

Abstract
ExsA is a transcriptional activator of the Pseudomonas aeruginosa type III secretion system (T3SS). The T3SS consists of >40 genes organized within 10 transcriptional units, each of which is controlled by the transcriptional activator ExsA. ExsA-dependent promoters contain two adjacent ExsA binding sites that when occupied protect the −30 to −70 region from DNase I cleavage. The promoters also possess regions bearing strong resemblance to the consensus −10 and −35 regions of σ70-dependent promoters. The spacing distance between the putative −10 and −35 regions of ExsA-dependent promoters, however, is increased by 4 to 5 bp compared to that in typical σ70-dependent promoters. In the present study, we demonstrate that ExsA-dependent transcriptional activation requires a 21- or 22-bp spacer length between the −10 and −35 regions. Despite the atypical spacing in this region, in vitro transcription assays using σ70-saturated RNA polymerase holoenzyme (RNAP-σ70) confirm that ExsA-dependent promoters are indeed σ70 dependent. Potassium permanganate footprinting experiments indicate that ExsA facilitates an early step in transcriptional initiation. Although RNAP-σ70 binds to the promoters with low affinity in the absence of ExsA, the activator stimulates transcription by enhancing recruitment of RNAP-σ70 to the PexsC and PexsD promoters. Abortive initiation assays confirm that ExsA enhances the equilibrium binding constant for RNAP while having only a modest effect on the isomerization rate constant.
Pseudomonas aeruginosa is an opportunistic pathogen of humans, causing acute and chronic infections in immunocompromised individuals (43, 44). A primary determinant of P. aeruginosa virulence is a type III secretion system (T3SS) (16, 55). The T3SS promotes tissue destruction and phagocytic avoidance through the action of several toxins that are translocated into eukaryotic host cells (4, 45). Mutants lacking a functional T3SS are attenuated for virulence in both tissue culture and animal infection models (3, 30).
The primary regulator of T3SS gene expression is ExsA (17, 53, 54). ExsA controls T3SS gene expression by directly binding to and activating transcription from all T3SS promoters (6, 31). ExsA is a member of the large family of AraC/XylS transcriptional regulators (17). The domain structure of these proteins generally consists of a conserved 100-amino-acid helix-turn-helix DNA binding domain located at the carboxy terminus and an amino-terminal dimerization and/or ligand binding domain (15). One way in which AraC/XylS family members can be distinguished from one another is by the type of bound ligand; ligands include sugars, small metabolites, urea, aromatic compounds, and proteins (20, 42). Family members responsive to protein ligands currently constitute a small group of AraC/XylS activators that regulate T3SS gene expression (42). Representatives of this subfamily are found in P. aeruginosa (ExsA), Salmonella enterica (InvF), and Shigella flexneri (MxiE) (8, 11, 12). Transcriptional activation by P. aeruginosa ExsA is antagonized by ExsD through a direct binding interaction (38). ExsD functions as an antiactivator by inhibiting the DNA binding activity of ExsA (48). Similarly, transcriptional activation by MxiE is antagonized by the OspD1 antiactivator through a direct binding interaction (40). In contrast, transcriptional activation by S. enterica InvF and S. flexneri MxiE is dependent upon protein coactivators that directly bind to their respective activators (11, 41).
Transcriptional start sites for several ExsA-dependent promoters have been mapped by primer extension (53, 54). The transcriptional start sites for the PexsD, PexoS, and Porf1 promoters are favorably positioned downstream from near-consensus −10 (TATAAT) and −35 (TTGACA) recognition hexamers typical of σ70-dependent promoters in both Escherichia coli and P. aeruginosa (14, 26). Atypical, however, is the apparent increase in spacing (21 to 22 bp) between the −10 and −35 elements of ExsA-dependent promoters compared to the optimal spacing of 17 bp for typical σ70-dependent promoters (6). Whether these near-consensus promoter sequences of ExsA-dependent promoters truly serve as recognition hexamers for σ70-saturated RNA polymerase holoenzyme (RNAP-σ70) is not known.
The DNA binding properties of ExsA have been characterized through genetic and biochemical studies (6, 7, 31). Purified ExsA is monomeric in solution and specifically binds to T3SS promoters with apparent equilibrium constants in the low nanomolar range (1 to 5 nM). Two distinct ExsA-promoter probe complexes are detected by electrophoretic mobility shift assays. Whereas the higher-mobility complex represents one ExsA monomer bound to the promoter probe, the lower-mobility complex represents two bound ExsA molecules (6). An alignment of all 10 ExsA-dependent promoters identified an ExsA consensus binding site that is centered around highly conserved guanine and cytosine nucleotides at the −47 and −45 positions, respectively (6). Further determinants for ExsA binding include a conserved adenine-rich region centered at the −51 position and several highly conserved nucleotides within the −35 region. The available data suggest that there are two distinct ExsA binding sites, one overlapping the putative −35 region (site 1) and a second consisting of the adenine-rich region (site 2) (6). In support of this, DNase I footprinting assays reveal an ExsA-dependent region of protection that begins at the −35 region and extends upstream to the −70 position relative to the transcriptional start site (6, 31). This region of protection is similar to that of other AraC family transcriptional activators such as RhaR and RhaS, both of which bind to two adjacent promoter sites (51). Furthermore, nucleotide substitutions within the adenine-rich, −47G/−45C, and −35 regions result in a significant decrease in ExsA-dependent transcriptional activation (6). The same substitutions, however, have variable effects on DNA binding. Whereas substitutions in the −47G/−45C or −35-like region significantly impair ExsA binding to sites 1 and 2, substitutions in the A-rich region inhibit binding only to site 2. These data suggest that two adjacent ExsA binding sites are required for full transcriptional activation and that binding of monomeric ExsA to site 1 is required for binding of the second ExsA molecule to site 2. Occupation of site 2 is dependent upon the amino terminus of ExsA (7), which likely includes a multimerization domain, typical of most AraC family members (15). This sequential monomer binding route of promoter complex assembly is a common protein-DNA interaction scheme that permits kinetic discrimination of specific and nonspecific DNA sequences (35).
Transcriptional activators generally function by recruiting RNAP to nonstandard promoters and/or facilitating isomerization to an open complex. In this study we characterized the mechanism of transcriptional activation by ExsA. Genetic data demonstrate that the extended spacing between the −10 and −35 regions of T3SS promoters is an essential determinant for ExsA-dependent activation. Whereas RNAP-σ70 binds to T3SS promoters inefficiently in the absence of ExsA, our data demonstrate that ExsA facilitates transcription by recruiting RNAP-σ70 to the PexsC and PexsD promoters, where it then forms an active open complex.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are summarized in Table S1 in the supplemental material. Escherichia coli strains were maintained on L-agar plates containing the following antibiotics as necessary: gentamicin, 15 μg/ml; ampicillin, 50 or 100 μg/ml; tetracycline, 10 μg/ml; kanamycin, 50 μg/ml; and spectinomycin, 50 μg/ml. P. aeruginosa strains were maintained on Vogel Bonner minimal medium (49) with the following antibiotics as indicated: gentamicin, 100 μg/ml; carbenicillin, 300 μg/ml; and tetracycline, 50 μg/ml. To assay for T3SS gene expression P. aeruginosa strains were grown with vigorous aeration at 30°C in tryptic soy broth supplemented with 100 mM monosodium glutamate, 1% glycerol, and 2 mM EGTA as indicated. β-Galactosidase assays were performed as previously described, and the reported values are the averages of those from at least three independent experiments (13).
Plasmid construction and promoter mutagenesis.
The primer sequences used to generate PCR products and the vectors into which each product was cloned are provided in Tables S2 and S3 in the supplemental material, respectively. Addition or deletion mutations in the PexoT and PexsD promoters were constructed using a two-step PCR. In the first step, megaprimers were generated by PCR using a forward primer incorporating the mutation and a common reverse primer. The megaprimers were gel purified (IBI Scientific, Peosta, IA) and used in a subsequent PCR with a common forward primer. The final PCR products were cloned as HindIII/EcoRI restriction fragments into mini-CTX-lacZ and integrated onto the PA103 chromosome as previously described (27). The PtacI constructs were generated by cloning annealed complementary oligonucleotides with KpnI/HindIII ends into mini-CTX-lacZ. Point mutations in the PtacI promoter were introduced by QuikChange site-directed mutagenesis (Stratagene). To limit β-galactosidase toxicity, E. coli subcloning strains were transformed with the LacIq-overexpressing plasmid pMS421 (21).
Purification of ExsAHis and RNAP.
ExsAHis was purified by metal affinity chromatography and shown to possess DNA binding activity by electrophoretic mobility shift assay as previously described (6). RNAP was purified from P. aeruginosa strain AK1012 (lacking expression of lipopolysaccharide O antigen) (33) as previously described (9) with the following modifications. Overnight cultures were diluted to an optical density at 600 nm (OD600) of 0.1 into 5 liters of tryptic soy broth and grown with shaking at 37°C. At an OD600 of 1.0, cells were harvested by centrifugation (10 min, 6,000 × g, 4°C), washed with 500 ml of 0.85% NaCl, collected by centrifugation, and resuspended in 60 ml purification buffer (20 mM Tris-HCl [pH 8.0], 0.05 mM EDTA, 1.7 mM phenylmethylsulfonyl fluoride, 0.3 mM dithiothreitol [DTT], and 5% glycerol) containing 0.1 M NaCl. Cells were lysed via passage through a French pressure cell, and unbroken cells were removed by centrifugation (30 min, 35,000 × g, 4°C). Polyethyleneimine (0.5% [wt/vol] final concentration; Sigma) was added to the soluble fraction and incubated at 4°C for 30 min with constant stirring. The precipitate was collected by centrifugation (30 min, 35,000 × g, 4°C) and washed with purification buffer containing 0.25 M NaCl using a Dounce homogenizer. Following centrifugation, RNAP was extracted with purification buffer containing 0.8 M NaCl. Insoluble material was removed by centrifugation, and solid ammonium sulfate (30% final concentration) was added to the soluble fraction and incubated at 4°C for 2 h with constant stirring. Insoluble material was removed by centrifugation, and ammonium sulfate (60% final concentration) was added to the soluble fraction and allowed to precipitate as described above. The precipitate was collected by centrifugation, homogenized in 11 ml of purification buffer containing 0.1 M NaCl, and dialyzed for 18 h at 4°C against 2 liters of purification buffer containing 0.1 M NaCl. Prior to heparin column chromatography, the material was subjected to ultracentrifugation (100,000 × g for 30 min at 4°C) to ensure solubility. Soluble material was loaded onto a 5-ml heparin column and developed with a linear elution gradient (0.1 to 1 M NaCl), and peak fractions (based on polymerase activity) were pooled. The heparin column procedure was repeated a second time, followed by a final purification using Superdex-300 gel filtration chromatography. Purified polymerase was stored at −20°C in purification buffer containing 0.1 M NaCl and 50% glycerol. The specific activity of RNAP was determined as described previously (2). One unit of RNAP activity is defined as the amount of enzyme required to incorporate 1 pmol of UMP into acid-precipitable material in 20 min. Protein samples were denatured in sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer and electrophoresed on 15% acrylamide denaturing gels. Gels were analyzed by Coomassie blue and silver staining methods.
Transcription templates.
Supercoiled plasmid templates containing the PexsD promoter fused to the rpoC terminator were generated and purified as described previously (7). Supercoiled minicircle templates were created by cloning PCR-generated PexsC and PexsD promoters (nucleotides [nt] −238 to +192 relative to the transcriptional start site) as SacI/KpnI fragments into pSA508 (10). The resulting plasmids add 29 additional bases upstream of the rpoC transcriptional terminator to generate 221-base transcripts from each promoter. Minicircle purification was as described previously (10) with the following modifications. T3SS promoter-containing pSA508 derivatives were introduced by transformation into E. coli strain SA1751 [λint+ xis439 cI857 (cro-chlA)ΔH1]. Transformants were grown in 800 ml of LB containing ampicillin (50 μg/ml) to an OD600 of 0.8 at 30°C, heat shocked at 42°C for 15 min, returned to 30°C in an ice-water bath, and grown at 30°C for an additional 30 min. Cells were harvested by centrifugation, and plasmid DNA was isolated with the Fast-Ion plasmid maxi kit (IBI Scientific, Peosta, IA). Transcription templates were subjected to agarose gel electrophoresis and visualized by methylene blue staining. Supercoiled DNA was excised and gel purified, and analytical samples were examined by agarose gel electrophoresis to confirm that the preparations were largely free of nicked template.
In vitro transcription assays.
Single-round transcription assays (20-μl final volume) were performed by incubating ExsAHis (35 nM) with supercoiled transcription templates (2 nM) at 25°C in 1× transcription buffer (40 nM Tris-HCl [pH 7.5], 50 mM KCl, 10 mM MgCl2, 0.01% Tween 20, and 1 mM DTT) containing the initiating nucleotides ATP and GTP (0.75 mM) for the PexsD and PexsC promoters, respectively. After 10 min, 25 nM E. coli RNAP holoenzyme (Epicentre, Madison, WI) or P. aeruginosa RNAP holoenzyme was added, and open complexes were allowed to form for 5 min at 30°C. Elongation was allowed to proceed by the addition of the remaining unlabeled nucleotides (0.75 mM each, including 5 μCi [α-32P]CTP) in 1× transcription buffer containing heparin (50-μg/ml final concentration). Reactions were stopped after 10 min at 30°C by the addition of stop buffer (20 μl) (98% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol). Samples were heated to 95°C for 5 min and electrophoresed immediately on 5% denaturing urea-polyacrylamide gels.
Transcriptional start site identification.
RNA was isolated from mid-log-phase (OD600 of 0.8) bacterial cells using RNAprotect reagent and an RNA mini isolation kit (Qiagen, Valencia, CA). Purified mRNA (200 ng) and gene-specific, antisense primers (positioned ∼500 bases downstream of the translational start sites) were used in reverse transcription reactions to generate cDNA for the PexsD and PexsC promoters with the SuperScript III first-strand synthesis system (Invitrogen). Reverse transcriptase (RT) reactions were allowed to proceed at 55°C for 30 min and were terminated by incubation at 70°C for 15 min. cDNA was purified using the MinElute PCR purification kit (Qiagen). The 5′ ends of the resulting cDNAs were identified using the PCR-based method of 5′ rapid amplification of cDNA ends (RACE) (46); 10 independent clones for each promoter were sequenced to confirm the start site.
Abortive initiation assays.
The steady-state properties of the abortive initiation assay have been described previously (39). Abortive initiation assays were performed with PexsD or PexsC supercoiled minicircle templates (2 nM) in the presence and absence of ExsAHis (35 nM) in 1× transcription buffer. The substrates for the abortive initiation reactions were as follows: for the PexsD promoter, 1 mM ATP, 1 mM UTP, and 0.33 μCi [α-32P]UTP to form pppApApApUpU and pppApApUpU; for the PexsC promoter, 1 mM GTP, 1 mM CTP, 1 mM UTP, and 0.33 μCi [α-32P]UTP to form pppGpCpUpUpU and pppCpUpUpU. Reaction mixtures including ExsAHis were incubated in 1× transcription buffer with template DNA for 10 min at 25°C prior to nucleotide/RNAP addition. To measure the lag time to open complex formation (τ), two separate reactions were performed for each of the seven RNAP concentrations tested (25, 28.6, 33.3, 40, 50, 66.7, and 100 nM). The first set of reaction mixtures contained template, ExsAHis (35 nM as indicated), and substrate nucleotides in 1× transcription buffer, and the reaction was initiated by the addition of RNAP. The second set of reaction mixtures contained template, ExsAHis (35 nM), and RNAP in 1× transcription buffer and were preincubated for 60 min at 30°C. Transcription was initiated by the addition of nucleotides. Both sets of reactions were allowed to proceed at 30°C. Samples were taken at various time points (1 to 120 min), the reactions were terminated with stop buffer, and the products were electrophoresed on denaturing 25% polyacrylamide gels. Gels were subjected to phosphorimaging and densitometry. The rate of abortive synthesis was calculated for reactions initiated with nucleotides by linear regression analysis (least squares). Curves plotted for reactions initiated by RNAP addition were analyzed by drawing a line through the curve but parallel to the reaction initiated by nucleotide addition. τobs was also obtained for these curves by linear regression analysis by solely using values 3 times greater than the initial estimate for τobs, as described previously (39), yielding results comparable to those with the first method. GraphPad Prism (GraphPad Software, Inc) was used to plot abortive initiation data and evaluate τobs.
Potassium permanganate footprinting.
Supercoiled minicircles carrying PexsC or PexsD were used as templates for the potassium permanganate footprinting reactions (57). Reaction mixtures containing ExsAHis were incubated for 10 min at 25°C to allow DNA binding in 1× potassium permanganate reaction buffer (40 mM Tris-HCl pH [7.5], 25 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.1% Tween 20, and 100 ng/ml bovine serum albumin). RNAP was added to the indicated concentrations, and open complex formation was allowed to proceed for 3 min at 25°C. Potassium permanganate (Sigma-Aldrich, St. Louis, MO) was immediately added (10 mM final concentration) and allowed to modify DNA for 1 min at 25°C. Reactions were stopped with termination buffer (0.5 M potassium acetate [pH 7.0], 1.5 M 2-mercaptoethanol, 5 mM EDTA), and products were purified with a PCR column purification kit (IBI Scientific) and eluted into 30 μl elution buffer (10 mM Tris-HCl [pH 8.5]). Modification by potassium permanganate was detected by primer extension. Primers (50 pmol) were end labeled with 50 μCi [γ-32P]ATP (Perkin-Elmer) and 10 U polynucleotide kinase (New England Biolabs, Ipswich, MA) as instructed by the manufacturer. Primer extension reactions (10 μl) were performed on the potassium permanganate-modified plasmids with 1 mM deoxynucleoside triphosphates, 0.5 μl end-labeled primer, and 1.25 units sequencing-grade Taq DNA polymerase (Promega, Madison, WI) in 1× sequencing buffer (50 mM Tris-HCl [pH 9.0], 2 mM MgCl2) under the following conditions: 1 cycle of 2 min at 94°C and 30 cycles of 0.5 min at 94°C, 0.5 min at 55°C, and 1 min at 72°C. Stop buffer (10 μl) was immediately added to each reaction mixture to terminate DNA synthesis. Dideoxy sequencing reactions for A and T were generated using the same thermocycling program and the following reaction components: 5 fmol PexsC/PexsD minicircle, 0.5 pmol labeled primer, 1.25 units sequencing-grade Taq DNA polymerase, 500 μM termination nucleotide (ddATP or ddTTP), and 20 μM elongation nucleotides (dCTP, 7-deaza-dGTP, dATP, and dTTP). Sequencing reactions were terminated with an equal volume of stop buffer and run alongside primer extension reactions on denaturing 6% polyacrylamide gels. Gels were dried and visualized by phosphorimaging, and analysis was performed with MultiGauge v3.0 software (Fujifilm).
RESULTS
The spacing between the −10 and −35 regions is critical for ExsA-dependent activation.
Each of the ExsA-dependent promoters used in this study (PexoT, PexsD, and PexsC) contains hexamers that match the consensus −10 and −35 regions of σ70-dependent promoters at 4/6 or 5/6 of the nucleotide positions (Fig. 1A; see Fig. 2A and 4). Nevertheless, it is not known whether the −10 and/or −35 region is truly recognized by RNAP-σ70. One reason for this uncertainty is that the −10 and −35 regions of ExsA-dependent promoters are separated by 21 or 22 bp, whereas optimal spacing for σ70-dependent promoters is 17 bp (2, 24, 26, 50). One interpretation of these data is that the increased spacing between the −10 and −35 regions prevents transcription in the absence of ExsA and that ExsA functions by overcoming the increased spacing. We predicted that if this was true, reducing the spacing of ExsA-dependent promoters to 17 bp would reconstitute a functional σ70-dependent promoter and eliminate the requirement for ExsA. To test this hypothesis, a series of lacZ transcriptional reporters was constructed in which the spacing between the −10 and −35 regions of PexoT was incrementally increased (22 to 24 bp) or decreased (16 to 20 bp) from the native spacing of 21 bp by inserting or deleting nucleotides at the −23 position relative to the start of transcription (Fig. 1A). The resulting reporters were integrated at a neutral chromosomal attachment site in wild-type PA103 and an exsA mutant (18). Strains were cultured under permissive (Ca2+-limiting) conditions for T3SS gene expression and assayed for β-galactosidase activity. Whereas expression of the native PexoT-lacZ reporter is exsA dependent, reporter derivatives in which the spacing was increased to 23 to 24 bp or decreased to 16 to 20 bp were largely inactive in either the absence or presence of exsA (Fig. 1B). The reporter having a spacing of 22 bp (PexoT-22-lacZ), however, retained exsA dependence, although activity was reduced ∼3-fold compared to the native PexoT-lacZ promoter. This finding is consistent with the fact that all ExsA-dependent promoters have a spacing of 21 or 22 bp (6) and suggests that the spacing distance between the putative −10 and −35 elements is critical for ExsA-dependent activation of T3SS promoters.
FIG. 1.

The spacing distance between the −10 and −35 regions is critical for ExsA-dependent activation of the PexoT promoter. (A) Diagram of the native and mutant PexoT-lacZ reporters. The PexoT16-lacZ-PexoT24-lacZ reporter constructs carry incremental 1-bp insertions (lowercase) or deletions (dash) between the putative −10 and −35 elements of the native PexoT promoter as indicated. A 14-bp segment of the native PexoT reporter was replaced with the complementary sequence (underlined), resulting in the PexoT-C-lacZ reporter. The −10 and −35 regions are indicated in bold. ExsA binding sites 1 and 2 are indicated by solid arrows. (B and C) Wild-type (wt) PA103 and the exsA::Ω mutant carrying the indicated reporters were cultured under inducing conditions for T3SS gene expression and assayed for β-galactosidase activity. The reported values (Miller units) are the averages from three independent experiments, and error bars represent the standard errors of the means.
FIG. 2.

Spacing requirements of the PexsD promoter and the role of the −10 and −35 regions. (A) Diagram of the native PexsD promoter and mutant derivatives, PtacI, and hybrid promoters derived from both PexsD and PtacI. The PexsD-17A-lacZ, PexsD-17B-lacZ, and PexsD-17C-lacZ reporters contain the indicated 4-bp deletions (dashes) between the −10 and −35 regions. To construct PexsD-tacI16-lacZ, the native 21-bp spacer region from PexsD was replaced with the 16-bp spacer from PtacI. The PtacI-PexsD(−35)-lacZ, PtacI-PexsD(−10)-lacZ, and PtacI-PexsD(−35/−10)-lacZ reporters were constructed by replacing the −10 and/or −35 regions of PtacI with the corresponding regions of PexsD. The putative −10 and −35 regions are indicated in bold. (B to D) Wild-type PA103 and the exsA::Ω mutant carrying the indicated reporters were cultured under T3SS-inducing conditions and assayed for β-galactosidase activity. The reported values represent the averages from three independent experiments, and error bars indicate the standard errors of the means.
FIG. 4.

Diagram of the Porf1, PexoS, PexsD, and PexsC promoters. The putative −10 and −35 regions are boxed. Transcription start sites previously mapped by primer extension (53, 54) are indicated with an asterisk. The start sites mapped in this study by RACE analysis and by abortive initiation assays are in bold or underlined, respectively. A putative transcriptional terminator from a transcript located upstream of the PexsC promoter is indicated by the double underline.
The lack of activity for the PexoT-17-lacZ reporter was somewhat surprising, as we expected that reducing the spacing to the σ70-dependent consensus of 17 bp might result in constitutive, ExsA-independent expression. We considered the possibility that the nucleotide sequence surrounding the −23 position is important for promoter activity. To test this possibility, we changed the nucleotides flanking the −23 position of the native PexoT-lacZ reporter to its complementary sequence while at the same time maintaining the spacing and G+C ratio of the native promoter (Fig. 1A). The resulting reporter (PexoT-C-lacZ) retained significant ExsA-dependent activity compared to the native PexoT reporter (Fig. 1C). This finding suggests that nucleotides adjacent to the −23 position are not essential for ExsA-dependent transcription and is consistent with the idea that the spacing distance between the putative −10 and −35 regions is a primary determinant of ExsA-dependent activation.
Another trivial explanation for the lack of constitutive PexoT-17-lacZ reporter activity is that the putative −10 and −35 hexamers of the PexoT promoter are inadequate for σ70-dependent activation. To test this possibility we used the PexsD promoter, since both the −10 and −35 regions match the σ70-dependent consensus sites at five of six positions. Deletions (4 bp) were separately introduced into the PexsD-lacZ reporter at three different locations, in each case reducing the spacing between the −10 and −35 regions from 21 bp to 17 bp (Fig. 2A). Compared to the native PexsD-lacZ construct, however, none of the modified reporters (PexsD-17A-lacZ, PexsD-17B-lacZ, and PexsD-17C-lacZ) had significant activity in either the absence or presence of exsA (Fig. 2B). To examine this further, the 21-bp spacer region from the PexsD-lacZ reporter was replaced with the 16-bp spacer region from the constitutive PtacI promoter (Fig. 2A). The PtacI promoter is active in P. aeruginosa (Fig. 2D) and has previously been studied both in vivo and in vitro (19). The PexsD-PtacI hybrid reporter (PexsD-tacI16), however, had very little activity in either the absence or presence of exsA (Fig. 2C). Finally, hybrid reporters were constructed by replacing the −10 and/or −35 region of the PtacI-lacZ reporter with the corresponding elements from the PexsD promoter (Fig. 2A). Whereas the native PtacI-lacZ reporter had significant activity in the absence of exsA, the hybrid PtacI-lacZ reporters containing the −10 and/or −35 region from the PexsD promoter demonstrated a 7- to 14-fold reduction in activity (Fig. 2D). These combined data indicate that even when properly spaced, the putative −10 and −35 hexamers of PexsD are suboptimal for σ70-dependent transcription and suggest that ExsA may function by recruiting RNAP to the promoter.
ExsA activates transcription from T3SS promoters in vitro.
To examine the mechanism of ExsA-dependent transcriptional activation, we developed a single-round in vitro transcription assay using ExsAHis purified from E. coli (Fig. 3A, lane 1) and RNAP-σ70 holoenzyme purified from P. aeruginosa (lane 2) or RNAP-σ70 holoenzyme from E. coli (obtained commercially). The specific activity of RNAP-σ70 isolated from P. aeruginosa was three- to fourfold lower than that of E. coli RNAP-σ70 (data not shown). The initial transcription template consisted of supercoiled plasmid DNA carrying the PexsD promoter fused to a strong transcriptional terminator (rpoCter). The plasmid template was preincubated with ExsAHis, RNAP-σ70, and the initiating nucleotide for transcription (ATP, as determined below) for 15 min. Ribonucleotides (including [α-32P]CTP) were then added in the presence of heparin (to prevent RNAP-σ70 from reinitiating), and transcripts were allowed to elongate for 10 min. RNAP-σ70 from either E. coli or P. aeruginosa generated the expected terminated transcript of 261 nt in an ExsA-dependent manner (Fig. 3B). Since the E. coli RNAP holoenzyme used for these studies is σ70 saturated and the P. aeruginosa RNAP holoenzyme isolated from log-phase cells is presumed to be largely σ70 saturated (Fig. 3A, lane 2), we conclude that ExsA-dependent promoters are σ70 dependent.
FIG. 3.

Purified ExsAHis activates transcription in vitro. (A) Silver-stained gel of ExsAHis purified from E. coli (lane 1) and σ70-RNAP purified from P. aeruginosa (lane 2). Molecular mass standards (in kDa) are indicated on the left. (B) Single-round in vitro transcription assays. ExsAHis (35 nM) was incubated with 2 nM supercoiled PexsD promoter template (pOM90-PexsD) at 25°C in the presence of rATP. After 10 min, RNAP from E. coli or P. aeruginosa (25 nM of each, normalized to specific activity of E. coli RNAP) was added, and the reaction mixture was incubated for 5 min at 30°C. Heparin and the remaining ribonucleotides (including 5 μCi [α32P]CTP) were immediately added, and the reaction mixture was incubated for 10 min at 30°C. Reactions were terminated, and the resulting products were electrophoresed on a 5% polyacrylamide-urea gel and subjected to phosphorimaging. The ExsA-dependent terminated transcript (261 nt) from the PexsD promoter is indicated. A transcript originating from an undetermined plasmid promoter is indicated with an asterisk. (C) Single-round in vitro transcription assays were performed as described above using the parental pSA508-PexsC/pSA508-PexsD plasmid templates or minicircles derived from the parental plasmids. The terminated transcript (221 nt) from these templates is indicated. A transcript originating from a weak plasmid promoter is indicated with an asterisk.
ExsA-independent transcription was not observed at high RNAP concentrations (200 nM) and at incubation times of as long as 2 h (data not shown). Because the transcription templates used in these experiments are self-replicating, supercoiled plasmids (∼5 kb in length), we hypothesized that in the absence of ExsA, strong constitutive plasmid promoters might outcompete the PexsD or PexsC promoter for RNAP occupancy. A similar result was previously observed for the gal promoters in E. coli and was addressed by constructing small supercoiled plasmid templates called minicircles (10). To generate PexsC and PexsD minicircles, the promoters were cloned upstream of the rpoC transcriptional terminator in the parental vector pSA508 (3.4 kb). The minicircles excise in vivo from the parental plasmid as supercoiled plasmids through a temperature-dependent recombination event. The resulting PexsC and PexsD minicircles (∼0.83 kb) consist solely of the cloned promoter fragments (0.43 kb), the rpoC transcriptional terminator, and residual plasmid sequences. Similar to the findings presented in Fig. 3B, the larger parental PexsC or PexsD plasmids were permissive for ExsA-dependent transcription, while ExsA-independent transcripts were undetectable (Fig. 3C). Minicircle templates derived from the parental plasmids, however, supported both ExsA-dependent and -independent transcription, although the amount of terminated transcript in the absence of ExsA was significantly reduced for both the PexsC and PexsD minicircle templates. Detection of ExsA-independent transcription from the minicircle templates is consistent with the possibility that strong promoters on the parental plasmid outcompete the PexsC and PexsD promoters for RNAP-σ70 occupancy and suggests that RNAP-σ70 binds to T3SS promoters poorly in the absence of ExsA.
Transcription from the PexsC and PexsD promoters initiates 8 to 9 nt downstream of near-consensus −10 TATA boxes.
Transcriptional start sites for several ExsA-dependent promoters were previously mapped by primer extension (53, 54). As expected of σ70-dependent promoters, the PexsD, PexoS, and Porf1 promoters initiate transcription 7 to 9 bp downstream of the −10 TATA box (Fig. 4). The PexsC start site, however, mapped to two adjacent nucleotides located ∼50 bases upstream of the putative −10 region. To resolve this apparent discrepancy, 5′ RACE and abortive initiation assays were used to reexamine the transcriptional start sites for the PexsC and PexsD promoters.
For the RACE assays, mRNA was isolated from wild-type PA103 and an exsA mutant grown under inducing conditions for T3SS gene expression (i.e., with EGTA). The mRNA was reverse transcribed into cDNA using gene-specific primers (exsC or exsD) and cloned into a plasmid vector for sequencing. At least 10 clones were sequenced for each promoter/RNA sample. Consistent with the previous primer extension data, the 5′ end of the exsD mRNA mapped to nucleotides located 7 to 9 bases downstream of the −10 region in both the wild type and the exsA mutant (Fig. 4). The 5′ end of the exsC mRNA, however, mapped to two distinct regions located 50 bp upstream and 8 to 9 bp downstream of the −10 region. Whereas the position of the former site is consistent with the previous primer extension data, the location of the latter site is nearly identical to the PexsD, PexoS, and Porf1 promoters with respect to the putative −10/−35 regions (Fig. 4). Subsequent studies (described below) indicate that the PexsC promoter initiates transcription at the second site.
An inherent limitation of 5′ RACE analysis is that the exact starting nucleotide cannot always be determined. To more precisely map the start sites, we analyzed abortive transcription products. Abortive RNA synthesis is thought to occur at all promoters and results from RNAP that initiates transcription but fails to clear the promoter and randomly aborts transcription within 15 to 20 nt (32). By starving an in vitro transcription reaction for one or more nucleotides and incorporating specific radiolabeled nucleotides, the length of the abortive transcripts can reveal the exact transcriptional start site. The abortive initiation assay mixtures for the PexsC promoter contained GTP, CTP, and radiolabeled UTP. Under these conditions, the putative start site located 50 bp upstream of the −10 region would generate a 10- to 12-nt transcript before terminating at an adenine, while the start site located 8 to 9 bp downstream of the −10 region would generate a 4- to 5-nt product. In the presence of ExsAHis, two abortive transcripts were generated (Fig. 5A, lane 4). The shorter product was nearly identical in size to the abortive transcript (4 nt, pppAAUU) from the well-characterized Pl8UV5 promoter when provided only ATP and radiolabeled UTP (data not shown). These data are consistent with the aborted PexsC transcripts representing pppCUUU (4 nt) and pppGCUUU (5 nt) and indicate that PexsC transcription initiates from the G and C nucleotides located 8 and 9 nt downstream of the −10 region (Fig. 4). The fact that the same products were not detected when the transcription reaction mixtures lacked UTP further supports this conclusion (Fig. 5A, lanes 1 and 2). The pppCUUU and pppGCUUU abortive transcripts were also were generated in the absence of ExsAHis, albeit to a much lesser extent and only after an extended incubation period (Fig. 5A, lane 6). The failure to detect the 10- to 12-nt product from the site located 50 bp upstream of the −10 region suggests that this start site mapped by primer extension and RACE may be an artifact.
FIG. 5.
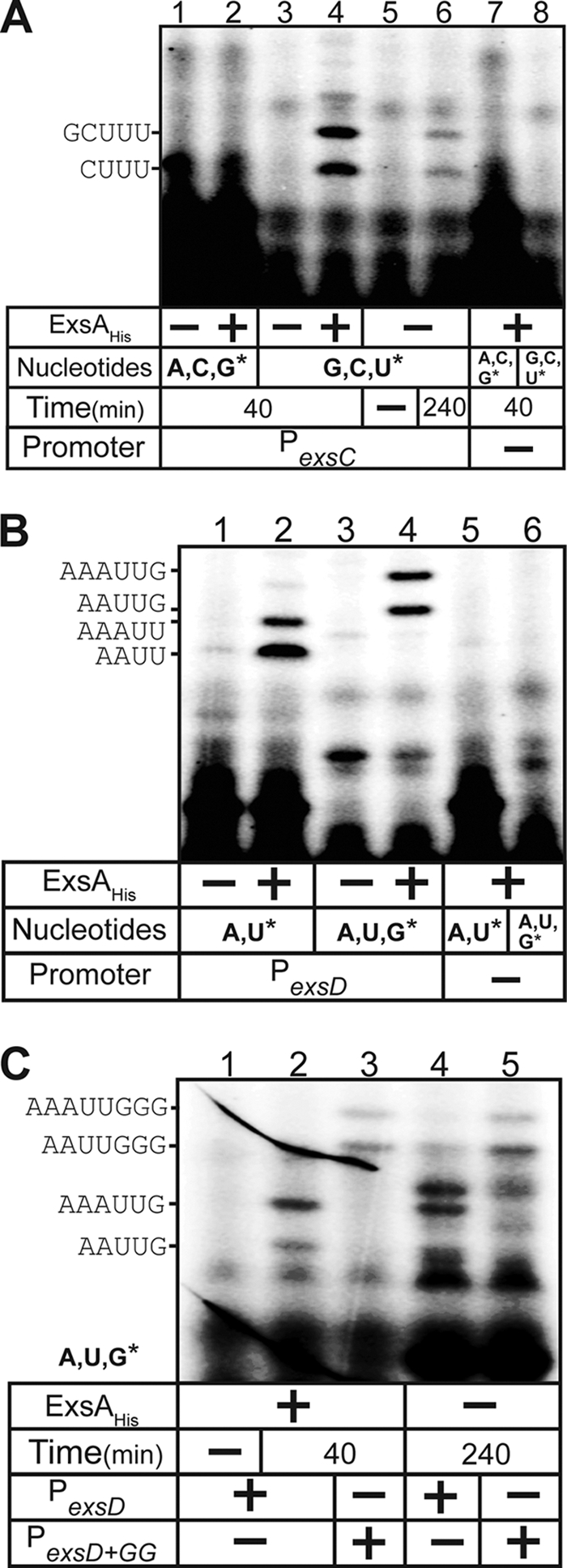
Abortive initiation assays for the PexsC and PexsD promoters. Reactions using the PexsC (A) or PexsD (B and C) minicircle templates were allowed to proceed for 40 min or 240 min, as indicated, with RNAP and substrate nucleotide sets (asterisks indicate labeled nucleotides) in the absence or presence of ExsAHis. Reactions were terminated, and the products were electrophoresed through a 25% denaturing polyacrylamide gel and visualized by phosphorimaging. Control reactions using a supercoiled minicircle template lacking T3SS promoters were performed with each substrate nucleotide set in the presence of ExsAHis. (C) Abortive initiation assays with the PexsD and modified PexsD+GG promoters in the absence or presence of ExsAHis. Abortive initiation reactions were allowed to proceed for 240 min in the presence of RNAP, unlabeled ATP/UTP, and labeled GTP. Control reactions in the presence of ExsAHis were incubated for 40 min.
Abortive initiation assays for the PexsD promoter that were limited to ATP and radiolabeled UTP generated two ExsA-dependent products (pppAAUU and pppAAAUU) (Fig. 5B, lanes 1 and 2), the shorter of which is identical in size to the aborted transcript generated by the Pl8UV5 promoter (data not shown). This finding is consistent with transcription initiating at the adenine nucleotides located 7 to 8 bp downstream of the −10 region (Fig. 4). To confirm this finding, the assays were repeated in the presence of ATP, UTP, and radiolabeled GTP, where only the terminal nucleotide would be labeled. As expected, the resulting abortive products (pppAAUUG and pppAAAUUG) were 1 nt longer (Fig. 5B, lane 2 versus lane 4) and ExsA dependent (lane 3 versus lane 4).
The ability to measure ExsA-independent abortive products can provide information regarding the mechanism of transcriptional activation by ExsA (discussed below). Unfortunately, extended incubation of RNAP-σ70 with the PexsD promoter in the absence of ExsA resulted in the appearance of background bands, which raised questions as to whether the abortive transcripts were truly arising from the PexsD promoter. To determine whether the aborted products were indeed derived from PexsD, a mutant promoter (PexsD+GG) in which two additional guanine nucleotides were added between nt +6 and +7 was generated. Compared to the wild-type PexsD promoter, the products generated from the PexsD+GG promoter (pppAAUUGGG and pppAAAUUGGG) were 2 bases greater in length (Fig. 5C, lane 2 versus lane 3) and, importantly, were clearly detected in the absence of ExsAHis if the incubation time was extended to 240 min.
ExsA promotes an early step in transcriptional initiation.
The most common rate-limiting steps during transcriptional initiation are closed and open complex formation (32). Closed complexes result from the binding of RNAP to the promoter; transition of the closed complex to an open complex involves unwinding of the −10 region of the promoter to single-stranded DNA (ssDNA). Open complex formation provides RNAP access to the template strand and is required for subsequent elongation of the transcript (32). The open complex can be detected with the DNA modification reagent potassium permanganate, which preferentially oxidizes pyrimidine bases in ssDNA (1). To determine whether ExsA is required for the initial steps in initiation of PexsC and PexsD transcription, the minicircles carrying these promoters were incubated with ExsAHis and/or RNAP-σ70 and then subjected to potassium permanganate modification. Modified minicircles were then used as templates in primer extension reactions with DNA polymerase and radiolabeled primers. DNA polymerase terminates transcription at bases oxidized by potassium permanganate, and the resulting enrichment of the terminated fragments indicates the regions of ssDNA. Incubation of the PexsC and PexsD minicircles with both ExsAHis and a low concentration of RNAP-σ70 (1.5 nM) resulted in strong permanganate modification of the −10 regions within each promoter (Fig. 6A and B). Weaker modification of the same regions could also be seen with RNAP-σ70 alone. These findings demonstrate that RNAP-σ70 can bind to the PexsD and PexsC promoters independent of ExsA and that ExsA facilitates transcriptional initiation by enhancing recruitment of RNAP-σ70 to the promoter and/or promoting isomerization to an open complex.
FIG. 6.

ExsA stimulates formation of open complexes as measured by potassium permanganate footprinting. (A and B) Supercoiled minicircles carrying the PexsC (A) or PexsD (B) promoter (1.6 nM) were incubated in the absence (−) or presence (+) of ExsAHis (30 nM) for 10 min. RNAP was added to the indicated concentrations and incubated for 3 min. Reaction mixtures were then treated with potassium permanganate (except for the control [KMnO4]), and the modified minicircles were used as templates in primer extension reactions with a radiolabeled coding-strand primer. Primer extension products were subjected to denaturing electrophoresis and phosphorimaging. Dideoxy sequencing reactions for A and T are indicated. The diagrams to the left show the transcriptional start sites (bold), the −10 regions (boxed), and the nucleotides modified by potassium permanganate (indicated with asterisks).
ExsA facilitates a rate-limiting step prior to open complex formation at the PexsC and PexsD promoters.
Potassium permanganate footprints indicate that ExsA functions at the level of transcription initiation, but this method cannot discern whether ExsA recruits RNAP and/or promotes isomerization to the open complex. The abortive initiation assay, however, can be used to estimate the isomerization rate constant and overall reaction rate for a given promoter by measuring the lag time to steady-state synthesis of abortive transcripts. To analyze the kinetics of transcription initiation at the PexsC and PexsD promoters, abortive transcripts were generated at various concentrations of RNAP-σ70 in the absence or presence of saturating concentrations of ExsAHis, and the lag time (τobs) to open complex formation was recorded for each RNAP-σ70 concentration on a τ plot. The resulting τ plots for both the PexsC and PexsD promoters show an inverse linear relationship between the lag time to open complex formation and the RNAP-σ70 concentration (Fig. 7A and B). The overall reaction rates for the PexsC and PexsD promoters increased 13- and 11-fold in the presence of ExsAHis, respectively (Table 1). In both cases, the stimulatory effect of ExsAHis resulted primarily from an increase in the equilibrium binding constant for RNAP-σ70 (five- to eightfold), although there was also a modest increase in the isomerization rate constant (∼2-fold). These data indicate that the primary mechanism by which ExsA stimulates transcription is through recruitment of RNAP-σ70 to the PexsC and PexsD promoters prior to open complex formation.
FIG. 7.

τ plots for the PexsC (A) and PexsD (B) promoters in the presence (open circles) and absence (closed circles) of ExsAHis. Values for τobs were calculated from abortive initiation assays measuring synthesis of the products pppGCUUU (PexsC) and pppAAAUU (PexsD). Calculated values for τobs were plotted on the ordinate as a function of reciprocal RNAP concentration.
TABLE 1.
Kinetic parameters of the PexsC and PexsD promoters
| Promoter | τ(s)a | k2 (s−1, 103)b | KB (M−1, 10−5)c | k2KB (M−1 s−1, 10−3)d |
|---|---|---|---|---|
| PexsC | ||||
| Without ExsAHis | 13.8 | 72 | 1.3 | 9.4 |
| With ExsAHis | 8.1 | 123 | 10 | 123 |
| PexsD | ||||
| Without ExsAHis | 52.7 | 18.9 | 3.4 | 6.4 |
| With ExsAHis | 22.6 | 44 | 16.5 | 72.6 |
Lag time to open complex formation.
Isomerization rate constant.
Equilibrium binding constant for RNAP.
Overall reaction rate for open complex formation.
DISCUSSION
In the present study we found that purified ExsAHis and RNAP-σ70 isolated from either P. aeruginosa or E. coli is sufficient to activate transcription from T3SS promoters in vitro. This is consistent with previous studies demonstrating that T3SS genes are expressed maximally during exponential growth phase (22, 29, 47) and with the fact that the −10 and −35 regions of T3SS promoters are similar to the σ70 consensus sequences from E. coli (21, 26) and P. aeruginosa (14). In addition, potassium permanganate footprinting assays reveal RNAP-σ70-dependent unwinding of the PexsD and PexsC −10 regions, and this was greatly enhanced in the presence of ExsAHis. Based on these data, we conclude that ExsA primarily utilizes RNAP-σ70 to activate T3SS gene expression. Our findings, however, do not preclude the possibility that alternative sigma factors might also be involved in ExsA-dependent gene expression.
5′ RACE was used to map the general region of the PexsC and PexsD transcription start sites in vivo, and abortive initiation products were used for more precise mapping in vitro. Although the PexsD start site matched previously published primer extension data, two apparent start sites were observed for the PexsC promoter. Whereas the first site is located upstream of the ExsA binding site, the second site is located downstream of the −10 element (Fig. 4). We believe that the latter site is the true PexsC start site, given its proximity to the ExsA binding site and RNAP recognition elements and its similarity to the positions of the PexsD, PexoS, and Porf1 start sites. The apparent upstream start site may result from transcriptional read-through from the upstream PpcrG promoter. In this regard it is worth noting the poly(dA) and poly(dT) tracts located just downstream of the start site mapped by primer extension and 5′ RACE (Fig. 4) and that both of those techniques rely upon RT extension from mRNA templates. RT is known to pause at sites of secondary structure and at poly(A) and poly(U) nucleotide runs (25, 34). We believe that the apparent upstream start site represents pausing of RT that results from either the poly(A)/poly(U) sequence or secondary structure associated with a transcriptional terminator. In either case this finding suggests that transcriptional read-through from the upstream PpcrG promoter, which is also ExsA dependent, contributes to expression of the exsCEBA operon and could represent another point at which ExsA expression levels are regulated.
AraC activates transcription by enhancing the RNAP equilibrium binding and the open complex isomerization rate constants (57). These activities have been attributed to protein-protein interactions between AraC and RNAP-σ70 (37). Given the similarities between ExsA and AraC with respect to the location of the activator binding site, we hypothesized that ExsA might activate transcription through similar mechanisms. Abortive initiation assays, however, indicate that ExsAHis only marginally altered the isomerization rate constant but had a significant effect on the equilibrium binding constant (five- to eightfold) at both the PexsC and PexsD promoters. These results indicate that ExsA functions primarily by enhancing recruitment of RNAP-σ70 to the promoter prior to open complex formation. The effect of ExsAHis on transcriptional initiation in vitro (11- to 13-fold) is much lower than that observed in vivo (100- to 1,000-fold) (38). The most likely explanation for this discrepancy is that the level of ExsA-independent transcription seen in vitro is artificially elevated due to the absence of competing promoters (Fig. 3D), a condition never seen in vivo. Other contributing factors include the inherent limitations of an in vitro assay, the use of minicircle transcription templates as a mimic for chromosomal DNA, and the absence of factors that may be required for maximal ExsA-dependent transcription. For example, ExsA may possess a coactivator, as is seen for the AraC family members (VirF and MxiE) that regulate T3SS gene expression in Salmonella enterica serovar Typhimurium and Shigella flexneri, respectively. Alternatively, some AraC family members function together with catabolite activator protein to regulate gene expression. It is interesting to note that the P. aeruginosa homolog of catabolite activator protein (Vfr) is required for T3SS gene expression through an undetermined pathway (52). Development of an in vitro transcription assay provides a means to test whether factors known to influence T3SS gene expression in P. aeruginosa do so by directly modulating ExsA-dependent activation.
ExsA-dependent promoters are unusual in that the putative −10 and −35 promoter elements are separated by 21 or 22 bp, compared to the 17 bp typical of σ70-dependent promoters. Employing a panel of promoter spacing mutants, we demonstrated that PexoT promoter activity shows a strict dependence on spacing of 21 or 22 bp. Increased spacing between the −10 and −35 regions has been described in only a few cases for transcription factors that activate σ70-dependent promoters (28, 56). The mechanism of Spo0A-dependent transcriptional activation in Bacillus subtilis closely parallels the ExsA situation, where in both cases the −10 and −35 sites are separated by 21 to 22 bp and the activator binding sites overlap with and extend upstream of the −35 regions. These observations initially raised concerns as to whether the −35 region was an authentic determinant for binding of RNAP-σA (the equivalent of RNAP-σ70 in B. subtilis) to Spo0A-dependent promoters (56). The findings that reducing the spacing between the −10 and −35 regions of the spoIIG promoter to 17 bp results in Spo0A-independent transcription and that RNAP-σA footprints the −35 site independently of Spo0A (5), however, indicate that the −35 region is recognized by RNAP-σA and that Spo0A activates transcription by suppressing the −10 and −35 spacing constraint. The current model proposes that RNAP binds to the spoIIG promoter independently of Spo0A through low-specificity interactions between RNAP-σA and the −35 region. Spo0A binding then repositions RNAP-σA 4 bp downstream of the −35 region such that region 2 of σA can interact with the −10 region resulting, in open complex formation (36).
While it is clear from permanganate footprinting experiments that the −10 regions of PexoT and PexsD isomerize to open complexes, the role of the putative −35 regions remains unclear. Permanganate footprints and abortive transcripts demonstrate that RNAP-σ70 is capable of binding to the PexoT and PexsD promoters in the absence of ExsA. In this regard, ExsA-dependent promoters are similar to Spo0A-dependent promoters in that RNAP can bind independently of the activator. Binding of RNAP-σ70 to the PexoT and PexsD promoters, however, was detected only in the absence of competing promoters and may not reflect the in vivo situation. A notable difference between the ExsA and Spo0A systems is the effect of altered spacing between the −10 and −35 regions. Based upon the Spo0A model, we hypothesized that reducing the spacing between the −10 and −35 regions of ExsA-dependent promoters to 17 bp would result in ExsA-independent activity. Both the PexoT and PexsD promoters, however, lacked ExsA-independent expression at the optimal spacing of 17 bp (Fig. 1B and 2B). This was particularly surprising for the PexsD promoter because the −10 and −35 regions closely match the σ70 consensus. Even more striking was the finding that the −10 and −35 regions of PexsD are poor substitutes for the corresponding elements of the PtacI promoter (Fig. 2D). These findings suggest that the −10 and −35 regions of PexsD function as poor recognition elements for RNAP-σ70 and support our conclusion that the primary role of ExsA is to facilitate recruitment of RNAP-σ70 to the promoter. Future experiments will focus on characterization of the ExsA-RNAP-σ70 interaction, the regions of σ70 that interact with T3SS promoters in both the presence and absence of ExsA, and whether the −35 region contributes to the binding of RNAP-σ70.
Supplementary Material
Acknowledgments
We thank Evan Brutinel and Mark Urbanowski for their suggestions regarding this work and Sankar Adhya and Dale Lewis for their generous gift of the minicircle transcription template system.
This study was supported by the National Institutes of Health (grant RO1-AI055042-06).
Footnotes
Published ahead of print on 28 August 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Akman, S. A., J. H. Doroshow, and M. Dizdaroglu. 1990. Base modifications in plasmid DNA caused by potassium permanganate. Arch. Biochem. Biophys. 282:202-205. [DOI] [PubMed] [Google Scholar]
- 2.Allan, B., and A. M. Kropinski. 1987. DNA-dependent RNA polymerase from Pseudomonas aeruginosa. Biochem. Cell Biol. 65:776-782. [DOI] [PubMed] [Google Scholar]
- 3.Apodaca, G., M. Bomsel, R. Lindstedt, J. Engel, D. Frank, K. E. Mostov, and J. Wiener-Kronish. 1995. Characterization of Pseudomonas aeruginosa-induced MDCK cell injury: glycosylation-defective host cells are resistant to bacterial killing. Infect. Immun. 63:1541-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbieri, J. T., and J. Sun. 2004. Pseudomonas aeruginosa ExoS and ExoT. Rev. Physiol. Biochem. Pharmacol. 152:79-92. [DOI] [PubMed] [Google Scholar]
- 5.Bird, T. H., J. K. Grimsley, J. A. Hoch, and G. B. Spiegelman. 1996. The Bacillus subtilis response regulator Spo0A stimulates transcription of the spoIIG operon through modification of RNA polymerase promoter complexes. J. Mol. Biol. 256:436-448. [DOI] [PubMed] [Google Scholar]
- 6.Brutinel, E. D., C. A. Vakulskas, K. M. Brady, and T. L. Yahr. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 68:657-671. [DOI] [PubMed] [Google Scholar]
- 7.Brutinel, E. D., C. A. Vakulskas, and T. L. Yahr. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191:3811-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brutinel, E. D., and T. L. Yahr. 2008. Control of gene expression by type III secretory activity. Curr. Opin. Microbiol. 11:128-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess, R. R., and J. J. Jendrisak. 1975. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry 14:4634-4638. [DOI] [PubMed] [Google Scholar]
- 10.Choy, H. E., and S. Adhya. 1993. RNA polymerase idling and clearance in gal promoters: use of supercoiled minicircle DNA template made in vivo. Proc. Natl. Acad. Sci. USA 90:472-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darwin, K. H., and V. L. Miller. 2000. The putative invasion protein chaperone SicA acts together with InvF to activate the expression of Salmonella typhimurium virulence genes. Mol. Microbiol. 35:949-960. [DOI] [PubMed] [Google Scholar]
- 12.Darwin, K. H., and V. L. Miller. 2001. Type III secretion chaperone-dependent regulation: activation of virulence genes by SicA and InvF in Salmonella typhimurium. EMBO J. 20:1850-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dasgupta, N., G. L. Lykken, M. C. Wolfgang, and T. L. Yahr. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53:297-308. [DOI] [PubMed] [Google Scholar]
- 14.Dominquez-Cuevas, P., and S. Marques. 2004. Compiling 70-dependent promoters, p. 319-344. In J.-L. Ramos (ed.), Pseudomonas, vol. 2. Virulence and gene regulation. Kluwer Academic/Plenum Publishers, London, United Kingdom. [Google Scholar]
- 15.Egan, S. M. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 184:5529-5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank, D. W. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26:621-629. [DOI] [PubMed] [Google Scholar]
- 17.Frank, D. W., and B. H. Iglewski. 1991. Cloning and sequence analysis of a trans-regulatory locus required for exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 173:6460-6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank, D. W., G. Nair, and H. P. Schweizer. 1994. Construction and characterization of chromosomal insertional mutations of the Pseudomonas aeruginosa exoenzyme S trans-regulatory locus. Infect. Immun. 62:554-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita, M., and A. Amemura. 1992. In vitro interactions of Pseudomonas RNA polymerases with tac and RNA I promoters. Biosci. Biotechnol. Biochem. 56:1644-1648. [DOI] [PubMed] [Google Scholar]
- 20.Gallegos, M. T., R. Schleif, A. Bairoch, K. Hofmann, and J. L. Ramos. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61:393-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grana, D., T. Gardella, and M. M. Susskind. 1988. The effects of mutations in the ant promoter of phage P22 depend on context. Genetics 120:319-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha, U., and S. Jin. 2001. Growth phase-dependent invasion of Pseudomonas aeruginosa and its survival within HeLa cells. Infect. Immun. 69:4398-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reference deleted.
- 24.Harley, C. B., and R. P. Reynolds. 1987. Analysis of E. coli promoter sequences. Nucleic Acids Res. 15:2343-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison, G. P., M. S. Mayo, E. Hunter, and A. M. Lever. 1998. Pausing of reverse transcriptase on retroviral RNA templates is influenced by secondary structures both 5′ and 3′ of the catalytic site. Nucleic Acids Res. 26:3433-3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawley, D. K., and W. R. McClure. 1983. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 11:2237-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoang, T. T., A. J. Kutchma, A. Becher, and H. P. Schweizer. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59-72. [DOI] [PubMed] [Google Scholar]
- 28.Hobman, J. L. 2007. MerR family transcription activators: similar designs, different specificities. Mol. Microbiol. 63:1275-1278. [DOI] [PubMed] [Google Scholar]
- 29.Hogardt, M., M. Roeder, A. M. Schreff, L. Eberl, and J. Heesemann. 2004. Expression of Pseudomonas aeruginosa exoS is controlled by quorum sensing and RpoS. Microbiology 150:843-851. [DOI] [PubMed] [Google Scholar]
- 30.Holder, I. A., A. N. Neely, and D. W. Frank. 2001. Type III secretion/intoxication system important in virulence of Pseudomonas aeruginosa infections in burns. Burns 27:129-130. [DOI] [PubMed] [Google Scholar]
- 31.Hovey, A. K., and D. W. Frank. 1995. Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 177:4427-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu, L. M. 2009. Monitoring abortive initiation. Methods 47:25-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jarrell, K., and A. M. Kropinski. 1977. The chemical composition of the lipopolysaccharide from Pseudomonas aeruginosa strain PAO and a spontaneously derived rough mutant. Microbios 19:103-116. [PubMed] [Google Scholar]
- 34.Klarmann, G. J., C. A. Schauber, and B. D. Preston. 1993. Template-directed pausing of DNA synthesis by HIV-1 reverse transcriptase during polymerization of HIV-1 sequences in vitro. J. Biol. Chem. 268:9793-9802. [PubMed] [Google Scholar]
- 35.Kohler, J. J., S. J. Metallo, T. L. Schneider, and A. Schepartz. 1999. DNA specificity enhanced by sequential binding of protein monomers. Proc. Natl. Acad. Sci. USA 96:11735-11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar, A., and C. P. Moran, Jr. 2008. Promoter activation by repositioning of RNA polymerase. J. Bacteriol. 190:3110-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin, R. G., and J. L. Rosner. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4:132-137. [DOI] [PubMed] [Google Scholar]
- 38.McCaw, M. L., G. L. Lykken, P. K. Singh, and T. L. Yahr. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46:1123-1133. [DOI] [PubMed] [Google Scholar]
- 39.McClure, W. R. 1980. Rate-limiting steps in RNA chain initiation. Proc. Natl. Acad. Sci. USA 77:5634-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsot, C., E. Ageron, C. Penno, M. Mavris, K. Jamoussi, H. d'Hauteville, P. Sansonetti, and B. Demers. 2005. A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol. Microbiol. 56:1627-1635. [DOI] [PubMed] [Google Scholar]
- 41.Pilonieta, M. C., and G. P. Munson. 2008. The chaperone IpgC copurifies with the virulence regulator MxiE. J. Bacteriol. 190:2249-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plano, G. V. 2004. Modulation of AraC family member activity by protein ligands. Mol. Microbiol. 54:287-290. [DOI] [PubMed] [Google Scholar]
- 43.Richards, M. J., J. R. Edwards, D. H. Culver, and R. P. Gaynes. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510-515. [DOI] [PubMed] [Google Scholar]
- 44.Richards, M. J., J. R. Edwards, D. H. Culver, R. P. Gaynes, et al. 1999. Nosocomial infections in medical intensive care units in the United States. Crit. Care Med. 27:887-892. [DOI] [PubMed] [Google Scholar]
- 45.Sato, H., and D. W. Frank. 2004. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53:1279-1290. [DOI] [PubMed] [Google Scholar]
- 46.Scotto-Lavino, E., G. Du, and M. A. Frohman. 2006. Amplification of 5′ end cDNA with ‘new RACE.’ Nat. Protoc. 1:3056-3061. [DOI] [PubMed] [Google Scholar]
- 47.Shen, D. K., D. Filopon, H. Chaker, S. Boullanger, M. Derouazi, B. Polack, and B. Toussaint. 2008. High-cell-density regulation of the Pseudomonas aeruginosa type III secretion system: implications for tryptophan catabolites. Microbiology 154:2195-2208. [DOI] [PubMed] [Google Scholar]
- 48.Thibault, J., E. Faudry, C. Ebel, I. Attree, and S. Elsen. 2009. The anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 284:15762-15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogel, H. J., and D. M. Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97-106. [PubMed] [Google Scholar]
- 50.Warne, S. E., and P. L. deHaseth. 1993. Promoter recognition by Escherichia coli RNA polymerase. Effects of single base pair deletions and insertions in the spacer DNA separating the −10 and −35 regions are dependent on spacer DNA sequence. Biochemistry 32:6134-6140. [DOI] [PubMed] [Google Scholar]
- 51.Wickstrum, J. R., J. M. Skredenske, A. Kolin, D. J. Jin, J. Fang, and S. M. Egan. 2007. Transcription activation by the DNA-binding domain of the AraC family protein RhaS in the absence of its effector-binding domain. J. Bacteriol. 189:4984-4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolfgang, M. C., V. T. Lee, M. E. Gilmore, and S. Lory. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4:253-263. [DOI] [PubMed] [Google Scholar]
- 53.Yahr, T. L., and D. W. Frank. 1994. Transcriptional organization of the trans-regulatory locus which controls exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 176:3832-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yahr, T. L., A. K. Hovey, S. M. Kulich, and D. W. Frank. 1995. Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J. Bacteriol. 177:1169-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yahr, T. L., and M. C. Wolfgang. 2006. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 62:631-640. [DOI] [PubMed] [Google Scholar]
- 56.York, K., T. J. Kenney, S. Satola, C. P. Moran, Jr., H. Poth, and P. Youngman. 1992. Spo0A controls the sigma A-dependent activation of Bacillus subtilis sporulation-specific transcription unit SpoIIE. J. Bacteriol. 174:2648-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang, X., T. Reeder, and R. Schleif. 1996. Transcription activation parameters at ara pBAD. J. Mol. Biol. 258:14-24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.