa). Figures 2 and 3 deal with washout in medium only. Figures 2A–C and 3 (microkinetic model): simulations deal with receptor binding and membrane release of the ligand (using k+ and different values for krel and k-). Figure 2D (exosite and rebinding models): simulations deal with receptor binding, exosite release and rebinding of the ligand (using k+, k- and kexo=‘kreb’).
Figure 2.
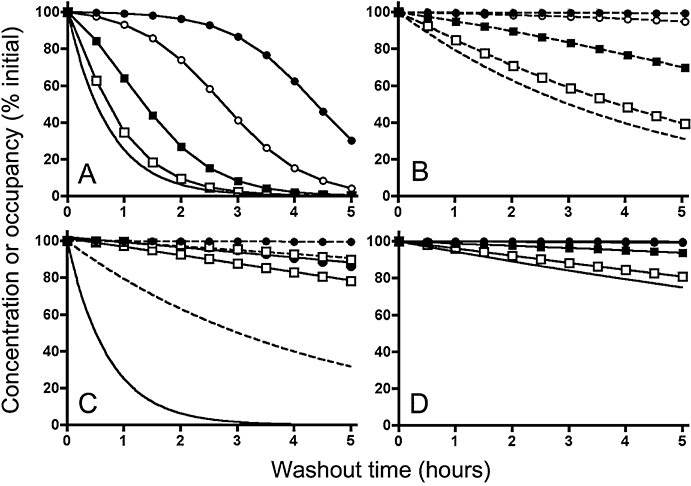
Decline in receptor (active site) occupancy by a ligand with time under washout conditions. (Panels A to C): microkinetic model depicted in Figure 1A; (Panel D): exosite and rebinding models depicted in Figures 1B and 1C, respectively. Description of the simulations is provided in the Materials and Methods section. The free ligand concentration decreases exponentially with time with a t1/2 of 30 min (solid curves in Panels A and C), 3 h (dashed curves in Panels B and C) or 12 h (solid curves in Panel D). Curves without symbols represent the free ligand concentration and those with symbols represent receptor occupancy. Symbols are the same in all panels and refer to initial levels of receptor occupancy: that is, 28.6% for open squares, 80% for closed squares, 97.6% for open circles and 99.8% for closed circles (corresponding to equilibrium binding at free ligand concentration/KD ratios of 0.4, 4, 40 and 400, respectively). Ligand dissociation from the receptors occurs with a t1/2 of 3 min (Panels A, B and D) or 12 h (Panel C). For the sake of comparison, initial ligand concentrations and receptor occupancies are normalized to 100%.
Figure 6.
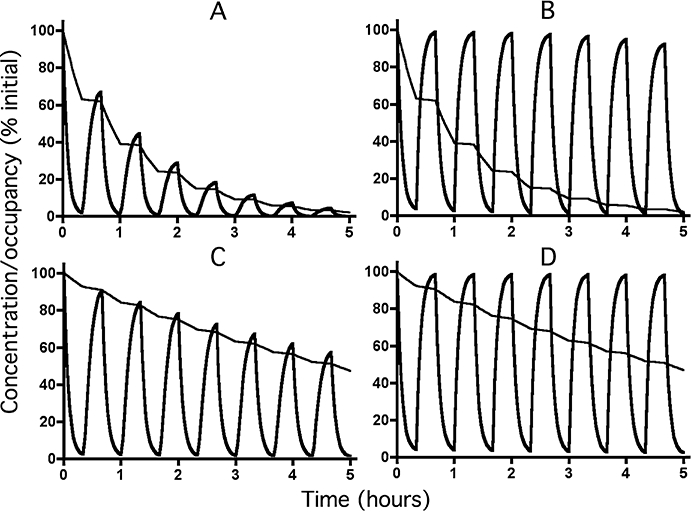
Decline in receptor occupancy (bold lines) by a fast-dissociating ligand (t1/2= 3 min) with time according to the rebinding model under reassertion conditions at low (28.6% for Panels A and C) and high (99.8% for Panels B and D) initial levels of occupancy. Simulations were carried out to mimic successive 20 min exposures to medium only and to medium containing a large excess (100 times its KD for Panels A and C and 10.000 times its KD for Panels B and D) of a fast-dissociating (t1/2= 1 min) competing ligand. The free ligand concentration (slim lines) decreases exponentially with time with a t1/2 of 12 h (‘kreb’= 0.00096 min−1, all panels) in medium only and with a t1/2 of 30 min (Panels A and B) or 3 h (panels C and D) when the medium contains competing ligand. For the sake of comparison, initial ligand concentrations and receptor occupancies are normalized to 100%.
Figure 3.
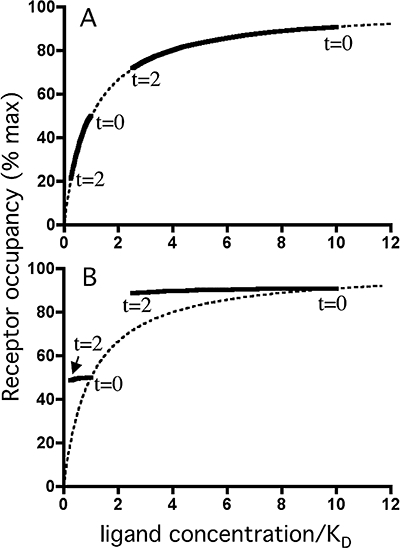
Decline in receptor occupancy by a ligand under washout conditions as a function of the free ligand concentration. Dotted lines: ligand saturation binding curves for a bimolecular ligand receptor interaction under equilibrium binding conditions. Solid lines are derived from simulations with the microkinetic model such as shown in Figure 2A to C and refer to receptor occupancies (in % of total receptor amount) as a function of the free ligand concentration (with KD values as unit) during washout. The free ligand concentration decreases exponentially with time with a t1/2 of 30 min and dissociation from the receptors occurs with a t1/2 of 3 min (Panel A) or 12 h (Panel B). For each panel, initial receptor occupancies amount 50% and 91% (at t = 0) and washout ends after 2 h (at t = 2), that is, when the free ligand concentration has declined 4-fold.
To reproduce reassertion in Figures 4–6, simulations start with a 20 min washout in medium only, followed with a 20 min treatment with an excess of competing ligand and repetitions of this cycle for a total period of 5 h. Figure 4 (microkinetic model): simulations deal with receptor binding of ligand and (when applicable) competing ligand and with membrane release of the ligand (using k+, k−, kc+, kc− and different values for krel). Figure 5 (exosite model): simulations deal with receptor binding of ligand and (when applicable) competing ligand and with exosite release of the ligand (using k+, k−, kc+, kc− and kexo). Figure 6 (rebinding model): simulations deal with receptor binding of ligand and (when applicable) competing ligand and release of the ligand from the rebinding ‘trap’ or membrane (using k+, k−, kc+, kc− and ‘kreb’ in medium only or different values for krel when antagonist is present).
Figure 4.
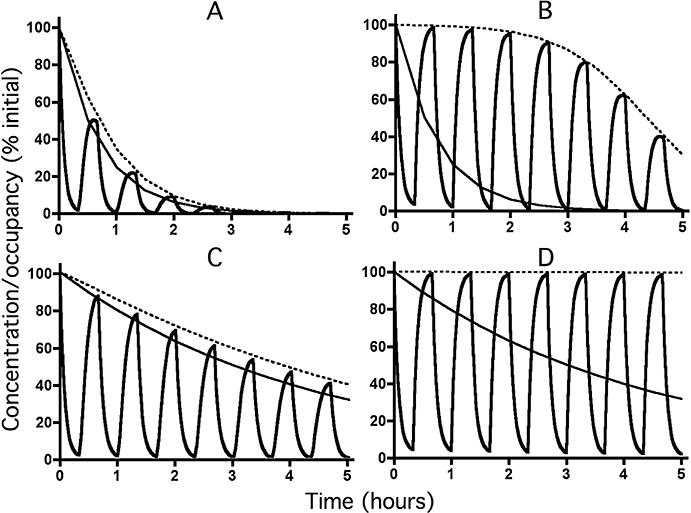
Decline in receptor occupancy (bold lines) by a fast-dissociating ligand (t1/2= 3 min) with time according to the microkinetic model under reassertion conditions at low (28.6% for Panels A and C) and high (99.8% for Panels B and D) initial levels of occupancy. Simulations were carried out to mimic successive 20 min exposures to medium only and to medium containing a large excess (100 times its KD for Panels A and C and 10.000 times its KD for panels B and D) of a fast-dissociating (t1/2= 1 min) competing ligand. The free ligand concentration (slim lines) decreases exponentially with time with a t1/2 of 30 min (Panels A and B) or 3 h (Panels C and D). Dotted lines correspond to receptor occupancy under washout only (i.e. curves in Figure 2A and B). For the sake of comparison, initial ligand concentrations and receptor occupancies are normalized to 100%.
Figure 5.
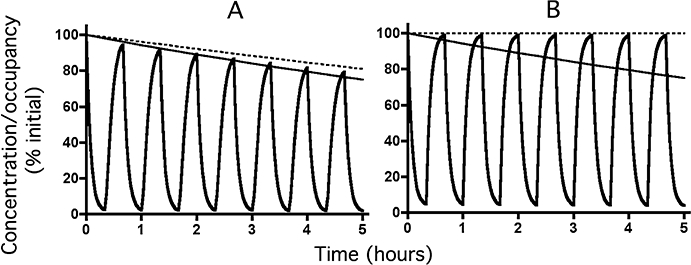
Decline in receptor active site occupancy (bold lines) by a fast-dissociating ligand (t1/2= 3 min) with time according to the exosite model under reassertion conditions at low (28.6% for Panel A) and high (99.8% for Panel B) initial levels of occupancy. Simulations were carried out to mimic successive 20 min exposures to medium only and to medium containing a large excess (100 times its KD for Panel A and 10.000 times its KD for Panel B) of a fast-dissociating (t1/2= 1 min) competing ligand. The free (i.e. exosite-bound) ligand concentration (slim lines) decreases exponentially with a t1/2 of 12 h (kexo= 0.00096 min−1). Dotted lines correspond to receptor occupancy under washout only (i.e. curves in Figure 2D). For the sake of comparison, initial ligand concentrations and receptor occupancies are normalized to 100%.
Materials
Dulbeccos' modifed Eagle's medium (DMEM) with glutamax, Leibowitz (L-15) with glutamax, geneticin, hygromycin and Dulbeccos' phosphate buffered saline (D-PBS) were purchased from Invitrogen (Paisley, UK). The radiolabelled CB1 cannabinoid receptor inverse agonist [3H]-taranabant (20 Ci·mmol−1) and rimonabant was from AstraZeneca (Mölndal, Sweden). Fatty acid free bovine serum albumin (BSA) was obtained from Sigma-Aldrich (St. Louis, MO, USA). The 24-Well poly-D-lysine coated plates were purchased from In Vitro (Stockholm, Sweden) and fetal bovine serum (FBS) was obtained from Hyclone (South Logan, UT, USA).
Cell culture
Human embryonic kidney cells stably expressing human Cannabinoid receptor 1 (hCB1r) and chimeric G protein, Gqi5, (HEK293s-hCB1r-Gqi5) (AstraZeneca, Mölndal, Sweden) were cultured in 175 cm2 flasks in DMEM with glutamax supplemented with 10% FBS, 600 µg·mL−1 geneticin and 300 µg·mL−1 hygromycin. The cells were cultured in 5% CO2 at 37°C. One day prior to the experiment, the cells were plated in 24-well plates (2 × 105 cells·well−1) and grown in DMEM with glutamax supplemented with 10% FBS.
[3H]-Taranabant dissociation binding
Prior to the experiment, cells were washed three times at room temperature with 500 µL·well−1 of L-15 with glutamax (binding buffer). Binding assays were performed at 37°C in binding buffer with 1% BSA. Cells were first incubated for 60 min with 2 nM [3H]-taranabant either alone (for total binding) or in the presence of 1µM of the unlabelled CB1 receptor antagonist rimonabant (for non-specific binding), washed three times with 750 µL·well−1 D-PBS with 1% BSA and subsequently exposed for the times indicated to 500 µL·well−1 fresh binding buffer either without or with an excess (1 µM) rimonabant, 1% (w/v) BSA or both together. Cells were then treated for 30 min at room temperature with 0.1 M NaOH and 0.5% Triton X-100 (500 µL·well−1). The radioactivity of the solutes was then counted for 5 min in presence of 3 mL scintillation liquid (Optiphase Hisafe from PerkinElmer; Boston, MA, USA) in a liquid scintillation counter.
Binding data analysis
Experimental values refer to specific binding and are represented as mean ± SEM of three independent experiments (each performed in triplicate). Non-specific binding of [3H]-taranabant after the initial incubation phase amounted 10% of its total binding. Mono-exponential dissociation curves were calculated by non-regression analysis with GraphPad PrismTM (San Diego, CA, USA).
Results and discussion
Two kinetic parameters are important for the microkinetic model (Figure 1A); the rate by which salmeterol dissociates from the receptor (k-) and the rate by which it is released from plasma membrane (krel). Based on the rapid reversal of the salmeterol effect when airway smooth muscle preparations are challenged with an excess of antagonist, receptor dissociation should be very rapid. Kinetic studies with radiolabelled salmeterol were never reported, but its dissociation rate from the β2-adrenoceptor has been indirectly estimated based on its potential to delay the association of the subsequently added radiolabelled antagonists [125I]-iodopindolol or [125I]-iodocyanopindolol. While initial studies pointed at irreversible and even (vs. the radioligand) non-competitive receptor binding of salmeterol (Jack, 1991; Nials et al., 1993b; Naline et al., 1994), subsequent studies pleaded in favour of fast reversibility and even competitivity (Clark et al., 1996; Teschemacher and Lemoine, 1999). The reason for this discrepancy has been attributed to the very high concentrations of salmeterol initially used (Teschemacher and Lemoine, 1999). As the latter binding studies with lower salmeterol concentrations are compatible with the relaxation experiments with respect to reversibility as well as competitivity (Ball et al., 1991), it is quite likely that dissociation of salmeterol from the β2-adrenoceptor takes place with a half-life of no more than a few minutes. In this respect, salmeterol release studies with synthetic membranes (Rhodes et al., 1992) led to the initial consensus that this agonist leaks out of the membrane with a half-life of about 30 min. More recently, Austin et al. (2003) observed a slower release of salmeterol from tracheal strips (t1/2 of 3 h).
The simulated data shown in Figures 2A–C and 3 refer to simple washout experiments (i.e. flushing salmeterol-preincubated receptor preparations with fresh medium) and those in Figure 4 refer to reassertion experiments (i.e. consecutively flushing salmeterol-preincubated receptor preparations with fresh medium and excess antagonist-containing medium). All were based on the inherent assumption that salmeterol molecules can only bind to the receptors when they are present in the membrane. The concentration of free ligand was thus set to decrease exponentially with the same rate as its release from the membrane.
Figures 2A and B compares the decline in receptor occupancy by a salmeterol-like ligand (k-= 0.23 min−1 for t1/2= 3 min) with time under washout conditions for different initial levels of receptor occupancy and for the presently known values of krel (0.023 min−1 for a release t1/2 of 30 min in Figure 2A and 0.0038 min−1 for a release t1/2 of 3 h in Figure 2B). Of note is that the receptor occupancy at each time point not only depends on the dissociation of the complexes but also on the association of free receptors with remaining free ligand. These simulations clearly show that the rate by which the occupancy declines is very much dependent on the initial level of occupancy. Indeed, when the free ligand concentration is at or below its equilibrium dissociation constant (KD) for the receptor, the initial occupancy will only be modest and it will decline almost as swiftly as the free ligand concentration. On the other hand, when the free ligand concentration is in large excess of its KD, the initial receptor occupancy will be nearly maximal and, within the same time period, it will decline much more slowly as the free ligand concentration. The reason for this initial occupancy-related effect can be explained by: (i) the fact that free and bound ligand concentrations remain in quasi-equilibrium during washout when k- > krel; and (ii) the hyperbolic shape of the saturation binding curve under equilibrium conditions, implying that, for example, halving the free ligand concentration will have a much smaller effect on occupancy when starting at a high level of occupancy than at a low level (Figure 3A). This latter characteristic implies that the time-wise decline in receptor occupancy is mainly dictated by krel at low initial free ligand concentrations. This explains why it is especially under those conditions that the decline receptor occupancy is appreciably slower when the simulations are based on release rate found by Austin et al. (2003) instead of the release rate advanced by Rhodes et al. (1992) (compare Figure 2B to 2A).
It is noteworthy that, the more the dissociation t1/2 increases, the slower receptor occupancy declines during washout. This trend is most pronounced at low levels of initial occupancy and when in k- < krel (i.e. for the very slow dissociating ligand) there is even no distinction any more when comparing the curves corresponding to: (i) the different initial occupancy situations; and (ii) the release rates found by Austin et al. (2003) and Rhodes et al. (1992) (Figure 2C). In this latter case, the occupancy versus free ligand relationship during washout produces a counterclockwise hysteresis when compared with the saturation binding curve at equilibrium, and this phenomenon is most apparent at low levels of initial occupancy (Figure 3B).
To deal with reassertion experiments, simulations were carried out to mimic time-wise variations in receptor occupancy by a salmeterol-like ligand when the ligand-preincubated receptor preparation is successively exposed to medium only and to an excess of a fast-dissociating (i.e. sotalol-like) competing ligand. Figures 4A and B apply to the release rate found by Rhodes et al. (1992) and to low and high initial levels of receptor occupancy, respectively. In both situations, there will be a rapid decline in its receptor occupancy in the presence of an excess of competing ligand and, after withdrawal of the competing ligand, receptor occupancy will be restored to a level corresponding to a control condition where no challenge with competing ligand took place (i.e. the simple washout condition shown in Figure 2A). This implies that, at low initial ligand concentrations (Figure 4A), the ‘reasserted’ receptor occupancy will gradually fade away after each challenge with competing ligand, while the ‘reasserted’ occupancy is quite persistent at higher initial ligand concentrations (Figure 4B). Corresponding simulations based on the release rate found by Austin et al. (2003) (Figures 4C and D) yield the same overall picture. Yet, because of the longer-lasting receptor occupancy under simple washout (compare Figure 2B to 2A), reassertion will also persist much longer at low initial ligand concentrations (compare Figure 4C to 4A).
Simulation studies were also performed to examine the impact of the ‘exosite’ model which stipulates that the lipophilic phenylalkoxyalkyl side chain of salmeterol is able to undergo a highly stable association with an accessory site (the ‘exosite’) located either in the immediate vicinity of the β2-adrenoceptor or even within the receptor molecule itself (Figure 1B). The same equations could be used as for the microkinetic model with the following considerations in mind: (i) the equation describing the reversible salmeterol- receptor interaction in the microkinetic model now describes the interaction between the saligenin head of salmeterol and the active site of the receptor (kinetic parameters k+ and k- remain unchanged); and (ii) the equation describing the release of salmeterol from the plasma membrane in the microkinetic model is now replaced by the much slower dissociation of the phenylalkoxyalkyl side chain- exosite complex [i.e. krel is replaced by kexo= 9.6 × 10−4 min−1 for t1/2= 12 h so that separate simulations dealing with the release rates found by Rhodes et al. (1992) and Austin et al. (2003) no longer need to be done]. The choice of such slow dissociation from the exosite is inspired by: (i) the fact that, regardless of its initial concentration, the action of salmeterol may persist for up to 12 h in washout experimens with airway smooth muscle preparations (Johnson et al., 1993; Nials et al., 1994); and (ii) the need for kexo to be substantially smaller than krel to mimick such situation (Vauquelin and Van Liefde, 2006; Tummino and Copeland, 2008).
Figure 2D compares the decline in receptor active site occupancy by a salmeterol-like exosite-binding ligand with time under washout conditions for different initial levels of active site occupancy. The comparison clearly shows that the active site occupancy is persistent regardless of the initial level of occupancy. This is in excellent agreement with the observed concentration-independent effect-duration of salmeterol under simple washout conditions. Reassertion experiments were simulated the same way as for the microkinetic model. Figures 5A and B show the simulated variations in receptor occupancy by a salmeterol-like exosite-binding ligand with time under reassertion conditions for a low and high initial level of active site occupancy, respectively. In both situations, the active site occupancy declines rapidly in the presence of an excess of competing ligand and, after withdrawal of the competing ligand, the occupancy will regain a level corresponding to the one observed under simple washout (Figure 2D). This implies that the ‘reasserted’ active site occupancy persists at all initial ligand concentrations.
We also explored the outcomes of an alternative model that is based on the concept of ‘rebinding’ or, in other words, the capability of dissociated ligands to reassociate to the same or other receptor molecules. In this respect, the classical assumption is that, once dissociated, free ligands are evenly distributed all over the aqueous (for hydrophilic ligands) or membrane compartment (for hydrophobic and ampiphilic ligands such as in the present case). Yet, from a biophysical perspective, rebinding is likely to be a more localized process wherein the diffusion rate of the free ligand plays a prominent role (DeLisi, 1981; Delisi and Wiegel, 1981; Goldstein et al., 1989). According to the model elaborated by these authors (Figure 7), binding between a ligand and its receptor should proceed according to a two-step process involving diffusion of the molecules to bring them together (or away from each other) to form an ‘encounter complex’ and the actual (also reversible) binding process. This model implies that when a ligand has just left its cognate receptor (and, hence, when both molecules are still in close proximity), it will have the ‘choice’ between either diffusing away or binding again. This ‘biophysical’ description of the rebinding phenomenon differs from the prevailing ‘classical’ one in two major ways; it is a localized phenomenon (so that its ‘amplitude’ does not strictly depend on the average ligand concentration in the aqueous or membrane compartment) and it will depend on the reaction forward rate constant rather than on the affinity of the ligand for its receptor.
Figure 7.
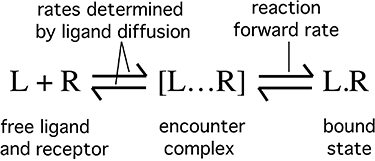
Schematic representation of drug (L) – receptor (R) binding with the intermediate formation of an encounter complex [L … R] from where the drug can either bind with the reaction forward rate constant or diffuse away from the receptor.
At the experimental level, rebinding will not only produce a decrease in the apparent dissociation rate of the ligand but eventually also halt this process even when some residual binding remains. This has clearly been shown to take place in intact cell binding studies for different radiolabelled AT1-type angiotensin II receptor antagonists (Fierens et al., 1999; Le et al., 2007). This is because the ‘apparent’ dissociation rate will decline with time as more free receptors become available for the rebinding process (Delisi and Wiegel, 1981; Gopalakrishnan et al., 2005). Sometimes, dissociation is even barely perceptible when rebinding is allowed to take place. This was recently shown in intact cell binding studies for the radiolabelled amphiphilic D2-type dopamine receptor antagonist spiperone (Packeu et al., 2008) and even more strikingly in the present experiments with the radiolabelled CB1-type cannabinoid receptor inverse agonist [3H]-taranabant (Kirkham, 2008). Figure 8 shows the time-wise decrease in binding of this radioligand from the CB1 receptors stably expressed in recombinant human embryonic kidney cells under different washout conditions. Whereas its apparent dissociation is only modest in medium, it is exacerbated upon addition of an excess of the unlabelled CB1 receptor inverse agonist rimonabant (by competing with liberated [3H]-taranabant for the receptor) as well as upon addition of BSA (by competing with the receptor for [3H]-taranabant and by preventing potential rebinding of this radioligand to non-receptor sites). The most pronounced effect is observed when the unlabelled ligand and BSA are given in combination, that is, when both mechanisms operate together.
Figure 8.
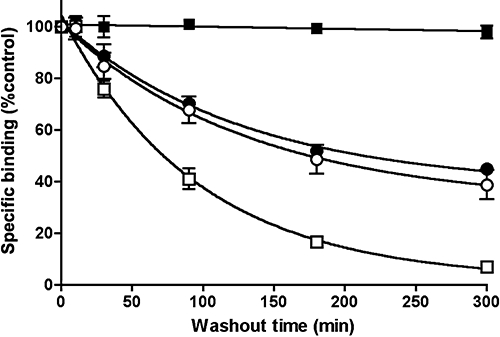
Dissociation of [3H]-taranabant from intact HEK293s-hCB1r-Gqi5 cells. Cells were incubated 2 nM [3H]-taranabant and its dissociation was initiated by washing of the cells and replacement with fresh medium only ( ), medium containing an excess (1 µM) of the unlabelled ligand rimonabant (•), medium containing 1% w/v bovine serum albumin (○) or medium containing both (□). Data are presented as specific (i.e. rimonabant-displaceable) binding and represent mean ± SEM of three independent experiments.
), medium containing an excess (1 µM) of the unlabelled ligand rimonabant (•), medium containing 1% w/v bovine serum albumin (○) or medium containing both (□). Data are presented as specific (i.e. rimonabant-displaceable) binding and represent mean ± SEM of three independent experiments.
Due to the present lack of tritiated salmeterol, similar data are presently unavailable for this ligand. Nevertheless, there are circumstantial theoretical indications for rebinding to take place in the case of such amphiphilic β2-adrenoceptor agonists like salmeterol. Important for the rebinding model is that the fate of the ligand is influenced by opposing factors like the ligand's diffusion rate (in favour of diffusing away when elevated), and the ‘reaction forward rate constant’ (in favour of rebinding when elevated). In this respect, certain amphiphilic drugs could be especially prone to undergo rebinding if they undergo slow diffusion within the lipid bilayer (McCloskey and Poo, 1986). High local receptor densities also favour rebinding as it increases the likelihood of dissociated ligands to bind to neighbouring receptors (DeLisi, 1981; Delisi and Wiegel, 1981; Goldstein et al., 1989; Posner et al., 1992; Andrews, 2004; Gopalakrishnan et al., 2005) and simulations indicate that rebinding is even more favoured when the ligand can undergo internal diffusion within a cluster of cell surface receptors (Gopalakrishnan et al., 2005). In this respect, β2-adrenoceptor signalling appears indeed to be spatially restricted and, at least in cardiomyocytes, to be concentrated in membrane microdomains like lipid rafts and caveolae (Okamoto et al., 1998; Steinberg, 2004). Finally, partitioning in the membrane also imposes conformational and orientational constraints to amphiphilic molecules (Rhodes et al., 1992; Schwyzer, 1995; Bader et al., 2001; Castanho and Fernandes, 2006). If the partitioning of salmeterol also optimizes its conformation for docking into the β2-adrenoceptor, it could favour its rebinding (and hence, its long-lasting action) by increasing the ‘reaction forward rate constant’.
Taken together, the rebinding model could be considered to be an extension of the microkinetic model as the ligand is either bound to the receptor or present in the lipid bilayer. However, the rebinding model could also be considered to represent a variant of the exosite model if one assumes that: (i) receptors behave as exosites for themselves and one-another as long as they are not occupied by competing foreign molecules; and (ii) persistent ligand binding under simple washout conditions is caused by the constant shuffling of salmeterol between receptors/exosites rather than by persistent occupation of a single receptor protein.
Assuming that a salmeterol-like molecule experiences efficient rebinding to the β2-adrenoceptors under simple washout conditions, its behaviour can be simulated with the equations for the microkinetic model provided that krel is replaced by a much smaller ‘kreb’. In this respect, ‘kreb’ was set equal to kexo (i.e. ‘kreb’= 9.6 × 10−4 min−1 for t1/2= 12 h) to grant the exosite binding and rebinding models the same opportunity to prolong receptor occupancy. Obviously, due to the use of the same equations and constants, simulations according to the rebinding model and the exosite model yield the same outcome (Figure 2D). This implies that the rebinding model also allows a concentration-independent effect duration of salmeterol under simple washout conditions. On the other hand, simulating reassertion experiments cannot be performed based on the equations for the rebinding model only. Indeed, when present at high concentration in the washout medium, competing ligands will quickly occupy all free receptor molecules. The rebinding process comes thereby to a halt and the liberated ligand molecules can only diffuse within the plasma membrane (Figure 1C– right panel). The behavior of salmeterol should only be governed by the microkinetic model under this condition (i.e. with krel= 0.023 min−1 for t1/2= 30 min or 0.0038 min−1 for a release t1/2 of 3 h). When the receptor occupancy of a salmeterol-like ligand under conditions of repeatedly adding and removing an excess of antagonist is simulated by alternating the microkinetic and rebinding models (Figure 1C), the ‘reasserted’ receptor occupancy will be quite persistent at high initial occupancy (Figures 6A and D) but gradually fade away at low initial occupancy (Figures 6B and D). The declining reassertion in these latter simulations is in fact caused by the need to invoke the microkinetic model for part of the time (i.e. when the competing ligand is present). This also explains why the reassertion is more persistent when the simulations are based on the release data by Austin et al. (2003) instead of those by Rhodes et al. (1992) (compare Figure 6D to 6B). For the same conditions, the fading is much slower when the simulations are based on the exosite model (Figure 5). This is because long-lasting exosite binding remains operative at all times.
A comparison between the outcomes of the present simulations and the experimental observations with airway smooth muscle preparations is greatly facilitated when examining the situations for high and low salemeterol concentrations one at the time. The comparison presented hereunder is obviously only relevant if at least two assumptions are correct. First, it is assumed that the experimental observations are genuine; that is, not biased by potential artifacts. Second, it is assumed that experimental observations on these intact tissues can be related to mechanisms that take place at the sub-cellular/molecular level. Both issues will be discussed further below.
The simplest picture is obtained when salmeterol initially occupies a large fraction of the receptors (i.e. when it is initially present at high concentrations). In this situation, most of the simulations agree with the experimental observations by predicting a long-lasting agonistic effect during washout as well as the persisting reassertion of this effect during repeated challenges with an antagonist (Figures 2B and D, 4D, 5B and 6B and D). Yet, of note is that the simulations based on the microkinetic model and the relatively fast release of salemeterol from synthetic membranes (t1/2= 30 min) yield somewhat less persistent receptor occupancy and reassertion (Figure 2A and 4B). Taken together, the present simulations suggest that the microkinetic model is already adequate to explain a long-lasting β2-adrenoceptor stimulation if the membrane can retain a high level of the agonist for a sufficiently long time. As long as this situation prevails, the participation of the more complex exosite and rebinding mechanisms should be overshadowed (but not ruled out per se).
A different picture emerges when the salmeterol concentration has declined so much that it starts to approach its Kd for the receptor. In contrast to the observed concentration-independent effect-duration of salmeterol under simple washout conditions (Johnson et al., 1993; Nials et al., 1994), the microkinetic model now predicts that the degree of receptor occupancy will start to parallel the free (i.e. membrane-associated) ligand concentration (Figure 3A). While the simulations according to a release t1/2 of 30 min (Figure 2A) are completely at odds with the observations, those according to a release t1/2 of 3 h (Figure 2B) fare better but still predict that the action of salmeterol should have dropped by over 90% after 12 h washout (i.e. after 4 release half-lives). The only way to make those simulations fit with the observations is to assume that salmeterol dissociates very slowly from the receptor's active site. Yet, this assumption can be refuted because of its incompatibility with the reassertion phenomenon. Hence, compared with the macrokinetic model, the rebinding and exosite models provide a much better explanation for the long-lasting dilatory effect at low salmeterol concentrations (Figure 2D). On the other hand, while simulations according to the microkinetic and rebinding models and a release t1/2 of 30 min (Figures 4A and 6A) fit well with the fading reassertion after just a few challenges with antagonist (Bergendal et al., 1996), the simulations according to exosite model (Figure 5A) and the microkinetic and rebinding models based on a release t1/2 of 3 h (Figures 4C and 6C) yield far to persistent reassertion.
The provisional conclusion of these simulations is that, under their present form, neither the microkinetic model nor the exosite model are capable to fully describe the kinetic properties of the salmeterol-mediated β2-adrenoceptor activation. A model that is based on the concept of rebinding (i.e. the capability of dissociated ligands to reassociate to the same or other receptor molecules) fares better, at least if one assumes salmeterol to be relatively rapidly released from the cell membrane. However, while this latter assumption is necessary for this model to reproduce the reassertion experiments by Bergendal et al. (1996), it is at odds with the relatively slow release of salmeterol from tracheal strips (t1/2 of 3 h) observed by Austin et al. (2003). In fact, this points at an incompatibility between these two experimental observations. Pending further confirmation/verification by dedicated experiments, we feel presently not qualified to rule out one of them. Yet, some critical considerations could be formulated about how Bergendal et al. (1996) interpreted their reassertion data and about the pervasiveness of the release kinetics by Austin et al. (2003). On the one hand, it cannot be excluded that the reassertion at low concentrations salmeterol is related to incomplete washout of the intermittently added antagonist instead of the proposed gradual drop of the salmeterol concentration in the membrane. If this were the case, it would give equal credit to the exosite and rebinding models for explaining the long-lasting β2-adrenoceptor activation at low concentrations of salmeterol. On the other hand, the release data for tracheal strips were obtained after treating the tissues with a β2-adrenoceptor-saturating concentration of salmeterol (i.e. 15 µM). This concentration is far higher than the one at which the reassertion was found to fade after repeated antagonist exposures (i.e. 0.01 µM) and even the one at which the release of salmeterol from artificial membranes was measured (i.e. 0.1 µM, Rhodes et al., 1992). Here again, it cannot be excluded that salmeterol alters the very architecture of the lipid bilayer when present at such high concentrations and that this affects the rate by which this agonist can be subsequently released. In that case, the rebinding model still offers the favoured explanation for the observed effects at low concentrations of salmeterol. Because of the incoherence between these key results, some ambiguity about the action of salmeterol remains. This partly arises from the fact that the dedicated experiments have only been reported once. There is certainly a need for many of the results to be confirmed independently, most preferably by all-inclusive studies combining duration of action-, release- and reassertion experiments on the same experimental system with both low and high concentrations of salmeterol. In this respect, reassertion experiments with low salmeterol concentrations should be especially useful as they represent a powerful kinetic approach to differentiate between the exosite and rebinding models.
The distinct rates of salmeterol release from artificial membranes and airway strips (30 min and 3 h, respectively) have been related by Austin et al. (2003) to differences in the surface area-to-volume ratio between both experimental systems. This certainly could influence the wash-in and washout kinetics but, whereas these authors assumed the tissue and surrounding liquid to constitute a homogenous phases between which salmeterol is able to partition with single rate constants, the situation is likely to be more complex. Indeed, diffusion of salmeterol is likely to be restricted in vascular smooth muscle tissue (Clark et al., 1996; Teschemacher and Lemoine, 1999) due to the need to pass the epithelial cell layer, tortuosities in interstitial spaces and repeated partitioning in cell membranes (Lovich et al., 2001; Hrabctová and Nicholson, 2004). According to this view, it is reasonable to assume that the relatively fast release of salmeterol from artificial membranes (Rhodes et al., 1992) reflects a one-time partitioning process and that the release of this agonist from airway strips (Austin et al., 2003) is slower because it stems from the combination of repeated partitioning and restricted diffusion. These considerations touch on an important question, namely, how relevant is it to link observations at the intact tissue/macroscopic scale (i.e. contraction studies) to those at the sub-cellular scale and, even more so, to explanations at the molecular scale? Yet, it is already for almost two decades that mechanisms that apply to the sub-cellular/molecular scale (microkinetic and exosite models as well as the present rebinding model) have been advanced to explain the long-lasting effect of salmeterol-based experimental observations with biological systems (i.e. contraction studies) that are maybe to complex for that purpose.
In conclusion, the present simulations suggest that the microkinetic model is sufficient to explain the ability of salmeterol to produce long-lasting dilatation of vascular tissues as well as the persistent reassertion thereof as long as the smooth muscle cell membranes harbour a high level of this agonist. It is only when this mechanism exhausts (i.e. at low agonist levels) that additional phenomena like rebinding and/or the presently proposed exosite binding start to play a preeminent role. Yet, many uncertainties remain and, in this respect, solid experimental proof still needs to be provided for one of these latter mechanisms to be operational in the case of salmeterol. Even the relevance of advancing mechanisms that apply to the sub-cellular/molecular scale to explain observations at the more complex tissue level could be questioned. One way to find out is to perform experiments with isolated intact β2-adrenoceptor-expressing cells to address ‘micoscopic’ aspects of salmeterol action and to compare the results thereof with those from corresponding airway smooth muscle contraction studies. Such comparison could provide clues for additional mechanisms that are particular to this macroscopic approach.
Glossary
Abbreviations:
- HEK293s-hCB1r-Gqi5
Human embryonic kidney cells stably expressing human Cannabinoid receptor 1
References
- Anderson GP. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–2160. doi: 10.1016/0024-3205(93)90729-m. [DOI] [PubMed] [Google Scholar]
- Anderson GP, Lindén A, Rabe KF. Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J. 1994;7:569–578. doi: 10.1183/09031936.94.07030569. [DOI] [PubMed] [Google Scholar]
- Andrews SS. Serial rebinding of ligands to clustered receptors as exemplified by bacterial chemotaxis. Phys Biol. 2004;2:111–112. doi: 10.1088/1478-3975/2/2/004. [DOI] [PubMed] [Google Scholar]
- Austin RP, Barton P, Bonnert RV, Brown RC, Cage PA, Cheshire DR, et al. QSAR and the rational design of long-acting dual D2-receptor/beta2-adrenoceptor agonists. J Med Chem. 2003;46:3210–3320. doi: 10.1021/jm020886c. [DOI] [PubMed] [Google Scholar]
- Bader R, Bettio A, Beck-Sickinger AG, Zerbe O. Structure and dynamics of micelle-bound neuropeptide Y: comparison with unligated NPY and implications for receptor selection. J Mol Biol. 2001;305:307–329. doi: 10.1006/jmbi.2000.4264. [DOI] [PubMed] [Google Scholar]
- Ball DI, Brittain RT, Coleman RA, Denyer LH, Jack D, Johnson M, et al. Salmeterol, a novel, long-acting beta 2-adrenoceptor agonist: characterization of pharmacological activity in vitro and in vivo. Br J Pharmacol. 1991;104:665–671. doi: 10.1111/j.1476-5381.1991.tb12486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach JR, Young CL, Stenton SC, Avery AJ, Walters EH, Hendrick DJ. A comparison of the speeds of action of salmeterol and salbutamol in reversing methacholineinduced bronchoconstriction. Pulm Pharmacol. 1993;5:133–135. doi: 10.1016/0952-0600(92)90031-b. [DOI] [PubMed] [Google Scholar]
- Bergendal A, Lindén A, Skoogh B-E, Gerspacher M, Anderson GP, Lolfdahl C-G. Salmeterol mediated reassertion of relaxation persists in guinea-pig trachea pretreated with aliphatic side chain structural analogues. Br J Pharmacol. 1996;117:1009–1015. doi: 10.1111/j.1476-5381.1996.tb16690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanho MARB, Fernandes MX. Lipid membrane-induced optimization for ligand–receptor docking: recent tools and insights for the ‘membrane catalysis’ model. Eur Biophys J. 2006;35:92–103. doi: 10.1007/s00249-005-0007-9. [DOI] [PubMed] [Google Scholar]
- Chester DW, Herbette LG, Mason RP, Joslyn AF, Triggle DJ, Koppel DE. Diffusion of dihydropyridine calcium channel antagonists in cardiac sarcolemmal lipid multibilayers. Biophys J. 1987;52:1021–1030. doi: 10.1016/S0006-3495(87)83295-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Allal C, Friedman J, Johnson MB. Stable activation and desensitization of beta 2-adrenergic receptor stimulation of adenylyl cyclase by salmeterol: evidence for quasi-irreversible binding to an exosite. Mol Pharmacol. 1996;49:182–189. [PubMed] [Google Scholar]
- Coleman RA, Johnson M, Nials AT, Vardey CJ. Exosites: their current status, and their relevance to the duration of action of long-acting beta 2-adrenoceptor agonists. Trends Pharmacol Sci. 1996;17:324–330. [PubMed] [Google Scholar]
- DeLisi C. The effect of cell size and receptor density on ligand-receptor reaction rate constants. Mol Immunol. 1981;18:507–511. doi: 10.1016/0161-5890(81)90128-0. [DOI] [PubMed] [Google Scholar]
- DeLisi C, Wiegel FW. Effect of nonspecific forces and finite receptor number on rate constants of ligand – cell bound-receptor interactions. Proc Natl Acad Sci U S A. 1981;78:5569–5572. doi: 10.1073/pnas.78.9.5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierens FLP, Vanderheyden PML, De Backer J-P, Vauquelin G. Binding of the antagonist [3H]candesartan to angiotensin II AT1 receptor-transfected Chinese hamster ovary cells. Eur J Pharmacol. 1999;367:413–422. doi: 10.1016/s0014-2999(98)00965-0. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Posner RG, Torney DC, Erickson J, Holowka D, Baird B. Competition between solution and cell surface receptors for ligand. Dissociation of hapten bound to surface antibody in the presence of solution antibody. Biophys J. 1989;56:955–966. doi: 10.1016/S0006-3495(89)82741-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan M, Forsten-Williams K, Nugent MA, Täuber UC. Effects of receptor clustering on ligand dissociation kinetics: theory and simulations. Biophys J. 2005;89:3686–3700. doi: 10.1529/biophysj.105.065300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SA, Spasoff AP, Coleman RA, Johnson M, Liggett SB. Sustained activation of a G-protein – coupled receptor via anchored agonist binding. J Biol Chem. 1996;271:24029–24035. doi: 10.1074/jbc.271.39.24029. [DOI] [PubMed] [Google Scholar]
- Herbette LG. Membrane pathways for drug/lon channel interactions: molecular basis for pharmacokinetic properties. Drug Devel Res. 1994;33:214–224. [Google Scholar]
- Hrabctová S, Nicholson C. Contribution of dead-space microdomains to tortuosity of brain extracellular space. Neurochem Int. 2004;45:467–477. doi: 10.1016/j.neuint.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Isogaya M, Yamagiwa Y, Fujita S, Sugimoto Y, Nagao T, Kurose H. Identification of a key amino acid of the beta2-adrenergic receptor for high affinity binding of salmeterol. Mol Pharmacol. 1998;54:616–622. [PubMed] [Google Scholar]
- Jack D. A way of looking at agonism and antagonism: lessons from salbutamol, salmeterol and other β-adrenoceptor agonists. Br J Clin Pharmacol. 1991;31:501–514. doi: 10.1111/j.1365-2125.1991.tb05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppsson AB, Lofdahl CG, Waldeck B, Widmark E. On the predictive value of experiments in vitro in the evaluation of the effect duration of bronchodilator drugs for local administration. Pulm Pharmacol. 1989;2:81–85. doi: 10.1016/0952-0600(89)90028-8. [DOI] [PubMed] [Google Scholar]
- Johnson M. Beta2-adrenoceptors: mechanisms of action of beta2-agonists. Paediatr Respir Rev. 2001;2:57–62. doi: 10.1053/prrv.2000.0102. [DOI] [PubMed] [Google Scholar]
- Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117:18–24. doi: 10.1016/j.jaci.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Johnson M, Coleman RA. Mechanisms of action of β2-adrenoceptor agonists. In: Busse WW, Holgate ST, editors. Asthma & Rhinitis. Cambridge: Blackwell; 1995. pp. 1278–1295. [Google Scholar]
- Johnson M, Butchers PR, Coleman RA, Nials AT, Strong P, Sumner MJ, et al. The pharmacology of salmeterol. Life Sci. 1993;52:2131–2143. doi: 10.1016/0024-3205(93)90728-l. [DOI] [PubMed] [Google Scholar]
- Kirkham TC. Taranabant cuts the fat: new hope for cannabinoid-based obesity therapies? Cell Metab. 2008;7:68–78. doi: 10.1016/j.cmet.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Le MT, Pugsley M, Vauquelin G, Van Liefde I. Angiotensin II AT1 antagonism and insurmountability: comparison of olmesartan and telmisartan. Brit J Pharmacol. 2007;151:952–962. doi: 10.1038/sj.bjp.0707323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindén A, Bergendal A, Ullman A. High concentrations of formoterol and salmeterol in the guinea-pig trachea – reassertion of smooth muscle relaxation after beta-blockade followed by washout. Am Rev Respir Dis. 1991;143:A749. [Google Scholar]
- Lovich MA, Creel C, Hong K, Hwang CW, Edelman ER. Carrier proteins determine local pharmacokinetics and arterial distribution of paclitaxel. J Pharm Sci. 2001;90:1324–1335. doi: 10.1002/jps.1085. [DOI] [PubMed] [Google Scholar]
- McCloskey MA, Poo MM. Rates of membrane – associated reactions: Reduction of dimensionality revisited. J Cell Biol. 1986;102:88–96. doi: 10.1083/jcb.102.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea KE, Hill SJ. Salmeterol, a long-acting β2-adrenoceptor agonist mediating cyclic AMP accumulation in a neural cell line. Br J Pharmacol. 1993;110:619–626. doi: 10.1111/j.1476-5381.1993.tb13856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea KE, Hill SJ. Comparison of duration of agonist action at beta 1- and beta 2- adrenoceptors in C6 glioma cells: evidence that the long duration of action of salmeterol is specific to the beta 2-adrenoceptor. Mol Pharmacol. 1996;49:927–923. [PubMed] [Google Scholar]
- Maesen FPV, Smeets JJ, Gubblemans HHL, Zveers PMGA. Bronchodilator effects of inhaled formoterol vs salbutamol over 12 hours. Chest. 1990;97:590–594. doi: 10.1378/chest.97.3.590. [DOI] [PubMed] [Google Scholar]
- Mason RP, Rhodes DG, Herbette LG. Re-evaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model of drug interactions with biological membranes. J Med Chem. 1991;34:869–877. doi: 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- Naline E, Zhang Y, Qian Y, Mairon N, Anderson GP, Grandordy B, et al. Relaxant effects and durations of action of formoterol and salmeterol on the isolated human bronchus. Eur Respir J. 1994;7:914–920. [PubMed] [Google Scholar]
- Nials AT, Coleman RA, Johnson M, Magnussen H, Rabe KF, Vardey CJ. Effects of β-adrenoceptor agonists in human bronchial smooth muscle. Br J Pharmacol. 1993a;110:1112–1116. doi: 10.1111/j.1476-5381.1993.tb13929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nials AT, Sumner MJ, Johnson J, Coleman RA. Investigations into factors determining the duration of action of the β2-adrenoceptor agonist, salmeterol. Br J Pharmacol. 1993b;108:507–515. doi: 10.1111/j.1476-5381.1993.tb12833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nials AT, Ball DI, Butchers PR, Coleman RA, Humbles AA, Johnson M, et al. Formoterol on airway smooth muscle and human lung mast cells: a comparison with salbutamol and salmeterol. Eur J Pharmacol. 1994;251:127–135. doi: 10.1016/0014-2999(94)90392-1. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing ‘preassembled signaling complexes’ at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Packeu A, De Backer J-P, Van Liefde I, Vanderheyden P, Vauquelin G. Antagonist- dopamine D2L- receptor interactions in intact cells. Biochem Pharmacol. 2008;75:2192–2203. doi: 10.1016/j.bcp.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Palmqvist M, Persson G, Lazer L, Rosenborg J, Larsson P, Lötvall J. Inhaled dry-powder formoterol and salmeterol in asthmatic patients: onset of action, duration of effect and potency. Eur Respir J. 1997;10:2484–2489. doi: 10.1183/09031936.97.10112489. [DOI] [PubMed] [Google Scholar]
- Posner RG, Lee B, Conrad DH, Holowka D, Baird B, Goldstein B. Aggregation of IgE-receptor complexes on rat basophilic leukemia cells does not change the intrinsic affinity but can alter the kinetics of the ligand-IgE interaction. Biochem. 1992;31:5350–5356. doi: 10.1021/bi00138a015. [DOI] [PubMed] [Google Scholar]
- Rhodes DG, Newton R, Butler R, Herbette L. Equilibrium and kinetic studies of the interactions of salmeterol with membrane bilayers. Mol Pharmacol. 1992;42:596–602. [PubMed] [Google Scholar]
- Rong Y, Arbabian M, Thiriot DS, Seibold A, Clark RB, Ruoho AE. Probing the salmeterol binding site on the beta 2-adrenergic receptor using a novel photoaffinity ligand, [(125)I]iodoazidosalmeterol. Biochem. 1999;38:11278–11286. doi: 10.1021/bi9910676. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. 100 years lock-and-key concept: are peptide keys shaped and guided to their receptors by the target cell membrane? Biopolymers. 1995;37:5–16. doi: 10.1002/bip.360370104. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. beta(2)-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J Mol Cell Cardiol. 2004;37:407–415. doi: 10.1016/j.yjmcc.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Swales N, Paterson G. The actions of salmeterol and two non-selective β-adrenoceptor agonists isoxuprine and nylidrin on β1- and β2-adrenoceptors. Br J Pharmacol. 1990;100:489P. [Google Scholar]
- Teschemacher A, Lemoine H. Kinetic analysis of drug-receptor interactions of long-acting beta2 sympathomimetics in isolated receptor membranes: evidence against prolonged effects of salmeterol and formoterol on receptor-coupled adenylyl cyclase. J Pharmacol Exp Ther. 1999;288:1084–1092. [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochem. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Morsing P, Fierens FL, De Backer JP, Vanderheyden PM. A two-state receptor model for the interaction between angiotensin II type 1 receptors and non-peptide antagonists. Biochem Pharmacol. 2001;61:277–284. doi: 10.1016/s0006-2952(00)00546-3. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I. From slow antagonist dissociation to long-lasting receptor protection. Trends Pharmacol Sci. 2006;27:355–359. doi: 10.1016/j.tips.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Voss H-P. Long-acting β2- adrenoceptor agonists in asthma: molecular pharmacological aspects. Thesis, Vrije Universiteit Amsterdam, The Netherlands.