

Graphical abstract
Keywords: Anthrax lethal factor, Quantitative high-throughput screening, Niclosamide, Diphyllin, Anthrax, Endocytosis
Abstract
Here, we report the results of a quantitative high-throughput screen (qHTS) measuring the endocytosis and translocation of a β-lactamase-fused-lethal factor and the identification of small molecules capable of obstructing the process of anthrax toxin internalization. Several small molecules protect RAW264.7 macrophages and CHO cells from anthrax lethal toxin and protected cells from an LF-Pseudomonas exotoxin fusion protein and diphtheria toxin. Further efforts demonstrated that these compounds impaired the PA heptamer pre-pore to pore conversion in cells expressing the CMG2 receptor, but not the related TEM8 receptor, indicating that these compounds likely interfere with toxin internalization.
1. Introduction
High-throughput screening (HTS) provides a platform for the identification of small molecules capable of modulating protein function and cellular phenotypes.1, 2 HTS of cellular pathway assays may identify a variety of small molecule compounds with different molecular targets which can be identified in subsequent mechanistic studies. Such pathway assays are particularly attractive when studying human pathogens, since they allow HTS to occur in a nonrestrictive biosafety level environment.
Recently Hobson et al. described such a cell-based pathway assay that monitors receptor-mediated endocytosis of a modified, non-toxic anthrax toxin lethal factor (LF)/protective antigen (PA) complex.3 The technology relies upon a LF-β-lactamase fusion protein (LF-β-Lac) that co-opts the native mechanism of LF internalization,4 whereby proteolysis of PA allows heptamerization of the cleaved PA, association with LF, internalization via endocytosis, and translocation of LF to the cytoplasm triggered by endosome acidification.5 Once internalized, β-lactamase hydrolysis of the cephalosporin-based fluorescein/coumarin fluorogenic substrate CCF2 disrupts intramolecular CCF2 FRET and changes emission from fluorescein (EM 530 nm) to coumarin (EM 460 nm) fluorescence. The number of blue (coumarin fluorescent) cells, and their proportion relative to green (fluorescein fluorescent) cells, indicates the extent of inhibition of LF-β-lac internalization. This system accurately reports on the cellular uptake processes by which cells become intoxicated by anthrax PA and LF, which together constitute anthrax lethal toxin (LT). It should be noted that the LF-β-Lac has no toxicity.
Humans contract anthrax through inhalation, ingestion, or cutaneous inoculation of endospores of the Gram-positive aerobic rod Bacillus anthracis to cause the disease entities known, respectively, as inhalation anthrax, intestinal anthrax, and cutaneous anthrax. The endospores germinate following introduction to the body to cause a productive infection with incubation times ranging from 12 h to two weeks, and mortality rates in untreated cases ranging from 20% to more than 90%. B. anthracis forms endospores under adverse conditions that are usually present in the soil where infected animals died. The bacterium is simple to culture, and sporulation can easily be induced in a controlled laboratory setting. The endospores of B. anthracis are highly resistant to chemical insults, heat exposure, and dehydration, and remain infectious even after prolonged storage. These properties, combined with the high morbidity and mortality of anthrax, makes B. anthracis a significant threat as a bioterrorism agent, as illustrated by the effects of the deliberate contamination of mail with endospores in the fall of 2001.
As a means to identifying chemical probes of the LF internalization process the NIH Chemical Genomics Center (NCGC), a member of the Molecular Libraries Initiative of the NIH Roadmap for Medical Research6 was engaged. The original assay design was miniaturized and screened against 70,094 compounds in 1536-well plate format at a minimum of 7 concentrations each, utilizing the quantitative high-throughput screening (qHTS)7 approach. Here we report the identification of several novel chemotypes from this screen which block internalization of anthrax LF and protect cells from LF action. Additionally, we provide evidence that the compounds disrupt the PA–LF constructs internalization process.
2. Results and discussion
2.1. Quantitative high-throughput screen
The originally reported assay,3 diagrammed in Figure 1 , was adapted and miniaturized to a 1536-well plate format. qHTS and data analysis were performed as previously described7 with modifications as noted in Section 4. A total of 70,094 compounds were tested at ⩾ 7 concentrations from 2.6 nM to 40 μM, and concentration–response curves were generated and classified for all compounds.7 From these data, 1170 compounds showing concentration-dependent inhibition of the β-lactamase catalyzed hydrolysis of CCF2 without effects on the green fluorescence channel (a proxy for cell number) and having efficacies >50% were identified and clustered for structural similarity. The dual fluorescence output design enabled the discrimination of translocation inhibitors from cytotoxic compounds, as the latter were clearly discernable as a decrease in green fluorescence without an increase in blue fluorescence. Compounds (211) representing 44 clusters of related compounds and singletons were prioritized based on compound curve classification, potency, and when applicable, structural series cluster size.
Figure 1.
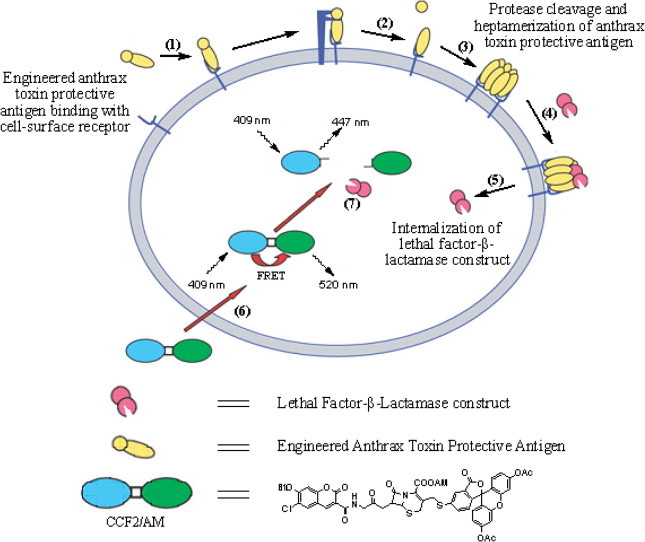
Principle of cell surface protease activity imaging assay. (1) PA binds to the high affinity cell surface receptors TEM8 or CMG2. (2) PA is cleaved by furin or furin-like pro-protein convertases. (3) The PA fragment that remains on the cell surface heptamerizes. (4) High-affinity binding sites for LF-β-Lac are generated by heptamerization of the PA. (5) LF-β-Lac is translocated to the cytoplasm after endocytosis of the PA–LF-β-Lac complex (not shown). (6) CCF2/AM is added to cells and diffuses into the cytoplasm where it is trapped by hydrolysis of its acetoxymethyl ester groups by non-specific cytoplasmic esterases. (7) LF-β-Lac hydrolyses the cephalosporin ring of CCF2, causing a shift in fluorescence emission from 520 nm (green light) to 447 nm (blue light) after excitation of cells at 409 nm. Blue fluorescence emission by a cell demonstrates successful translocation of LF-β-Lac to the cytoplasm. Reproduced from Ref. 3.
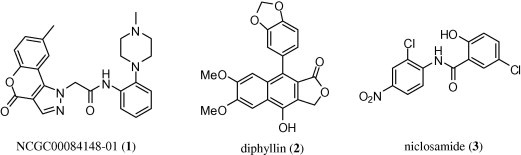
As an initial exploration into the numerous actives, and in order to validate the screening assay as a reliable predictor of protection against anthrax intoxication, 30 compounds with high confidence qHTS concentration–response curves (Class 1.1)7 and/or known biological activities (niclosamide, homoharringtonine, GF 109203, PD 404182, cucurbitacin I, proscillaridin A, and bisacodyl) were chosen for follow-up. Fresh powder samples of these 30 compounds were obtained and serially diluted into 24 concentrations; EC50 values were determined using the primary screen protocol in 1536-well plate format. The compounds were then further tested in 96-well format as a comparative experiment to the qHTS confirmatory assays, with results determined both by epifluorescence microscopy and plate reader quantification. Among the most compelling confirmed actives identified were a novel chromeno[4,3-c]pyrazol-4(1H)-one (NCGC00084148-01) (1), the lignan diphyllin (2) and the teniacide niclosamide (3) (Fig. 2 ).
Figure 2.
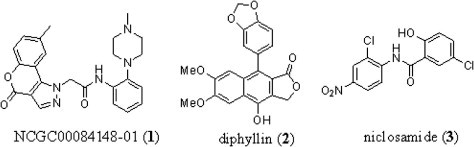
Chemical structure of anthrax toxin internalization inhibitors identified through the qHTS. Structures of NCGC00084148-01 (1), diphyllin (2) and niclosamide (3).
2.2. Protective effect on RAW264.7 macrophages from lethal toxin
Compounds (30) with confirmed activity in the qHTS assay were entered into follow-up studies to determine their native LF-protective effects on macrophages and their mechanisms of action, since the primary assay may detect compounds acting via a variety of mechanisms including inhibition of β-lactamase.3 First, selected compounds were assayed for their ability to protect RAW264.7 macrophages from the effects of anthrax lethal toxin. RAW264.7 cells were treated with varying amounts of a combination of PA and LF in the presence of increasing concentrations of compounds. Following incubation, cell viability was assessed using a colorimetric MTT reduction assay. Of the 30 compounds tested, seven offered significant protection from LF activity. Diphyllin (2) at 3 μM afforded nearly complete protection against the LF–PA-mediated cell killing, while NCGC00084148-01 (designated 148-01) (1) and niclosamide (3) provide the same protection at ∼10 μM (Fig. 3 ). The known kinase inhibitor GF 1092038 was also found to have protective effects on RAW264.7 cells; however, GF 109203 did not achieve the same potency or efficacy and was noted to be cytotoxic at concentrations above 10 μM. Additionally, we assayed the alkaloid and potent myelosuppressive homoharringtonine9 (designated 1A/61951), the KDO-8-P synthase inhibitor PD 40418210 (designated K4607) and 2-(4-methylpyrimidin-2-yl)benzo[d]isothiazol-3(2H)-one (designated 889-01). None of these other leads from the primary screen were noted as having protective effects on RAW264.7 cells. Given the strong protective effect demonstrated by diphyllin (2), we ascertained the protective effects of this agent across a concentration range of LT and after a 45 min pre-incubation of LT. The results show that diphyllin (2) is effective as a protective agent up to 1000 ng/mL concentrations of LT and demonstrates protective effects even after 45 min pre-exposure to LT (Fig. 4 ).
Figure 3.
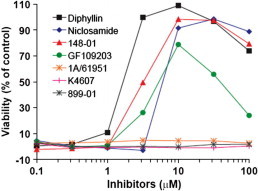
Effects of inhibitors on cytotoxicity of RAW264.7 cells by anthrax lethal toxin. RAW264.7 cells that were pre-incubated with various concentrations of the inhibitors for 30 min were treated with 100 ng/ml PA and 100 ng/ml LF for 4 h. Cell viability then was measured by adding MTT. Among 30 compounds tested (only seven shown in the figure) NCGC00084148-01 (listed as 148-01) (1), diphyllin (2) and niclosamide (3) and GF 109203 showed clear protection to RAW264.7 cells from LT. GF109203 displayed cytotoxicity when used at high concentrations (>10 μM)].
Figure 4.
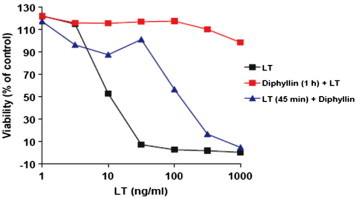
Effects of diphyllin (2) on cytotoxicity of anthrax lethal toxin toward RAW264.7 cells. Viability of RAW264.7 cells after treatment with toxin either in the absence of inhibitor, with a 1 h pre-incubation of cells with diphyllin (2) (10 μM), and with diphyllin (2) added 45 min after toxin addition.
2.3. Protective effect on CHO cells from the fusion toxin FP59
Next we determined if NCGC00084148-01 (1), diphyllin (2) and niclosamide (3) protected cells from killing by interfering with delivery of LF proteins to their cytoplasmic targets. For this purpose, CHO cells were incubated with PA and FP59 in the presence of the putative inhibitors. FP59 is a fusion protein consisting of the PA-binding LF amino-terminal fragment and the catalytic domain of Pseudomonas exotoxin A (PE). FP59 kills cells by ADP-ribosylating the diphthamide residue on elongation factor-2 (EF-2). Diphthamide is a unique post-translationally modified histidine residue found only in EF-2, and conserved from archaebacteria to humans, serving as the target for diphtheria toxin (DT) and PE.11, 12 These studies showed that NCGC00084148-01 (1) diphyllin (2) and niclosamide (3) were all capable of cell protection at micromolar concentrations (data not shown) indicating a potentially shared mechanism of action with the protective effects of these agents versus LT.
2.4. Protective effect on CHO cells from diphtheria toxin (DT)
Next, NCGC00084148-01 (1), diphyllin (2) and niclosamide (3) were studied for their ability to block the actions of DT in CHO cells. This toxin utilizes a different receptor, does not require PA for endocytosis, and exits from endosomes at an earlier stage than LF.13 Interestingly, diphyllin (2) and niclosamide (3) provided complete protection (100% cell viability) at approximately 5 μM while NCGC00084148-01 (1) and GF 109203 produced approximately 75% and 60% cell viability at a higher concentration (10 μM), respectively (Fig. 5 ). This strongly suggests that the mechanism by which NCGC00084148-01 (1), diphyllin (2) and niclosamide (3) block the toxic effects of LF does not involve interfering with the binding of PA and/or LF to its receptors, but rather the common process of toxin internalization, including endosome acidification and endocytosis.
Figure 5.
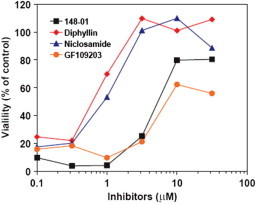
Protective effect of the compounds on CHO cells exposed to diphtheria toxin. Viability of CHO cells in the presence of diphtheria toxin (25 ng/mL) and increasing amounts of NCGC00084148-01 (listed as 148-01) (1), diphyllin (2) and niclosamide (3) and GF 109203.
2.5. Blocking of PA heptamer prepore-to-pore conversion
That NCGC00084148-01 (1), diphyllin (2) and niclosamide (3) protect RAW264.7 cells from the effects of PA and LF, and CHO cells from PA + FP59 and diphtheria toxin suggests that these compounds act on the process of endocytosis, or endosomal acidification just prior to pore formation and toxin translocation. It has previously been shown that pore formation is triggered by an acidification of the endosomal pH.14 To discriminate among these possibilities, we used PA receptor-deficient CHO PR230 cells transfected with either tumor endothelial cell marker 8 (TEM8) (PR230 TEM8-T4 cells) or capillary morphogenesis gene 2 product (CMG2) (PR230 CMG2-C4 cells).15, 16 Both TEM8 and CMG2 are well characterized PA receptors, but they differ significantly in their pH dependence for pore formation and LF translocation.15 The TEM8 receptor requires a relatively mild acidic environment (pH 6.0–6.5) to allow PA pore formation, while the CMG2 receptor requires a relatively low pH (5.5–6.0). Conversion of the SDS/heat-sensitive PA heptamer prepore to the SDS/heat-resistant functional LF-conductive pore requires acidic pH in endosomes.
The TEM8- and CMG2-expressing cells were treated with PA and LF in the presence of NCGC00084148-01 (1), diphyllin (2) and niclosamide (3), GF 109203 or the known vacuolar (H+)-ATPase (V-ATPase) inhibitor bafilomycin. PA binding, proteolytic processing, PA heptamer formation, and PA-dependent LF binding to cells were assessed by SDS–PAGE followed by Western blotting using PA and LF antibodies as described previously.5 None of the compounds blocked cellular binding, proteolytic processing, heptamerization of PA, or PA-mediated LF binding. In contrast, all four compounds disrupted heptamer prepore-to-pore conversion in CMG2-expressing cells to a degree comparable to bafilomycin, with markedly less effects on TEM8 expressing cells (Fig. 6 ). Interestingly, diphyllin displayed a modest effect on TEM8 cells. This trend was mirrored in the viabilities of the cells in the presence of each agent across a concentration gradient (Fig. 7 ). These results suggest that the protective effect of these agents is the result of preventing endosome acidification (most likely through the inhibition of V-ATPase action) or blocking of toxin trafficking to endosomes of increased acidity. It is possible to differentiate between these mechanisms through the use of pH sensors that allow direct quantification of endosomal acidity (these experiments were not performed). Importantly, Collier and coworkers recently demonstrated that selective mutagenesis of phenylalanine 427 of PA (a crucial residue for membrane transport of PA) produced variable effects on PA function.17 Selected mutants (F427K, F427R, F427D, F427G) were found to mitigate the pH-induced conformational change associated with pore formation. Other mutations (F427H, F427S, F427T) did not affect prepore-to-pore conversion but inhibited translocation of the LF construct. This study clearly demonstrates that small molecule intervention of cellular entry of the PA–LF construct can occur through different mechanisms.
Figure 6.
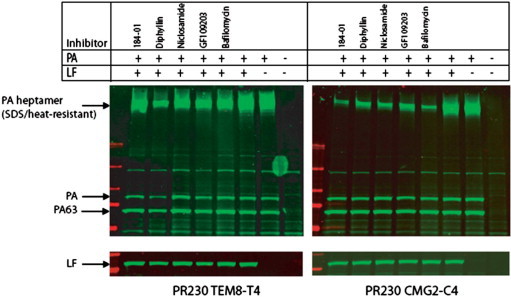
Effects of the inhibitors on anthrax toxin binding, proteolytic processing, and heptamer formation on CHO cells. Anthrax toxin receptor-deficient CHO PR230 cells transfected with either TEM8 (PR230 TEM8-T4, left panel) or CMG2 (PR230 CMG2-C4, right panel) were pre-treated with 10 μM of NCGC00084148-01 (listed as 148-01) (1), diphyllin (2) and niclosamide (3) and GF 109203, or 0.2 μM of Bafilomycin A1 for 30 min. Then the cells were incubated with 1 μg/ml PA and 1 μg/ml LF for 1 h at 37 °C, washed to remove unbound toxin, and lysed using modified RIPA buffer. Cell lysates boiled in SDS sample buffer were separated by SDS-PAGE, followed by Western blotting using PA antiserum (no. 5308) or LF antiserum (no. 5309) for the upper and lower images, respectively.
Figure 7.
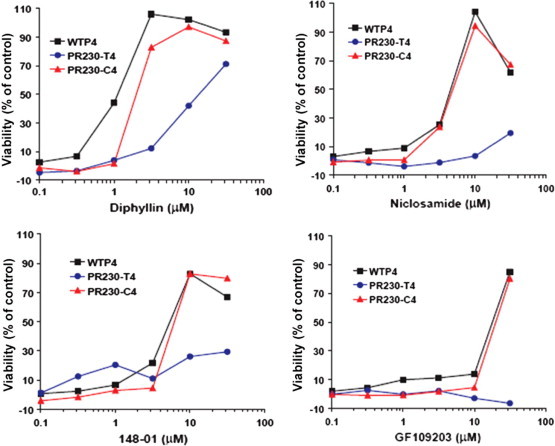
Effects of inhibitors on viability of PR230 TEM8-T4 and PR230 CMG2-C4 cells treated with PA and LF. Protective effects by NCGC00084148-01 (listed as 148-01) (1), diphyllin (2) and niclosamide (3) and GF 109203 mirror the heptamer formation results by protecting PR230 CMG2-C4 cells, but not PR230 TEM8-T4 cells, indicating a mechanism of action that blocks late-stage vacuolar release but not early stage vacuolar release of LF complex. The cells were pre-incubated with drugs for 30 min, then 100 ng/ml PA + 100 ng/ml FP59 (final concentration) were added and incubated for 3 h. After that the toxin/durg-containing media were replaced with regular growth media containing 10 mM of ammonium chloride and incubated for 48 h as described in Section 4.5.
Interestingly, it has been demonstrated that V-ATPases are important mediators of cellular entry by a number of viruses, including Ebola and the Severe Acute Respiratory Syndrome coronavirus (SARS-CoV),18 and the ability of bafilomycin to block endosomal acidification and limit viral entry into cells is well known.19, 20 Recently, Diphyllin (2) was noted to be a potent inhibitor of V-ATPase activity and was further shown to affect bone resorption in osteoclast models.21 This study is consistent with our findings and yields insight into this particular mechanism for inhibition of LF internalization. That niclosamide (3) potentially acts as a V-ATPase inhibitor is possibility relevant to recent reports suggesting niclosamide may serve as a therapy for SARS-CoV infection.21 Niclosamide, found here to disrupt heptamer prepore-to-pore conversion in the pH sensitive CHO-CMG2 cell line, eliminated SARS-CoV viral antigen synthesis at modest concentrations (1.56 μM).22 The mechanism of niclosamide’s effect on viral replication is unresolved; though modifications of niclosamide’s core 2-chloro-4-nitroaniline substructure lead to inhibitors of the SARS 3CL protease23 (niclosamide had only weak inhibitory activity (IC50 40 μM)23, 24 against this target). The possibility that niclosamide (3) does act on V-ATPase is intriguing and may yield insight into its effect on viral entry. It should be noted that niclosamide (CID: 4477) appears as an active compound numerous times in the Pubchem online database.
The discovery of the novel chromeno[4,3-c]pyrazol-4(1H)-one (NCGC00084148-01) (1) is also of interest. It has been hypothesized that the presence of highly basic tertiary amine moieties such as those present in NCGC00084148-01 (1) may directly neutralize endosomal pH; for example, GF109203 inhibits anthrax toxin entry into cells.25 However, in this study, we found no such correlation between highly basic compounds and activity within the primary screen, and analogues of NCGC00084148-01 (1) that retained the piperizine moiety but differed in other significant ways were inactive in our experiments, suggesting that at least under these conditions these compounds do not act via direct neutralization of endosomal pH.
3. Conclusion
Anthrax remains a significant bioterrorism threat, given its ease of propagation and the physical resistance properties of its endospores. While some vaccines and treatments are available, these are frequently ineffective or associated with significant adverse effects. Thus, effective and rapidly acting treatments for anthrax disease are urgently needed. High-throughput screening of large chemical libraries can identify leads for such programs, but is costly and cumbersome to perform in restrictive biosafety level environments. Screening of pathogen pathways reconstituted in non-pathogenic cell types, coupled with rapid follow-up testing of active compounds in the pathogenic organism, offers an attractive alternative to direct pathogen screening. The assay used here takes advantage of the complex process of anthrax intoxication (Fig. 1) to allow the screening of a large chemical library for compounds that affect multiple steps of the intoxication cascade.
A quantitative high-throughput screen for small molecules capable of modulating the internalization of anthrax lethal factor was accomplished. Follow-up studies allowed the rapid triage of compounds that were inactive in physiological anthrax intoxication assays. These studies successfully identified compounds with activity not only in the original assay but also on LF-induced cell killing. Of the 30 compounds identified in the primary assay and tested in native LF cell killing assays, 7 (23%) protected against LF-induced macrophage killing. The clinically used teniacide niclosamide, (3) the natural product diphyllin, (2) and a novel chromeno[4,3-c]pyrazol-4(1H)-one (NCGC00084148-01) (1) all protected macrophages and CHO cells from cell death induced by PA combined with LF and diphtheria toxin. Since the primary assay detects compounds acting via a variety of mechanisms, a series of mechanism of action studies were undertaken on three selected active compounds, NCGC00084148-01 (1), diphyllin (2) and niclosamide (3). All three compounds were found to protect CHO cells from the fusion toxin FP59 and from cell killing by diphtheria toxin (which utilizes a different receptor and does not require PA for endocytosis), suggesting that the compounds interfere with toxin internalization. Blockade of PA heptamer prepore-to-pore conversion was then measured in two cell lines where endosomal pH requirements differ. All three compounds influenced heptamer prepore-to-pore conversion at the same concentrations which affect cell viability of each cell line and to a degree comparable to the known vacuolar (H+)-ATPase (V-ATPase) inhibitor bafilomycin. These results suggest a mechanism for NCGC00084148-01 (1), diphyllin (2) and niclosamide (3) that is directly linked to endosome acidification (either inhibition of V-ATPase function or blocking of toxin internalization and/or trafficking to endosomes of increased acidity). These compounds and their derivatives promise to be useful in the study of anthrax pathogenesis and the assay will be useful for identifying additional anthrax protective compounds acting via alternative mechanisms.
4. Experimental
4.1. General procedures
Anthrax toxin LF-β-Lac was generated and purified as described.3 Recombinant B. anthracis LF, PA, and FP59 were made as previously described.11 CCF2/AM (Coumarin cephalosporin fluorescent acetoxymethyl ester) and its loading buffers were purchased from Invitrogen (Carlsbad, CA). Cervix epithelial cancer line ME-180 was from American Type Culture Collection (Manassas, VA) and cultured in DMEM with 10% FBS (Fetal Bovine Serum, Gibco-BRL, Gaithersburg, MD) and antibiotics.
4.2. Assay miniaturization and quantitative high-throughput screen
Cells and reagents were prepared as described,3 and the assay was optimized in a miniaturized 1536-well plate format. The qHTS was performed using a fully automated screening system in a single run, according to the following protocol: 5 μL of ME-180 cells (approximately 2000 cells) were dispensed into each well of the 1536 well plates and allowed to incubate for 18 h in a 37 °C/5% CO2 environment. The screening library of 70,094 compounds was composed of compounds collected from several sources: 1279 compounds from LOPAC (Sigma-Aldrich, St. Louis, MO), 1120 compounds from Prestwick (Prestwick Chemical Inc., Illkirch, France), 280 purified natural products from TimTec (Newark, DE), three 1000-member combinatorial libraries from Pharmacopeia (Princeton, NJ), 973 compounds from Tocris (Bristol, UK), 1981 compounds from the National Cancer Institute, 718 compounds from Boston University Center for Chemical Methodology and Library Development, and the rest were from the NIH Molecular Libraries Small Molecule Repository. The library was prepared at a minimum of 7 concentrations in DMSO as described.7 All compounds were dispensed via pintool, to a final concentration range of 2.6 nM–40 μM. A combination of engineered LF-β-lac and PA (1 μg/mL) or engineered LF-β-lac alone (as a control) was then added, and the mixture incubated for 2 h at 37 °C/5% CO2. The FRET β-lactamase substrate CCF2/AM was then added to a final concentration of 5 μM, and following a 4-h incubation at room temperature, plates were read in an EnVision plate reader (PerkinElmer, Boston MA), with excitation at 405 nm and emission detection at 460 nm (blue channel) and 530 nM (green channel).
4.3. Analysis of qHTS data
Screening data was processed using in-house developed software. Percent activity was computed from the median values of the uninhibited, or neutral, control set and a set of control wells containing no LF/PA. For assignment of plate concentrations and sample identifiers, ActivityBase (ID Business Solutions Ltd, Guildford, UK) was used for compound and plate registrations. An in-house database was used to track sample concentrations across plates. Correction factors were generated from control assay plates containing vehicle (DMSO) only inserted uniformly throughout the screen to monitor background systematic variation in assay signal potentially resulting from issues with reagent dispensers or decrease in enzyme specific activity. Curve fitting was performed using an in-housed developed algorithm. A four parameter Hill equation was fitted to the concentration–response data by minimizing the residual error between the modeled and observed responses. Outliers were identified and masked by modeling the Hill equation and determining if the differences exceeded those expected from the noise in the assay.
The curve classification used is the same as described previously,7 where concentration–response curves are classified based on efficacy, potency, and statistical fit. Curves are classified as negative or positive depending on whether they exhibit signal decrease (apparent inhibition) or increase (apparent activation). Translocation inhibitors are noted as negative response curves in the blue emission channel, and as flat responses in the green channel. Natively fluorescent molecules were noted to stimulate both channels. A negative response in both channels typically indicates non-specific fluorophore quenching or non-specific mechanisms of inhibition, such as cellular apoptosis. The ratio of the green and blue channels normalizes for variation in cell number, is a more robust measure of compound performance, and further allows compounds to be classified as potential inhibitors of LF translocation.
Once an active set of compounds was identified, hierarchical agglomerative clustering with a 0.7 Tanimoto cutoff was performed using Leadscope (Leadscope Inc., Columbus, OH) fingerprints, which are ideally suited for two-dimensional scaffold-based based clustering. For each cluster, maximal common substructures (MCS) were extracted, and a manual step of trimming the MCSs was performed to create a list of scaffolds. This clustering step typically has overlapping compounds and thus can lead to overlapping MCSs. This list of trimmed scaffolds is abridged to a canonical set. Each scaffold is then represented as a precise definition to indicate number of attachments, ring size variability, etc. All filters were then relaxed to include all negative assay data. In the initial clustering, a set of singletons was found. These compounds were reported upon separately with their individual activity profiles. SAR series and singletons were finally ranked by their activity profile, including the average potency and efficacy of the series. The primary screening results can be viewed in the PubChem database (AIDs 912 and 942, http://pubchem.ncbi.nlm.nih.gov/).
4.4. Fluorescent imaging assay
ME-180 cells were seeded at a density of 10,000 cells per well in black walled 96-well plates. After overnight incubation, the cells were washed twice by phenol red-free DMEM and incubated with 13 nM (1 μg/ml) PA and 18 nM (1 μg/ml) LF-β-Lac for 0.5 h at 37 °C; the compounds were then added at final concentrations of 50, 10, 2 and 0.4 μM to duplicate wells for each concentration. After a1 h incubation, the cells were washed twice with DMEM and loaded with 1.5 μM CCF2/AM using the alternative substrate loading solution from Invitrogen. Cells were incubated for 4 h at room temperature in the dark. Cells were visualized and photographed using a Zeiss Axiovert 200 M inverted microscope with Zeiss Axiovision software (Carl Zeiss, Jena, Germany). For acquisition of blue fluorescence, excitation filter HQ405/20 nm bandpass, and emitter filter HQ460/40 nm bandpass were used. For green fluorescence, HQ405/20 nm bandpass, and emitter filter HQ530/30 nm bandpass were used. All filters were purchased from Chroma Technology (Rockingham, VT). To quantify the signals, plates were then read in a Wallac Victor 2 plate reader (Perkin Elmer, Wellesley, MA). The following filters were used in dual read mode: CW-lamp filter 400DF10, emission filters F460 for blue fluorescence and F535 for green fluorescence. The ratios of blue/green were calculated for each concentration, the wells with PA plus LF-β-Lac were used as positive control and the wells with LF-β-Lac only as negative control. The blue/green ratio (B/G ratio) at each concentration was normalized to the average ratio of positive controls.
4.5. Cytotoxicity assays
Cytotoxicity assays were performed as previously described.26 RAW264.7 cells (American Type Culture Collection) were grown in DMEM with 10% FBS, 2 mM Glutamax, 2 mM Hepes, and 50 μg/ml gentamycin (all from Invitrogen) at 37 °C in a 5% CO2 atmosphere. CHO cells were grown in as previously described.27 Compounds in DMSO (in serial dilutions) were added to cells for 30 min, followed by addition of the combinations of PA and LF or PA and FP59 or DT as indicated. For RAW264.7 macrophages, cells were incubated for 3 h at 37 °C, followed by addition of 3-[4,5-dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide (MTT) to a final concentration of 0.5 mg/ml. After additional 45 min incubation, all medium was removed and cells were dissolved in 50 μL of 0.5% (wt/vol) SDS/25 mM HCl, in 90% (vol/vol) isopropyl alcohol. Plates were vortexed, and the oxidized MTT was measured at A 570 by a microplate reader. For CHO cells, cells were incubated with PA and FP59 or DT for 3 h, followed by changing the compound/toxin-containing medium with regular growth medium containing 10 mM ammonium chloride thereafter to block the toxin-entry into the cytosol of the cells.15 After incubation for 48 h, MTT cell viability measurement was performed as described above. Percent viability was calculated as a percentage of medium-treated controls. EC50 values for protection curves were calculated by using prism 4.0 software (graphpad, San Diego).
4.6. Analysis of PA heptamer formation
This assay was performed essentially as previously described.5 Briefly, cells were cultured in 24-well plates to confluence, washed, and incubated in serum-free DMEM with 1 μg/ml PA. The cells were washed five times to remove unbound PA. Cells were lysed in 100 μl/well of modified radioimmune precipitation lysis buffer (RIPA buffer: 50 mM Tris–HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml each of aprotinin, leupeptin, and pepstatin) on ice for 10 min. Equal amounts of protein from cell lysates and equal volumes of the conditioned media were separated by PAGE using 4–20% gradient Tris-glycine gels (Novex). Western blotting was performed using a polyclonal PA antibody (no. 5308) and a polyclonal LF antibody (no. 5309).
Acknowledgments
We thank Carleen Klumpp and Sam Michael for assistance with the robotic screen, and Adam Yasgar and Paul Shinn for assistance with compound management. This research was supported by: the Intramural Research Programs of the National Institute of Dental and Craniofacial Research, National Institute of Allergy and Infectious Diseases, and National Human Genome Research Institute; by the NIAID Support of Intramural Biodefense Research from ICs other than NIAID, and by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research.
References and notes
- 1.Inglese J., Johnson R.L., Simeonov A., Xia M., Zheng W., Austin C.P., Auld D.S. Nat. Chem. Biol. 2007;3:466. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 2.Walsh D.P., Chang Y.-T. Chem. Rev. 2006;106:2476. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]
- 3.Hobson J.P., Liu S., Rønø B., Leppla S.H., Bugge T.H. Nat. Methods. 2006;3:259. doi: 10.1038/nmeth862. [DOI] [PubMed] [Google Scholar]
- 4.Moayeri M., Leppla S.H. Curr. Opin. Microbiol. 2004;7:19. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Liu S., Leppla S.H. J. Biol. Chem. 2003;278:5227. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- 6.Austin C.P., Brady L.S., Insel T.R., Collins F.S. Science. 2004;306:1138. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 7.Inglese J., Auld D.S., Jadhav A., Johnson R.L., Simeonov A., Yasgar A., Zheng W., Austin C.A. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11473. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis P.D., Elliott L.H., Harris W., Hill C.H., Hurst S.A., Keech E., Kumar M.K.H., Lawton G., Nixon J.S., Wilkinson S.E. J. Med. Chem. 1992;35:994. doi: 10.1021/jm00084a004. [DOI] [PubMed] [Google Scholar]
- 9.Witte R.S., Hsieh P., Elson P., Oken M.M., Trump D.L. Invest. New Drugs. 1996;14:409. doi: 10.1007/BF00180819. [DOI] [PubMed] [Google Scholar]
- 10.Brick M.R., Holler T.P., Woodard R.W. J. Am. Chem. Soc. 2000;122:9334–9335. [Google Scholar]
- 11.Liu S., Leppla S.H. Mol. Cell. 2003;12:603. doi: 10.1016/j.molcel.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Liu S., Milne G.T., Kuremsky J.G., Fink G.R., Leppla S.H. Mol. Cell Biol. 2004;24:9487. doi: 10.1128/MCB.24.21.9487-9497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yates S.P., Jørgensen R., Andersen G.R., Merrill A.R. Trends Biochem. Sci. 2006;2:123. doi: 10.1016/j.tibs.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Rainey G.J.A., Wigelsworth D.J., Ryan P.L., Scobie H.M., Collier R.J., Young J.A.T. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13278. doi: 10.1073/pnas.0505865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen K.-H., Liu S., Bankston L.A., Liddington R.C., Leppla S.H. J. Biol. Chem. 2007;282:9834. doi: 10.1074/jbc.M611142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu S., Leung H.J., Leppla S.H. Cell. Microbiol. 2007;9:977. doi: 10.1111/j.1462-5822.2006.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J., Lang A.E., Aktories K., Collier R.J. Proc. Natl. Acad. Sci. U.S.A. 2009;105:4346. doi: 10.1073/pnas.0800701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Science. 2005;308:1643. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsh M., Helenius A. Cell. 2006;124:729. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayer N., Schober D., Prchla E., Murphy R.F., Blass D., Fuchs R. J. Virol. 1998;72:9645. doi: 10.1128/jvi.72.12.9645-9655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sørensen M.G., Henriksen K., Neutzsky-Wulff A.V., Dziegiel M.H., Karsdal M.A. J. Bone Miner. Res. 2007;22:1640. doi: 10.1359/jbmr.070613. [DOI] [PubMed] [Google Scholar]
- 22.Wu C.-J., Jan J.-T., Chen C.-M., Hsieh H.-P., Hwang D.-R., Liu H.-W., Liu C.-Y., Huang H.-W., Chen S.-C., Hong C.-F., Lin R.-K., Chao Y.-S., Hsu J.T.A. Antimicrob. Agents Chemother. 2004;48:2693. doi: 10.1128/AAC.48.7.2693-2696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shie J.-J., Fang J.-M., Kuo C.-J., Kuo T.-H., Liang P.-H., Huang H.-J., Yang W.-B., Lin C.-H., Chen J.-L., Wu Y.-T., Wong C.-H. J. Med. Chem. 2005;48:4469. doi: 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- 24.Wen C.-C., Kuo Y.-H., Jan J.-T., Liang P.-H., Wang S.-Y., Liu H.-G., Lee C.-K., Chang S.-T., Kuo C.-J., Lee S.-S., Hou C.-C., Hsiao P.-W., Chien S.-C., Shyur L.-F., Yang N.-S. J. Med. Chem. 2007;50:4087. doi: 10.1021/jm070295s. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez A.M., Thomas D., Gillespie E.J., Damoiseaux R., Rogers J., Saxe J.P., Huang J., Manchester M., Bradley K.A. Antimicrob. Agents Chemother. 2007;51:2403. doi: 10.1128/AAC.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S., Bugge T.H., Leppla S.H. J. Biol. Chem. 2001;276:17976. doi: 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- 27.Liu S., Redeye V., Kuremsky J.G., Kuhnen M., Molinolo A., Bugge T.H., Leppla S.H. Nat. Biotechnol. 2005;23:725. doi: 10.1038/nbt1091. [DOI] [PMC free article] [PubMed] [Google Scholar]