

Abstract
G protein coupled receptors and tropomyosin-related kinase (Trk) receptors have distinct structure and transducing mechanisms; therefore, cross-talk among them was unexpected. Evidence has, however, accumulated showing that tonic adenosine A2A receptor activity is a required step to allow synaptic actions of neurotrophic factors, namely upon synaptic transmission at both pre- and post-synaptic level as well as upon synaptic plasticity. An enhancement of A2A receptor tonus upon ageing may partially compensate the loss of TrkB receptors, rescuing to certain degree the facilitatory action of brain derived neurotrophic factor in aged animals, which might prove particularly relevant in the prevention of neurodegeneration upon ageing. A2A receptors also trigger synaptic actions of other neurotrophic factors, such as glial derived neurotrophic factor at dopaminergic striatal nerve endings. The growing evidence that tonic adenosine A2A receptor activity is a crucial step to allow actions of neurotrophic factors in neurones will be reviewed and discussed in the light of therapeutic strategies for neurodegenerative diseases.
Keywords: adenosine, A2A receptors, neurotrophic factors, brain derived neurotrophic factor, neurodegenerative diseases, Parkinson's disease, Hungtington's disease, Alzheimer's disease
Introduction
G protein coupled receptors (GPCRs) are structurally very different from the receptors for neurotrophic factors. GPCRs are a single peptide chain that crosses the lipid bilayer sevenfold, being therefore also known as seven transmembrane domain receptors. They may operate as monomeric, as well as homo- or hetero- dimers, but a monomeric protein per se, once activated, has the ability to interact with a G protein and to trigger a transducing pathway, which most frequently involves changes in adenylate cyclase activity or phosphoinositide metabolism.
Tropomyosin-related kinase (Trk) receptors are a distinct class of membrane receptors, each receptor being a dimer of two identical peptide chains, which share the extracellular binding site for the neurotrophin. These receptors have an intracellular catalytic domain that possesses enzymatic capability and initiates a cascade of phosphorylation steps that involve at least three distinct pathways, the PLCγ pathway involved in fast signalling, the MEK/MAP Kinase mostly involved in differentiation, the PI3K/Akt survival pathway, among others (Kaplan and Miller, 2000; Lee et al., 2002).
Due to structural dissimilarities, interactions between these so distinct classes of membrane receptors were therefore less expected to occur than between two GPCRs. However, three different lines of evidence triggered the interest of evaluating a possible cross-talk between adenosine receptors, which are GPCRs, and receptors for neurotrophic factors, namely:
The ability of cyclic adenosine 3′,5′-monophosphate (cyclic AMP) to gate the facilitatory action of brain derived neurotrophic factor (BDNF) on transmission in developing synapses (Boulanger and Poo, 1999a), together with the well-known positive coupling of adenosine A2 receptors to the adenylate cyclase/cyclic AMP transducing system in the brain (van Calker et al., 1979).
The ability of depolarization to trigger a facilitatoty action of BDNF on transmission in developing synapses (Boulanger and Poo, 1999b), together with the well-known depolarization-induced adenosine release (Latini and Pedata, 2001).
The ability of adenosine A2A receptors to transactivate TrkB BDNF receptors (Lee and Chao, 2001).
In this paper the so far accumulated evidence that tonic adenosine A2A receptor activity is a crucial step to allow actions of neurotrophic factors in neurones will be reviewed and discussed in the light of therapeutic strategies for neurodegenerative diseases.
Neurotrophic factors and their receptors
It is now widely accepted that the influence of neurotrophins on the nervous system spans from neuronal development (Lewin and Barde, 1996) and survival (Murer et al., 2001), to activity-dependent forms of synaptic plasticity (McAllister et al., 1999; Nagappan and Lu, 2005). The neurotrophin family, in mammals, includes nerve growth factor (NGF), BDNF, neurotrophin-3 (NT-3) and neurotrophin 4/5 (NT-4/5). Each neurotrophin shows binding specificity for a Trk receptor, such as TrkA for NGF, TrkB for BDNF and TrkC for NT-3. Neurotrophins also bind to another receptor, the p75. This receptor binds to nearly equal affinity to BDNF, NGF, NT-3 and NT-4. In contrast to Trk receptors, which have a well-known survival and trophic role according to the cascades of phosphorylation steps that are activated, the p75 has no intrinsic kinase property and has activities ranging from trophism to apoptosis (see Blöchl and Blöchl, 2007).
Receptors for other neurotrophic factors, such as the GFRα1/Ret for glial derived neurotrophic factor (GDNF) operate in a way similar to that of Trk receptors, with binding of GDNF to a GFRα1/RET complex to initiate the phosphorylation cascades (Bespalov and Saarma, 2007).
Lack of neurotrophic factors has been involved in several neurodegenerative diseases, namely GDNF in Parkinson's disease (see Bespalov and Saarma, 2007), NGF and/or BDNF in amyotrophic lateral sclerosis (ALS) and Alzheimer's disease (AD) (see e.g. Schulte-Herbrüggen et al., 2007).
Adenosine as an ubiquitous neuromodulator
There are two ways for extracellular adenosine accumulation (Figure 1), one derived from released adenosine 5′-triphosphate (ATP), followed by metabolism into adenosine through a cascade of ectonucleotidases; the other through the equilibrative nucleoside transporters (ENTs). Transport inhibition, therefore, usually leads to an enhancement of extracellular adenosine levels, in particular during high neuronal activity as under this condition substantial amounts of ATP are released together with neurotransmitters.
Figure 1.

Main mechanisms involved in the control of extracellular levels of adenosine in neuronal cells. Adenosine (ADO) can be formed extracellularly from the breakdown of released ATP through a cascade of ectoenzymes, the last step being hydrolysis of AMP by ecto-5′-nucleotidase. Adenosine is also released as such through equilibrative transporters; as the intracellular adenosine concentrations are kept low, mainly due to the activity of adenosine kinase (3), and as considerable amounts of adenosine are formed from released ATP, the main direction of adenosine transport is inwards. However, under some conditions (e.g. low oxygen, low glucose, depolarization), the intracellular adenosine levels rise and outward transport of adenosine occurs. Interconversion of adenosine into S-adenosyl-homocysteine (SAH) or deamination into inosine (INO) also contributes to the low intracellular adenosine levels. AMP, adenosine 5′-monophosphate; ATP, adenosine 5′-triphosphate.
An enhancement of extracellular adenosine levels can also be achieved by inhibition of intracellular enzymes that are responsible for keeping intracellular adenosine concentrations low, such as adenosine kinase that phosphorylates adenosine into AMP. Inhibition of this enzyme selectively amplifies extracellular adenosine concentrations at cell and tissue sites where adenosine release occurs. The therapeutic antiepileptic potential of adenosine kinase inhibition or of its underexpression in implanted cells has been highlighted recently (Li et al., 2007).
Other mechanisms that lead to pronounced changes in the extracellular levels of adenosine are depolarization, and in this case most of the adenosine seems to be released through ENTs (Latini and Pedata, 2001). Similarly, hypoxia, ischaemia or glucose deprivation are highly efficient adenosine releasing stimuli (Fowler, 1993; Frenguelli et al., 2007), and this occurs even before signs of anoxic depolarization. Extracellular accumulation of adenosine during low-energy and oxygen availability is highly important to protect cells. Indeed, adenosine, by activating membrane located inhibitory A1 receptor, is a well-known neuroprotective agent under ischaemic situations (de Mendonça et al., 2000). At synapses, this neuroprotection is directly related to the ability of adenosine A1 receptors to inhibit synaptic transmission during the insult (Sebastião et al., 2001) and this is most probably due to a concerted inhibitory action upon glutamate release at the presynaptic level and upon N-methyl-D-aspartate (NMDA) receptor activation at the post-synaptic level. Adenosine A1 receptor-related mechanisms are involved in the preconditioning-induced neuroprotection (Nakamura et al., 2002; Pugliese et al., 2003).
High-frequency neuronal firing, which leads to an enhancement of ATP release (most of it probably together with neurotransmitters) and subsequent extracellular adenosine formation, is another way to enhance extracellular adenosine levels. As recently pointed out (Bekar et al., 2008), the increase in extracellular adenosine caused by high-frequency neuronal firing may be one of the major causes of the beneficial influence of deep brain stimulation in some pathological conditions.
Four different adenosine receptors have been identified and cloned up to now, namely, the A1, A2A, A2B and A3 receptors. All are GPCRs that lead not only to changes in second messenger levels but also to modulation of ion channels, such as calcium and potassium channels. Through these processes, adenosine modulates neuronal activity, pre-synaptically by inhibiting or facilitating transmitter release, post-synaptically by affecting the action of other neurotransmitters and non-synaptically by hyperpolarizing or depolarizing neurones (see e.g. Ribeiro et al., 2003). The A1 and A2A receptors are high-affinity receptors, whereas the A2B is a low-affinity one. The A1 is mostly an inhibitory receptor, whereas the A2A receptors are mostly excitatory, being positively coupled to adenylate cyclase. For detailed pharmacological characteristics of the different adenosine receptors, see for example Fredholm et al. (2001).
The past few years have brought new insights in our understanding of the role of tripartite synapses, with its pre- post- and glial component, in neurological diseases (Halassa et al., 2007). Adenosine and ATP are among the most relevant players in neuron–glia communication (Fields and Burnstock, 2006). ATP has a dual role as it acts upon its own receptors, mostly of the P2Y subtype, which are abundant in astrocytes and are relevant for calcium signalling; ATP is a substrate of ectonucleotidases, leading to adenosine formation, which then operates its own receptors. Actions of adenosine upon glial cell functions include modulation of glycogen metabolism, neurotransmitter transporters, astrogliosis and astrocyte swelling (Daréet al., 2007). Adenosine receptors at oligodendrocytes regulate white matter development and myelinization (Fields and Burnstock, 2006; Daréet al., 2007).
Adenosine as a modulator of other neuromodulators
Besides its direct pre- and post- synaptic actions on neurones, adenosine is rich in nuances of priming, triggering and braking the action of several neurotransmitters and neuromodulators. Adenosine was, therefore, proposed as a fine tuner, considering that in this way, adenosine is a partner of a very sophisticated interplay between its own receptors and with receptors for other neurotransmitters and/or neuromodulators. Several possibilities exist for this interplay, either at the transducing system level (Sebastião and Ribeiro, 2000) or as a consequence of receptor–receptor heteromerization (Ferréet al., 2007), greatly expanding the number of receptor combination possibilities to modulate cell signalling.
It is not difficult to envisage the different possibilities adenosine receptors may use to interact with other GPCRs. Dimerization of GPCRs, either homo- or heteromerization, is nowadays easily accepted since the strong evidence that GABAB receptors are dimers of two seven transmembrane domain GABAB receptor molecules (White et al., 1998). First hints of A2A/D2 heterodimers in the striatum were several years a head (Ferréet al., 1991) and their functional relevance is now firmly established (see Ferréet al., 2007). Futhermore, GPCRs most frequently share α subunits, not to speak of the βγ subunits, which are common to all G proteins and may change activation equilibrium of other GPCRs. Last, but not least, there are many possibilities of cross-talk with related transduction pathways that may have several kinases and other key molecules in common. Adenosine receptors can also interact with ionotropic receptors with putative implications for neuroprotection, plasticity and learning, as it is the case of AMPA and NMDA glutamate receptors, as well as nicotinic acetylcholine receptors (nAChRs). Some of these interactions involve cyclic AMP – mediated ionotropic receptor phosphorylation followed by enhanced desensitization; others involve more complex transduction pathways. For a detailed review on interactions between adenosine receptors and other GPCRs as well as between adenosine receptors and ionotropic receptors, see Sebastião and Ribeiro (2009).
Interaction between adenosine receptors and receptors for neurotrophic factors
As mentioned above, it is known for several years that presynaptic depolarization (Boulanger and Poo, 1999b), which increases extracellular adenosine levels, as well as enhancement of intracellular cAMP (Boulanger and Poo, 1999a), the most frequent adenosine A2 receptor transducing pathway, triggers synaptic actions of BDNF. On the other hand, adenosine A2A receptors are able to transactivate TrkB receptors in the absence of the neurotrophin (Lee and Chao, 2001). This transactivation requires long-term incubation with GPCR agonists and receptor internalization (Rajagopal et al., 2004) and it is not yet clear whether it operates the same mechanism as the more recently identified ability of adenosine A2A receptors to trigger and promote synaptic and survival actions of neurotrophic factors.
Evidence has been accumulated clearly showing that adenosine A2A receptor activation is a crucial requisite for the functioning of neurotrophic receptors at synapses. This has been shown for the facilitatory actions of BDNF on synaptic transmission (Diógenes et al. 2004; Tebano et al., 2008) and on long term potentiation (LTP) (Fontinha et al., 2008) at the CA1 area of the hippocampus as well as for the action of GDNF at striatal dopaminergic nerve ending (Gomes et al., 2006) and cortico-striatal pathway (Gomes et al., 2009). Interestingly, A2A receptors, adenosine A2A receptors and TrkB BDNF receptors can coexist in the same nerve ending as the facilitatory action of adenosine A2A receptors upon TrkB-mediated BDNF action is also visible at the neuromuscular junction (Pousinha et al., 2006), a single nerve ending synapse model.
The ability of BDNF to facilitate synaptic transmission is dependent on the age of the animals (Diógenes et al., 2007) and this may be related to the degree of activation of adenosine A2A receptors by endogenous adenosine at different ages. Thus, in infant animals, that is, immediately after weaning, to trigger a BDNF facilitatory action, it is necessary to increase the extracellular levels of adenosine, either by inhibiting adenosine kinase or by a brief depolarization (Diógenes et al. 2004; Pousinha et al., 2006) or by inducing high-frequency neuronal firing, such as LTP-inducing paradigms (Fontinha et al., 2008); in all cases, the actions of BDNF are lost by blocking adenosine A2A receptors with selective antagonists. The actions of BDNF are also blocked by inhibition of Trk phosphorylation, but the Trk phosphorylation inhibitor does not prevent A2A receptor-mediated facilitation of synaptic transmission (Pousinha et al., 2006), indicating that the A2A receptor operates upstream of TrkB activation. In adult animals, BDNF per se, through TrkB receptor activation, can facilitate synaptic transmission but this effect is also fully lost with blockade of adenosine A2A receptors (Diógenes et al., 2007) or in A2A receptor knockout mice (Tebano et al., 2008). Nicotinic α-7 cholinergic currents in GABAergic hippocampal neurons are inhibited by BDNF, and this also requires co-activation of adenosine A2A receptors (Fernandes et al., 2008). Inhibition of GABA transporters (GAT) of the predominant neural subtype, GAT1, by BDNF does not fully depend upon co-activation of A2A receptors, as it is not abolished by A2A receptor blockade; however, A2A receptor activation can facilitate this BDNF action (Vaz et al., 2008).
A2A receptors, due to their ability to enhance excitotoxicity phenomena, including glutamate release and action, are mostly regarded as promoters of neuronal death. However, in some cases, such as cultured retinal neurones, A2A receptors have been shown to protect neurones against glutamate-induced excitotoxicity (Ferreira and Paes-de-Carvalho, 2001). Whether this is due to the ability of A2A receptors to facilitate actions of neurotrophic factors, as it has been shown to occur in relation to A2A receptor-mediated neuroprotection of motor neurones (Wiese et al., 2007), requires further investigation. It is worthwhile to note that while Wiese et al. (2007) reported a TrkB-mediated enhancement of survival of injured facial motor neurons in vivo, TrkB receptor activation by BDNF may render spinal cord cultured motor neurons more vulnerable to insult (Mojsilovic-Petrovic et al., 2006). However, another recent study reported an A2A receptor-mediated increase in TrkB signalling through Akt, leading to strengthened synaptic pathways to phrenic motoneurons, as well as to increased breathing in unanesthetized rats, and improved breathing in rats with cervical spinal injuries (Golder et al., 2008). Interestingly enough, A2A receptor antagonism prevented both the favourable (Wiese et al., 2007) and the deleterious (Mojsilovic-Petrovic et al., 2006) TrkB-mediated actions.
Activation of adenosine A2A receptors enhances NGF-induced neurite outgrowth in PC12 cells and rescues NGF-induced neurite outgrowth impaired by blockade of the mitogen-activated protein kinase cascade, an action that requires protein kinase A (PKA) activation (Cheng et al., 2002). Furthermore, activation of adenosine A2A receptors, through Trk-dependent and phosphatidylinositol 3-kinase/Akt-mechanisms, promotes PC12 cell survival after NGF withdrawal (Lee and Chao, 2001). A similar A2A receptor-mediated neuroprotection mechanism has been shown to occur in hippocampal neurones after BDNF withdrawal (Lee and Chao, 2001). Contrasting with A2A receptors which usually promote the actions of neurotrophic factors, adenosine A1 receptors inhibit neurite outgrowth of cultured dorsal root ganglion neurons, both in the absence and in the presence of NGF (Thevananther et al., 2001). It is worthwhile to note that while synaptic actions of adenosine, such as modulation of neurotransmitter release, are visible within minutes after adenosine receptor activation, the trophic neuroprotective adenosine actions might involve prolonged or even tonic activation of adenosine receptors.
Besides interactions at the neurotrophin receptor level, adenosine receptor activation may also induce release of neurotrophic factors. Thus, the expression and/or release of NGF are enhanced by activation of A2A receptors in microglia (Heese et al. 1997) and by activation of A1 receptors in astrocytes (Ciccarelli et al., 1999). Adenosine A2B receptors in astrocytes are also able to enhance GDNF expression (Yamagata et al., 2007). In the whole hippocampus, A2A receptors are required for normal BDNF levels (Tebano et al., 2008). Interestingly, in a mice model of Hungtington's disease, A2A receptors are also required to keep striatal BDNF levels close to those obtained in wild-type mice (Potenza et al., 2007).
Interactions among purinergic, growth factors and cytokine signalling are relevant to regulate neuronal and glial maturation as well as development. In neuronal-dependent glial maturation, both ATP and adenosine purinoceptors are involved (Fields and Burnstock, 2006). The extracellular adenosine levels attained during high-frequency neuronal firing are sufficient to stimulate adenosine receptors in oligodendrocyte ancestor cells inhibiting their proliferation and stimulating their differentiation into myelinating oligodendrocytes (Stevens et al., 2002) but unfortunatelly, the nature of the adenosine receptor involved was not identified in this work. In premyelinating Schwann cells, A2A receptors activate phosphorylation of extracellular signal-regulated kinases (ERKs), namely ERK1/2, and inhibit Schwann cell proliferation without arresting differentiation (Stevens et al., 2004).
Physiological and pathophysiological implications
Brain derived neurotrophic factor has an important role upon synaptic plasticity even in the adult hippocampus (McAllister et al., 1999). BDNF expression and release (Hartmann et al., 2001); (Balkowiec and Katz, 2002), as well as release of adenosine (Pazzagli et al., 1993), or of its precursor ATP (Wieraszko et al., 1989), is more pronounced upon depolarization and during physiologically relevant patterns of neuronal activity, namely those that induce hippocampal LTP. Accordingly, released ATP (Cunha et al., 1996) and high-frequency neuronal stimulation (Correia-de-Sáet al., 1996) favours A2A receptor activation. Therefore, high neuronal activity seems to create ideal physiological conditions for the interplay between adenosine A2A and TrkB receptors to occur. The finding that the facilitatory action of BDNF upon LTP in the CA1 area of the hippocampus is fully lost upon blockade of adenosine A2A receptors as well as upon depletion of extracellular adenosine (Fontinha et al., 2008) highlights the A2A receptor as a new physiologic partner, to the TrkB signalling processes that influences synaptic plasticity phenomena.
Another way A2A receptors have to influence BDNF-related plasticity is through the interplay with the homopentameric α-7 subtype of nAChR, which is particularly relevant for transmitter release and plasticity (Gray et al., 1996; Ji et al., 2001) due to its high-calcium permeability. Adenosine, through A2A receptors, and BDNF, through TrkB receptors, exert double control over α-7-nicotinic currents at GABAergic interneurons in the hippocampus, as it can be concluded from the finding that blockade of A2A receptors abolishes the BDNF-induced current inhibition (Fernandes et al., 2008). As postsynaptic α-7 nAChR-mediated inputs to GABAergic interneurons regulate inhibition within the hippocampus, A2A receptors by allowing the inhibition of cholinergic currents by BDNF might temporarily relieve GABAergic inhibition, therefore facilitating plasticity phenomena.
A decrease in levels and/or action of neurotrophic factors have been implicated in the pathophysiological mechanisms of many diseases of the nervous system, such as Alzheimer's disease (AD), Parkinson's disease, Huntington's disease, diabetic neuropathies, ALS and even depression, therefore making the use of the naturally occurring neurotrophic factors promising for treatment of these disorders (Castrén et al., 2007; Schulte-Herbrüggen et al., 2007) (Figure 2). However, until now the pharmacological administration of neurotrophic factors in vivo has not been easy as these molecules are unable to cross the blood brain barrier, making invasive application strategies like intracerebroventricular infusion necessary. As pointed out by Thoenen and Sendtner (2002), repeated failures of clinical trials using neurotrophic factors claim for improved methods for regulated local supply of these substances to specific populations of neurons together with a more detailed knowledge of the signal transduction pathways activated by neurotrophins via their receptors. The evidence that adenosine A2A receptors trigger or facilitate actions of neurotrophins upon synaptic strength and neuronal survival opens a new therapeutic strategy (Figure 2), as there are adenosine A2A receptor agonists that cross the blood brain barrier, which can be explored as tools to potentiate neurotrophic actions in the brain. The expression (Cunha et al., 1995) and functioning (Rebola et al., 2003) of A2A receptors in the forebrain increases with age, whereas the number of TrkB receptors is markedly lower in the hippocampus of aged rats (Silhol et al., 2005). The increase in the adenosine A2A receptor tonus partially compensates the loss of TrkB receptors upon ageing, rescuing to certain degree the facilitatory action of BDNF in aged animals (Diógenes et al., 2007). This might prove particularly important in the prevention of neurodegeneration, as neurodegenerative diseases are most frequent upon ageing; furthermore, it reinforces the therapeutic potential of adenosine-related therapies to influence the actions of neurotrophic factors in old subjects. Interestingly, daily administration of the A2A receptor agonist, CGS 21680 delays progressive deterioration of motor performance, huntingtin aggregation and increase in striatal choline levels in a transgenic mouse model (R6/2) of Huntington's disease (Chou et al., 2005). This animal model involves genetic mutation of Huntingtin, therefore most probably, a reduction of striatal BDNF levels. Indeed, there is strong evidence that a major contributing pathway to striatal degeneration in Huntington's disease is an impairment of anterograde transport BDNF from the cortex to the striatum, due to loss of function of mutated huntingtin (Zuccato et al., 2001; Baquet et al., 2004; Gauthier et al., 2004; Strand et al., 2007). How the low BDNF signalling can be compensated by A2A receptor activation deserves detailed investigation.
Figure 2.
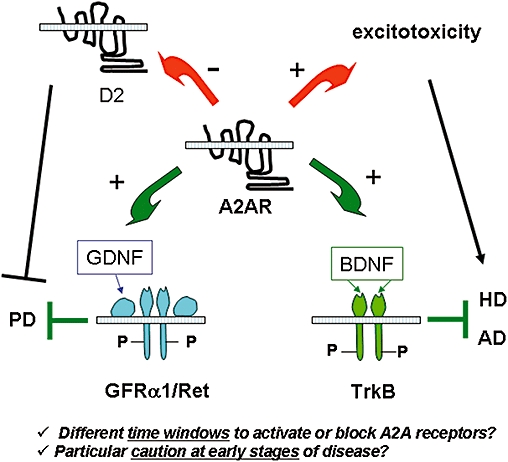
Interplay between adenosine A2A receptors and receptors for neurotrophic factors and its putative influence. Beneficial influences that may contribute to slow down disease progression are represented by the green arrows; those that operate as aggravating factors are represented by red arrows. A2A receptors facilitate actions of GDNF in the striatum as well as the actions of BDNF in the hippocampus. These neurotrophic factors and their receptors (GFRα/Ret for GDNF; and TrkB for BDNF) may slow down progression of Parkinson's disease (PD) or Alzheimer's disease (AD) and this is probably most relevant in early stages of the disease, where neurotrophic influences start to diminish. However, A2A receptors negatively interact with dopamine D2 receptors in medium spiny striatal neurones; under late stages of the disease, blockade of A2A receptors may be advantageous, to relieve this negative interaction that constitutes an aggravating factor in PD. A2A receptors, though facilitation of glutamate release, may also enhance excitotoxicity phenomena, and this might be particularly relevant when neuronal death involves enhanced glutamate signalling. Therefore, both the timing and the nature of neuronal injury may determine whether blockade or activation of adenosine A2A receptors is desirable to protect neurones. BDNF, brain derived neurotrophic factor; GDNF, glial derived neurotrophic factor; Trk, tropomyosin-related kinase.
A particular mention has to be made about epilepsy, where neurotrophic factors have been considered both harmful, being causal mediators in the development of acquired epileptic syndromes, and eventually useful to treat epilepsy-associated damage (Simonato et al., 2006; Scharfman and Hen, 2007). On the top of this controversy, we can add discrepant findings of both anti-convulsive (Huber et al., 2002) and pro-convulsive (Zeraati et al., 2006) adenosine A2A receptor-mediated actions, the pro-convulsive being much more expected due to the usually excitatory nature of these receptors. The evaluation of a putative interplay between A2A and TrkB receptors in epileptogenic conditions could help and provide hints to solve some of these discrepancies.
Results from clinical and basic studies have demonstrated that stress and depression decrease BDNF expression and neurogenesis, leading to the neurotrophic hypothesis of depression (Castrén et al., 2007; Kozisek et al., 2008). As A2A receptor activation may have anti-depressive action (Kaster et al., 2004), one may, therefore, speculate that the ability of A2A receptors to facilitation the actions BDNF may contribute to the antidepressive actions of adenosine. It is worthwhile to note that deep brain stimulation, now widely used by neurosurgeons to treat tremor and other movement disorders, as well as in a number of psychiatric diseases, including obsessive-compulsive disorders and depression (Larson, 2008), produces its effects by inducing the release of ATP which is subsequently converted extracellularly to adenosine (Bekar et al., 2008). Whether adenosine, through facilitation of BDNF actions, contributes to the antidepresssive properties of deep brain stimulation also awaits further evaluation.
Finally, the cross-talk between adenosine A2A receptors and receptors for neurotrophins also points to the need of caution about therapies with A2A receptor antagonists in neurodegenerative diseases, as it has been proposed for Parkinson's disease to ameliorate L-DOPA-induced dyskinesias (see Morelli et al., 2007). Indeed, the identification of postsynaptic A2A/D2 receptor interactions in the striatum together with the findings that A2A receptor antagonists are neuroprotective in Parkinson's disease models (Chase et al., 2003) and increase dopamine synthesis from L-DOPA (Golembiowska and Dziubina, 2004) led to the proposal for the use of A2A receptor antagonists in Parkinson's disease. On the other hand, neurotrophic factors, in particular GDNF, may be a potential therapeutic approach in the management of Parkinson's disease (Love et al., 2005; Patel et al., 2005). Enhancing GDNF actions, as it is the case of adenosine A2A receptor agonists (Gomes et al., 2006) might also be of high therapeutic interest. In any case, the finding that GDNF actions on dopamine release in the striatum are prevented by A2A receptor antagonism (Gomes et al., 2006) points towards the need for further studies on the consequences of long-term therapy with A2A receptor blockers in neurodegenerative diseases where neurotrophic factors may play a beneficial role. One issue that should be explored in the future is the optimal time window for combined beneficial effects for neurotrophic factors and adenosine A2A receptor agonists/antagonists. Perhaps, in the late stages of neurodegenerative diseases, A2A receptor antagonists may be advantageous to prevent and/or attenuate diskenesias; however, in the early stages, where neurones are struggling for life and an enhancement of neurotrophic factors is highly desirable, A2A receptor antagonists should be avoided and perhaps A2A agonists could be considered to potentiate neurotrophic influences.
Conclusions
The neuromodulator adenosine, through A2A receptor activation, has profound influence upon the actions of neurotrophic factors. Due to the role of neurotrophic factors upon neuronal survival, neuronal plasticity and neuronal differentiation, the adenosine-induced control of neurotrophic factors opens new windows of adenosinergic influence on neuronal cells and novel therapeutic perspectives in neuronal dysfunction.
Acknowledgments
The work in the authors' laboratory is supported by research grants from Fundação para a Ciência e Tecnologia (FCT), Gulbenkian Foundation and European Union (COST B30).
Glossary
Abbreviations:
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- BDNF
brain derived neurotrophic factor
- ENTs
equilibrative nucleoside transporters
- GAT
GABA transporters
- GDNF
glial derived neurotrophic factor
- GPCRs
G protein coupled receptors
- LTP
long term potentiation
- nAChRs
nicotinic acetylcholine receptors
- NT-3
neurotrophin-3
- NT-4/5
neurotrophin 4/5
- NGF
nerve growth factor
- Trk
tropomyosin-related kinase
Conflicts of interest
None.
References
- Balkowiec A, Katz D. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekar L, Libionka W, Tian GF, Xu Q, Torres A, Wang X, et al. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med. 2008;14:17–19. doi: 10.1038/nm1693. [DOI] [PubMed] [Google Scholar]
- Bespalov MM, Saarma M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol Sci. 2007;28:68–74. doi: 10.1016/j.tips.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Blöchl A, Blöchl R. A cell-biological model of p75NTR signaling. J Neurochem. 2007;102:289–305. doi: 10.1111/j.1471-4159.2007.04496.x. [DOI] [PubMed] [Google Scholar]
- Boulanger L, Poo M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999a;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- Boulanger L, Poo M. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nature Neurosci. 1999b;2:346–351. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- van Calker D, Müller M, Hamprecht B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem. 1979;33:999–1005. doi: 10.1111/j.1471-4159.1979.tb05236.x. [DOI] [PubMed] [Google Scholar]
- Castrén E, Võikar V, Rantamäki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol. 2007;7:18–21. doi: 10.1016/j.coph.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Chase TN, Bibbiani F, Bara-Jimenez W, Dimitrova T, Oh-Lee JD. Translating A2A antagonist KW6002 from animal models to Parkinsonian patients. Neurology. 2003;61:S107–S111. doi: 10.1212/01.wnl.0000095223.08711.48. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Shih HM, Chern Y. Essential role of cAMP-response element-binding protein activation by A2A adenosine receptors in rescuing the nerve growth factor-induced neurite outgrowth impaired by blockage of the MAPK cascade. J Biol Chem. 2002;277:33930–33942. doi: 10.1074/jbc.M201206200. [DOI] [PubMed] [Google Scholar]
- Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, et al. CGS21680 attenuates symptoms of Huntington's disease in a transgenic mouse model. J Neurochem. 2005;93:310–320. doi: 10.1111/j.1471-4159.2005.03029.x. [DOI] [PubMed] [Google Scholar]
- Ciccarelli R, Di Iorio P, Bruno V, Battaglia G, D'Alimonte I, D'Onofrio M, et al. Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia. 1999;27:275–281. [PubMed] [Google Scholar]
- Correia-de-Sá P, Timóteo MA, Ribeiro JA. Presynaptic A1 inhibitory/A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J Neurophysiol. 1996;76:3910–3919. doi: 10.1152/jn.1996.76.6.3910. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Constantino MC, Sebastião AM, Ribeiro JA. Modification of A1 and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. Neuroreport. 1995;6:1583–1588. doi: 10.1097/00001756-199507310-00029. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Correia-de-Sá P, Sebastião AM, Ribeiro JA. Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. Br J Pharmacol. 1996;119:253–260. doi: 10.1111/j.1476-5381.1996.tb15979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daré E, Schulte G, Karovic O, Hammarberg C, Fredholm BB. Modulation of glial cell functions by adenosine receptors. Physiol Behav. 2007;92:15–20. doi: 10.1016/j.physbeh.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Diógenes MJ, Fernandes CC, Sebastião AM, Ribeiro JA. Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. J Neurosci. 2004;24:2905–2913. doi: 10.1523/JNEUROSCI.4454-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diógenes MJ, Assaife-Lopes N, Pinto-Duarte A, Ribeiro JA, Sebastião AM. Influence of age on BDNF modulation of hippocampal synaptic transmission, interplay with adenosine A2A receptors. Hippocampus. 2007;17:577–585. doi: 10.1002/hipo.20294. [DOI] [PubMed] [Google Scholar]
- Fernandes CC, Pinto-Duarte A, Ribeiro JA, Sebastião AM. Postsynaptic action of brain-derived neurotrophic factor attenuates alpha7 nicotinic acetylcholine receptor-mediated responses in hippocampal interneurons. J Neurosci. 2008;28:5611–5618. doi: 10.1523/JNEUROSCI.5378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, von Euler G, Johansson B, Fredholm BB, Fuxe K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc Natl Acad Sci USA. 1991;88:7238–7241. doi: 10.1073/pnas.88.16.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007;30:440–446. doi: 10.1016/j.tins.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Ferreira JM, Paes-de-Carvalho R. Long-term activation of adenosine A(2a) receptors blocks glutamate excitotoxicity in cultures of avian retinal neurons. Brain Res. 2001;900:169–176. doi: 10.1016/s0006-8993(01)02279-x. [DOI] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Fontinha BM, Diógenes MJ, Ribeiro JA, Sebastião AM. Enhancement of long-term potentiation by brain-derived neurotrophic factor requires adenosine A(2A) receptor activation by endogenous adenosine. Neuropharmacology. 2008;54:924–933. doi: 10.1016/j.neuropharm.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Changes in extracellular adenosine levels and population spike amplitude during graded hypoxia in the rat hippocampal slice. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:73–78. doi: 10.1007/BF00168775. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Wigmore G, Llaudet E, Dale N. Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J Neurochem. 2007;101:1400–1413. doi: 10.1111/j.1471-4159.2006.04425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, et al. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci. 2008;28:2033–2042. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golembiowska K, Dziubina A. Striatal adenosine A(2A) receptor blockade increases extracellular dopamine release following l-DOPA administration in intact and dopamine-denervated rats. Neuropharmacology. 2004;47:414–426. doi: 10.1016/j.neuropharm.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Gomes CA, Vaz SH, Ribeiro JA, Sebastião AM. Glial cell line-derived neurotrophic factor (GDNF) enhances dopamine release from striatal nerve endings in an adenosine A2A receptor-dependent manner. Brain Res. 2006;1113:129–136. doi: 10.1016/j.brainres.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Gomes CA, Simões PF, Canas PM, Quiroz C, Sebastião AM, Ferré S, et al. GDNF control of the glutamatergic cortico-striatal pathway requires tonic activation of adenosine A receptors. J Neurochem. 2009;108:1208–1219. doi: 10.1111/j.1471-4159.2009.05876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse, roles for gliotransmission in health and disease.Trends. Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. The EMBO Journal. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heese K, Fiebich BL, Bauer J, Otten U. Nerve growth factor (NGF) expression in rat microglia is induced by adenosine A2a-receptors. Neurosci Lett. 1997;231:83–86. doi: 10.1016/s0304-3940(97)00545-4. [DOI] [PubMed] [Google Scholar]
- Huber A, Güttinger M, Möhler H, Boison D. Seizure suppression by adenosine A(2A) receptor activation in a rat model of audiogenic brainstem epilepsy. Neurosci Lett. 2002;329:289–292. doi: 10.1016/s0304-3940(02)00684-5. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kaster MP, Rosa AO, Rosso MM, Goulart EC, Santos AR, Rodrigues AL. Adenosine administration produces an antidepressant-like effect in mice: evidence for the involvement of A1 and A2A. receptorsNeurosci Lett. 2004;355:21–24. doi: 10.1016/j.neulet.2003.10.040. [DOI] [PubMed] [Google Scholar]
- Kozisek ME, Middlemas D, Bylund DB. Brain-derived neurotrophic factor and its receptor tropomyosin-related kinase B in the mechanism of action of antidepressant therapies. Pharmacol Ther. 2008;117:30–51. doi: 10.1016/j.pharmthera.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Larson PS. Deep brain stimulation for psychiatric disorders. Neurotherapeutics. 2008;5:50–58. doi: 10.1016/j.nurt.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini S, Pedata F. Adenosine in the central nervous system, release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci USA. 2001;98:3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Chao MV. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 2002;13:11–17. doi: 10.1016/s1359-6101(01)00024-7. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Barde Y-A. Physiology of the neurotrophins. Annu Rev. Neuroscience. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A Segschneider M, et al. Suppression of kindling epileptogenesis by adenosine releasing stem cell derived brain implants. Brain. 2007;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med. 2005;11:703–704. doi: 10.1038/nm0705-703. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Ver Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- de Mendonça A, Sebastião AM, Ribeiro JA. Adenosine, does it have a neuroprotective role after all? Brain Res Rev. 2000;33:258–274. doi: 10.1016/s0165-0173(00)00033-3. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Jeong GB, Crocker A, Arneja A, David S, Russell DS, et al. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci. 2006;26:9250–9263. doi: 10.1523/JNEUROSCI.1856-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli M, Di Paolo T, Wardas J, Calon F, Xiao D, Schwarzschild MA. Role of adenosine A2A receptors in parkinsonian motor impairment and l-DOPA-induced motor complications. Prog Neurobiol. 2007;83:293–309. doi: 10.1016/j.pneurobio.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB, mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nakakimura K, Matsumoto M, Sakabe T. Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A1 receptor antagonist. J Cereb Blood Flow Metab. 2002;22:161–170. doi: 10.1097/00004647-200202000-00004. [DOI] [PubMed] [Google Scholar]
- Patel NK, Bunnage M, Plaha P, Svendsen CN, Heywood P, Gill SS. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD, a two-year outcome study. Ann. Neurol. 2005;57:298–302. doi: 10.1002/ana.20374. [DOI] [PubMed] [Google Scholar]
- Pazzagli M, Pedata F, Pepeu G. Effect of K+ depolarization, tetrodotoxin, and NMDA receptor inhibition on extracellular adenosine levels in rat striatum. Eur J Pharmacol. 1993;234:61–65. doi: 10.1016/0014-2999(93)90706-n. [DOI] [PubMed] [Google Scholar]
- Pousinha PA, Diogenes MJ, Ribeiro JA, Sebastião AM. Triggering of BDNF facilitatory action on neuromuscular transmission by adenosine A2A receptors. Neurosci Lett. 2006;404:143–147. doi: 10.1016/j.neulet.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Potenza RL, Tebano MT, Martire A, Domenici MR, Pepponi R, Armida M, et al. Adenosine A(2A) receptors modulate BDNF both in normal conditions and in experimental models of Huntington's disease. Purinergic Signal. 2007;3:333–338. doi: 10.1007/s11302-007-9066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese AM, Latini S, Corradetti R, Pedata F. Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus, role of adenosine receptors. Br J Pharmacol. 2003;140:305–314. doi: 10.1038/sj.bjp.0705442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Sebastião AM, de Mendonca A, Oliveira CR, Ribeiro JA, Cunha RA. Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J Neurophysiol. 2003;90:1295–1303. doi: 10.1152/jn.00896.2002. [DOI] [PubMed] [Google Scholar]
- Ribeiro JA, Sebastião AM, de Mendonça A. Adenosine receptors in the nervous system, pathophysiological implications. Prog. Neurobiol. 2003;68:377–392. doi: 10.1016/s0301-0082(02)00155-7. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Hen R. Is more neurogenesis always better? Science. 2007;315:336–338. doi: 10.1126/science.1138711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Herbrüggen O, Braun A, Rochlitzer S, Jockers-Scherübl MC, Hellweg R. Neurotrophic factors – a tool for therapeutic strategies in neurological, neuropsychiatric and neuroimmunological diseases? Curr Med Chem. 2007;14:2318–2329. doi: 10.2174/092986707781745578. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Fine-tuning neuromodulation by adenosine. Trends Pharmacol Sci. 2000;21:341–346. doi: 10.1016/s0165-6147(00)01517-0. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Adenosine receptors and the central nervous system. In: Wilson CN, Mustafa SJ, editors. Adenosine Receptors in Health and Disease, Handbook of Experimental Pharmacology. Berlin Heidelberg: Springer-Verlag; 2009. 193 DOI 10.1007/978-3-540-89615-9-16. [Google Scholar]
- Sebastião AM, de Mendonça A, Moreira T, Ribeiro JA. Activation of synaptic NMDA receptors by action potential-dependent release of transmitter during hypoxia impairs recovery of synaptic transmission on reoxygenation. J. Neurosci. 2001;21:8564–8571. doi: 10.1523/JNEUROSCI.21-21-08564.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhol M, Bonnichon V, Rage F, Tapia-Arancibia L. Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience. 2005;132:613–624. doi: 10.1016/j.neuroscience.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Simonato M, Tongiorgi E, Kokaia M. Angels and demons, neurotrophic factors and epilepsy. Trends Pharmacol Sci. 2006;27:631–638. doi: 10.1016/j.tips.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine, a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855–868. doi: 10.1016/s0896-6273(02)01067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Ishibashi T, Chen JF, Fields RD. Adenosine, an activity-dependent axonal signal regulating MAP kinase and proliferation in developing Schwann cells. Neuron Glia Biol. 2004;1:23–34. doi: 10.1017/s1740925x04000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Baquet ZC, Aragaki AK, Holmans P, Yang L, Cleren C, et al. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebano MT, Martire A, Potenza RL, Grò C, Pepponi R, Armida M, et al. Adenosine A(2A) receptors are required for normal BDNF levels and BDNF-induced potentiation of synaptic transmission in the mouse hippocampus. J Neurochem. 2008;104:279–286. doi: 10.1111/j.1471-4159.2007.05046.x. [DOI] [PubMed] [Google Scholar]
- Thevananther S, Rivera A, Rivkees SA. A1 adenosine receptor activation inhibits neurite process formation by Rho kinase-mediated pathways. Neuroreport. 2001;12:3057–3063. doi: 10.1097/00001756-200110080-00015. [DOI] [PubMed] [Google Scholar]
- Thoenen H, Sendtner M. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci. 2002;5(Suppl):1046–1050. doi: 10.1038/nn938. [DOI] [PubMed] [Google Scholar]
- Vaz S, Cristóvão-Ferreira S, Ribeiro JA, Sebastiao AM. Brain-derived neurotrophic factor inhibits GABA uptake by the rat hippocampal nerve terminals. Brain Res. 2008;1219:19–25. doi: 10.1016/j.brainres.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Wieraszko A, Goldsmith G, Seyfried TN. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Research. 1989;485:244–250. doi: 10.1016/0006-8993(89)90567-2. [DOI] [PubMed] [Google Scholar]
- Wiese S, Jablonka S, Holtmann B, Orel N, Rajagopal R, Chao MV, et al. Adenosine receptor A2A-R contributes to motoneuron survival by transactivating the tyrosine kinase receptor TrkB. Proc Natl Acad Sci USA. 2007;104(43):17210–17215. doi: 10.1073/pnas.0705267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, et al. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Hakata K, Maeda A, Mochizuki C, Matsufuji H, Chino M, et al. Adenosine induces expression of glial cell line-derived neurotrophic factor (GDNF) in primary rat astrocytes. Neurosci Res. 2007;59:467–474. doi: 10.1016/j.neures.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Zeraati M, Mirnajafi-Zadeh J, Fathollahi Y, Namvar S, Rezvani ME. Adenosine A1 and A2A receptors of hippocampal CA1 region have opposite effects on piriform cortex kindled seizures in rats. Seizure. 2006;15:41–48. doi: 10.1016/j.seizure.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]