

Abstract
There have been no recent advances in drug development for mood disorders in terms of identifying drug targets that are mechanistically distinct from existing ones. As a result, existing antidepressants are based on decades-old notions of which targets are relevant to the mechanisms of antidepressant action. Low rates of remission, a delay of onset of therapeutic effects, continual residual depressive symptoms, relapses, and poor quality of life are unfortunately common in patients with mood disorders. Offering alternative options is requisite in order to reduce the individual and societal burden of these diseases.
The glutamatergic system is a promising area of research in mood disorders, and likely to offer new possibilities in therapeutics. There is increasing evidence that mood disorders are associated with impairments in neuroplasticity and cellular resilience, and alterations of the glutamatergic system are known to play a major role in cellular plasticity and resilience. Existing antidepressants and mood stabilizers have prominent effects on the glutamate system, and modulating glutamatergic ionotropic or metabotropic receptors results in antidepressant-like properties in animal models. Several glutamatergic modulators targeting various glutamate components are currently being studied in the treatment of mood disorders, including release inhibitors of glutamate, N-methyl-D-aspartate (NMDA) antagonists, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) throughput enhancers, and glutamate transporter enhancers. This paper reviews the currently available knowledge regarding the role of the glutamatergic system in the etiopathogenesis of mood disorders and putative glutamate modulators.
Keywords: AMPA, antidepressant, bipolar disorder, depression, drug development, glutamate, metabotropic, NMDA
INTRODUCTION
Mood disorders (particularly bipolar disorder (BPD) and major depressive disorder (MDD)) are among the most severe of all psychiatric disorders. They affect the lives of millions worldwide and are often incapacitating, leading to significant psychosocial dysfunction. Mood disorders typically commence in childhood [1] and usually progress unabated into adulthood [2]. For many individuals suffering from these illnesses, the course is characterized by recurrent mood episodes with residual mood symptoms, functional impairment, cognitive deficits, and disability [3]. Compared to other medical illnesses, the World Health Organization’s (WHO) Global Burden of Disease project ranked MDD within the top five causes of disability in 1990, and projected that by 2020 it would be the second leading cause of disability worldwide. BPD also ranked among the most disabling of all non-communicable illnesses [4].
Fortunately for many patients suffering from these recurrent mood disorders, a variety of pharmacological treatments are available. For acute manic episodes and its subtypes, these options consist mainly of lithium, anticonvulsants (e.g., valproate) and both typical and atypical antipsychotic agents [5–7]. However, for most patients with BPD, monotherapy is often insufficient and combination treatment is required [8]. Furthermore, it has become apparent in recent years that these options are far from adequate in treating acute episodes of the illness, relapses, and recurrences, and in restoring premorbid functioning. For long-term prophylaxis, far fewer agents are available; they include lithium, aripiprazole, and lamotrigine, but of these only lamotrigine has been shown to help prevent depressive relapses [9]. In addition, some patients are unable to tolerate existing therapies for BPD, which leads to either frequent changes in medications or to non-adherence [10–12].
In the treatment of MDD, available antidepressants are based on theories developed and popularized over 40 years ago [13], and drug development in mood disorders research noticeably lags behind other areas in medicine [14, 15]. Drugs that are mechanistically distinct from previous ones are still not available for either MDD or BPD and are urgently needed [16]. The dearth of knowledge of the precise molecular and cellular basis of these complex disorders is perhaps a large reason for the lack of progress in developing novel therapeutics that are more effective, better tolerated, and act more rapidly than current drug therapies.
Two recently completed effectiveness studies in mood disorders highlight the limitations in terms of efficacy of our current pharmacological treatments. The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) found that only approximately one-third of patients with MDD achieved remission with an adequate trial (i.e., dose, duration) of a conventional antidepressant (i.e., citalopram) after approximately 10–14 weeks of treatment [17]. This work highlights that many individuals could benefit from better treatments than those currently available, and emphasizes that response and remission rates with any given antidepressant treatment have remained essentially unchanged since these drugs were first introduced in the early 1960s. Thus, regardless of whether patients are prescribed monoamine oxidase inhibitors, tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), or newer serotonin and norepinephrine reuptake inhibitors (SNRIs), response rates range between 60–70%. Moreover, in most double-blind, placebo-controlled studies, three to six weeks of antidepressant administration are usually required to produce a therapeutic response; for remission the time period is even greater. Given the difficulties in adherence to treatment and the risk of self-injurious behavior, particularly in the first month of starting an antidepressant [18], the need for rapid-acting antidepressants with high response rates remains as pressing now as it was several decades ago.
One contemporary theory that has prominent support in the scientific community is that mood disorders arise from alterations in cellular resilience and neuroplasticity [19]. This theory is supported in part by evidence generated from both neuroimaging and postmortem studies. Structural imaging studies in patients with recurrent mood disorders have consistently reported small reductions in the gray matter volume of brain regions implicated in mood disorders such as the orbital, medial, and dorsolateral prefrontal cortex, and left subgenual prefrontal cortex (reviewed in [20, 21]). Similarly, neuropathological investigations have consistently found reductions in cortex volume, glial cell counts, and/or neuron size in the subgenual prefrontal cortex, orbital cortex, dorsal anterolateral prefrontal cortex, amygdala, basal ganglia, and dorsal raphe nuclei (reviewed in [22, 23]). Based on the conjecture that abnormalities in neuroplasticity and resilience are likely implicated in mood disorders, different theories have been postulated to account for these findings, including impairment in neurotrophins [24], bioenergetics [25], and glutamatergic function [19, 26]. A comprehensive review of emerging experimental therapeutics and theories of impairments in neuroplasticity and cellular resilience in mood disorders is summarized elsewhere [27]. This review will focus on the glutamatergic system.
Glutamate is one of the key excitatory neurotransmitters in the mammalian brain and has been under study for several decades for a range of neuropsychiatric illnesses [28].This excitatory amino acid was first proposed as a neurotransmitter as early as 1959. It is now known that neurons employing excitatory amino acids for neurotransmission comprise approximately one-third of the neurons in the forebrain and that these neurons are non-uniformly dispersed in the central nervous system (CNS). High densities of glutamate neurons reside in cortex and in subcortical structures such as hippocampus, caudate nucleus, thalamic nuclei, and in the cerebellum [29].
With regards to mood disorders, the role of the glutamatergic system was first pursued several decades ago [30, 31] mainly in the form of preclinical studies. Early reports described the action of antidepressants on glutamatergic receptors and the antidepressant-like effects of N-methyl-D-aspartate (NMDA) antagonists in animal models. These preclinical data were complemented by a preliminary study that found than an NMDA antagonist had rapid antidepressant effects in MDD[32]. However, for unclear reasons, work on glutamate in mood disorders remained at a lull until recently, when a series of preclinical and clinical studies “re-discovered” the importance of glutamate in mood disorders; it is now a very active area of research.
Work at several different levels from in vitro to human clinical studies indicates that alterations in the modulation of glutamate neurotransmission are likely fundamental aspects of the etiopathophysiology of recurrent mood disorders. Human assay studies, which provide a window into glutamatergic function in patients with mood disorders, consist of measuring glutamate in the peripheral (serum) and central (cerebral spinal fluid (CSF), brain) tissue of individuals with mood disorders. Evidence from these studies points to a glutamatergic dysfunction in patients with mood disorders. Whether this is a primary disturbance, an epiphenomenon, or a consequence of the illness remains to be determined. Although enlightening, there are only so many assumptions that can be made from these results because of methodological limitations (reviewed in [26]). Taken together, however, this small body of work suggests that abnormalities are apparent within the glutamatergic system of patients with mood disorders, but the magnitude and extent of the abnormalities still need to be clarified. Elevated glutamate concentrations in the occipital cortex of depressed patients with reduced levels in the anterior cingulate cortex appear to be the most consistent findings in patients with mood disorders. Other MRS imaging studies show changes in Glx brain levels in different brain regions; Glx is an in vivo measure of brain glutamate content that predominantly reflects glutamate content but also contains glutamine and GABA components. The key findings in humans and animals are summarized in Table 1.
Table 1.
Glutamate levels in brain of patients with mood disorders and in animal models of depression
| • | Decreased Glx levels in the ACC of severely depressed patients vs. controls |
| • | Decreased Glx levels in the DLPFC of depressed patients. Levels were found to increase following response to treatment with ECT |
| • | Significant reductions of Glx content in two regions of the PFC in subjects with MDD |
| • | Glx levels reduced in ACC but not OCC in pediatric MDD. Reanalysis of the spectra suggest the decrease is specifically related to a 23% reduction in glutamate |
| • | Small elevations in gray matter Glx in patients in the depressed and mixed phases of BPD. Differences in gray matter Glx greatest in BPD II subjects. These differences partially normalized following treatment with lithium. |
| • | Elevated levels of Glx found in the frontal lobe and basal ganglia of medication-free depressed children with BPD compared to a control group |
| • | Glx levels in a small group of mixed medicated and non-medicated children with BPD failed to show any significant difference from healthy comparison subjects in the ACC |
| • | Higher Glx/Cr ratios in the contralesional frontal cortex of patients experiencing depression immediately after a first ischemic stroke located outside the frontal lobes compared to non-depressive first ischemic stroke patients |
| • | Glutamate levels were found to be elevated in the frontal cortex in brains from patients with BPD and MDD |
| • | Significantly increased cortical glutamate in the occipital region of 29 depressed subjects. Increased glutamate concentrations especially evident in a subgroup of MDD subjects with melancholic features |
| • | Significant elevations of OCC Glu and Gln levels in medication-free recovered MDD subjects. Elevated Glx content in the OCC of remitted medication-free BPD subjects |
| • | Increased spectroscopic glutamate/GABA in rats. Treatment with desipramine or ECT decreased the ratio of these chemicals in the hippocampus and PFC |
ACC: anterior cingulated cortex; BPD: bipolar disorder; Cr: creatine; DLPFC: dorsolateral prefrontal cortex; ECT: electroconvulsive therapy; GABA: gamma-aminobutyric acid; Gln: glutamine; Glu: glutamate; Glx: an in vivo measure of brain Glu content that predominantly reflects Glu content but also contains Gln and GABA components; MDD: major depressive disorder; OCC: occipital cortex; PFC: prefrontal cortex
The next section provides an overview of the anatomy and physiology of the glutamate system in order to better understand the specific areas of dysfunction of this excitatory amino acid system.
A Brief Overview of Glutamate Neurotransmission
Glutamate neurotransmission requires three distinct cell types that comprise the typical glutamate synapse: an astrocyte, a presynaptic neuron, and a postsynaptic neuron (Figure 1) [33]. Glutamate is produced from α-ketoglutarate, an intermediate in the tricarboxylic acid cycle (TCA) of intermediary metabolism. Glutamate is then packaged into secretory vesicles in the presynaptic neuron by a family of at least three vesicular glutamate transporters (VGLUT1-VGLUT3) and released into the synapse from the neuron in an activity-dependent process through interactions with soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins (Figure 1; [34, 35]). The excitatory amino acid is then released pre-synaptically into the synaptic cleft. Synaptic glutamate can stimulate both metabotropic and ionotropic glutamate receptors, located in receptor-specific distributions on pre- and postsynaptic neurons, as well as on astrocytes [36, 37]. Glutamate receptor subtypes (Figure 1) include a group of pharmacologically distinct ligand-gated ion channels (NMDA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors) and the eight G-protein coupled metabotropic receptors (see Figure 1 and its legend). There is no process by which glutamate is metabolized; its concentrations are tightly regulated by glutamate re-uptake transporters localized on glia and neurons. The major glutamate transporter proteins found in the CNS include excitatory amino acid transporters EAAT1 (or GLAST-1), EAAT2 (or GLT-1), and EAAT3 (or EAAC1); EAAT2 is the most predominantly expressed form in the forebrain. EAAT1 and EAAT2 are found primarily in glial cells while EAAT3 is localized principally in neurons and EAAT4 in cerebellum (for review, see [33]).
Figure 1. Pathophysiological basis and potential therapeutic targets for mood disorders involving glutamatergic neurotransmission.
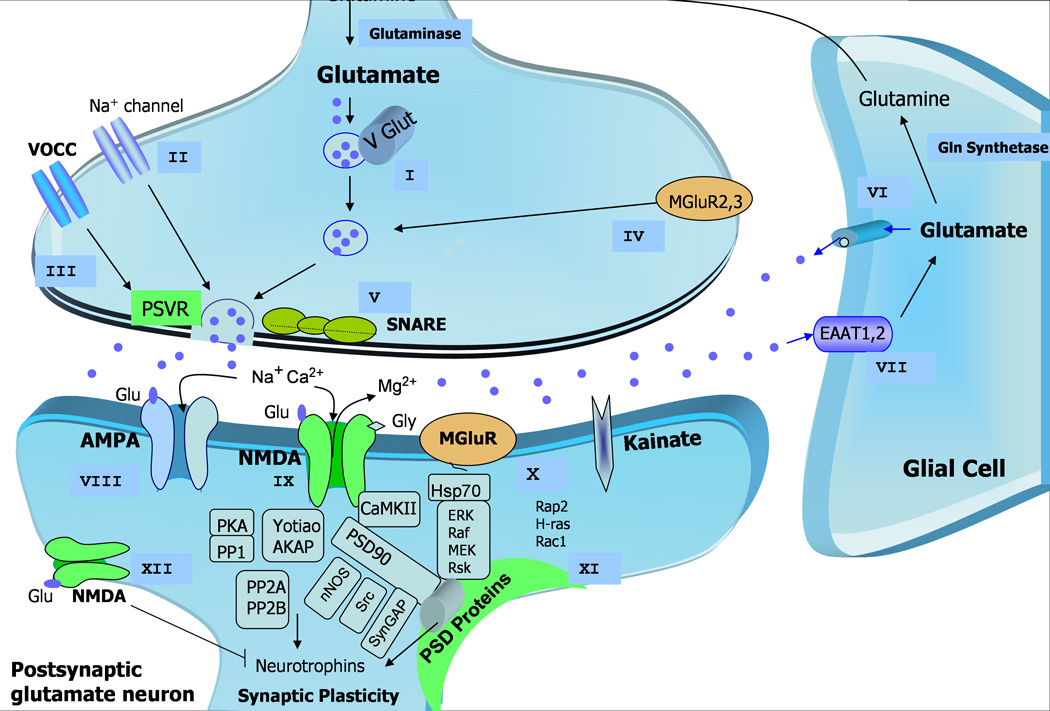
After uptake by glial cells, glutamate is transformed into glutamine, which in turn returns to the presynaptic neuron to be reconverted into glutamate by the enzyme glutaminase or synthesized de novo from glucose via the Krebs cycle (not shown). Glu is packaged into presynaptic vesicles and released in the synapse to target diverse postsynaptic receptors, including both ionotropic receptors — AMPA, NMDA, and kainate receptors — as well as mGluRs. These and other promising therapeutic targets are described as presynaptic (I–V), glial (VI, VII) and postsynaptic Glu targets (VIII–XII):
I- Regulation of presynaptic vesicular release of Glu. Glu is packaged into presynaptic vesicles by the VGLUTs, which critically modulates glutamate concentration in the synaptic vesicles and its consequent release in the synaptic cleft.
II- Voltage-dependent Na+ channel regulation that controls Glu release. Attenuation of voltage-activated sodium channel activity reduces the exaggerated action potential in the presynaptic terminal, thus limiting Glu release.
III- Presynaptic voltage-operated release of Glu regulates calcium accumulation in the synaptic terminal at the same time that it controls glutamate release.
IV- Up- and down-regulation of presynaptic group II mGluR modulation have anxiolytic and antidepressant activity.
V- Presynaptic vesicular Glu release from the neuron is directly regulated by the SNARE proteins.
VI- Direct effects of extrasynaptic Glu release from glial cells.
VII- Glu clearance from the extracellular space via high-affinity EAATs presented mainly in glial cells, which may have altered expression, thus facilitating accumulation of Glu in the synapse.
VIII- AMPA receptor regulation. Downregulation in AMPA receptor expression in different brain areas is found in mood disorders, which may be reversed by treatment with antidepressants, which also increase AMPA receptor trafficking.
IX- Synaptic NMDA receptor regulatory effects. Altered expression and functional polymorphisms have been demonstrated in mood disorders, which may increase the NMDA-receptor-mediated Ca2+ influx and Glu levels. Downregulating these targets and thus decreasing Glu levels may be beneficial in the therapeutics of mood disorders.
X- Group I mGluR modulation.
XI- Regulation in the expression and function of PSD proteins, also comprising glutamate receptors and anchor proteins, may represent an important therapeutic target because centralized Glu effects are modulated by diverse receptor subtypes and interactions with various signaling and scaffolding proteins in the PSD.
XII- extrasynaptic NMDA receptor modulation.
AMPA (-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid); Glu (Glutamate); Gly (Glycine); EAATs (excitatory amino-acid transporters); mGluRs (metabotropic Glu receptors); NMDA (N-methyl-D-aspartate); PSD (postsynaptic density); PSVR (presynaptic voltage-operated release); SNARE (soluble N-ethylmaleimide-sensitive factor attachment receptor); VGLUTs (vesicular Glu transporters);
The physiological processes regulating the activity of glutamate are still poorly understood, but phosphorylation of the transporters by protein kinases may differentially regulate glutamate transporters and therefore glutamate reuptake. Glutamate taken up by astrocytes is then converted to glutamine by the enzyme glutamine synthetase. Glutamine then exits the glial cell and is transported back to neurons where it is oxidized back into glutamate by the enzyme glutaminase and repackaged into vesicles for release (Figure 1). Recovered glutamate may enter the TCA cycle via conversion to α-ketoglutarate by glutamate dehydrogenase, thus completing the glutamate-glutamine cycle (for review, see [33]). It is important that glutamate synaptic concentrations be tightly regulated as there is evidence that it could rise to excitotoxic levels rapidly after an insult (e.g., trauma, ischemia) and that glutamate transporter function could be altered under these circumstances; thus a variety of components (i.e., packaging, release, reuptake) are intimately involved in tightly regulating glutamate synaptic concentrations.
The next section reviews key elements of brain glutamatergic architecture that might be putative targets for drug development with the goal of more precisely modulating glutamate. The drug targets within this system include the vesicular glutamate transporters (VGLUTs), the glutamate release process, glutamate ionotropic (NMDA, AMPA, and kainate) and metabotropic receptors, and the glutamate reuptake transporters. For each putative target we summarize pertinent information on its physiology, evidence for the modification of the target by antidepressants and mood stabilizers, any antidepressant-like effects achieved by modulating the target, and any relevant human studies. Key findings are summarized in Table 2.
Table 2.
Relevant findings at the ionotropic and metabotropic receptors in mood disorders
| RECEPTOR SUBTYPES | FINDINGS |
|---|---|
| Ionotropic Glutamate | |
| AMPA | Plasticity and neurotransmission. Increased binding in the striatum of BPD subjects. Riluzole increases AMPA trafficking. AMPA potentiators produce antidepressant effects. |
| GluR1 | Decreased expression in the striatum in BPD. Antidepressants and ECT increase GluR1 expression in dentate and hippocampus. Fluoxetine increases phosphorylation of Ser831 and Ser845. Chronic treatment with mood stabilizers decreases synaptic expression in hippocampus. |
| GluR2 | Decreased expression in the DLPFC in BPD and MDD. |
| GluR3 | Decreased expression in MDD. |
| GluR4 | None reported |
| NMDA | Reduced receptor binding in BPD. NMDA antagonists have antidepressant properties. Ketamine produces rapid antidepressant effects in humans and animal models. |
| NR1 | Decreased transcript expression found in BPD subjects. NR1 subunit expression is lower in the temporal cortex in MDD and BPD. The gene coding for the NR1 subunit (GRIN1) showed two polymorphisms associated with BPD |
| NR2 A–D | Lower NR2A transcript expression in BPD. Polymorphisms in the GRIN2B (gene coding for NR2B) linked to BPD with psychosis. Experimental use of NR2B antagonists as antidepressants is being explored. |
| NR3 A–B | None reported |
| Kainate | Neuromodulation |
| GluR5 | Knockout did not lead to manic behaviors as occurred with GluR6 knockout. |
| GluR6 | Regulates manic behavior. GRIK2 subunit is implicated in BPD and schizophrenia. |
| GluR7 | GRIK3 may be a susceptibility gene in MDD. |
| KA1 | GRIK4 subunit involved in MDD. GRIK4 were 23% less likely to experience non-response to citalopram. |
| KA2 | None reported |
| Metabotropic Glutamate | |
| Group I (excitatory G-protein coupled) | |
| mGlu1 (A–D) | Cerebellar localization, postsynaptic Glu regulation of synaptic plasticity including LTP and LTD. Strong anxiolytic effects in preclinical models. |
| mGlu5 (A–B) | Present in postsynaptic neurons and glial cells. SP in hippocampus and amygdala. Anxiolytic effects in animal models. |
| Groups II and III (inhibitory G-protein coupled) | |
| mGlu2 | Presynaptic. Abundant in the limbic system. Regulates LTD. Potential antidepressant effects using antagonists and anxiolytic effects using agonists. |
| mGlu3 | Abundant in glia, where it is linked to neurotrophin release. mGluR3-targeted drugs have both antidepressant and anxiolytic properties. |
| mGlu4 (A–B) | Pre- and postsynaptic. Linked to motor learning. |
| mGlu6 | Retinal localization. |
| mGlu7 (A–B) | Pre and postsynaptic. Lower affinity for Glu. Presynaptic is more active. |
| mGlu8 (A–B) | Mainly in presynapses. |
AMPA:-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; BPD: bipolar disorder; DLPFC: dorsolateral prefrontal cortex; ECT: electroconvulsive therapy; Glu: glutamate; LTD: long-term depression; LTP: long-term potentiation; MDD: major depressive disorder; NMDA: N-methyl-D-aspartate)-receptor
Vesicular Glutamate Transporter (VGLUTs)
The study of VGLUTs in mood disorders is in its earliest stages of investigation and is only beginning to be understood. A series of compounds that target VGLUTs are in development that should provide insight into the nature and extent of their role in the pathophysiology of mood disorders and its treatment [38].
Physiology
An essential initial step in glutamate neurotransmission is the concentration of glutamate into synaptic vesicles before release from the presynaptic terminal. Three isoforms of a vesicular glutamate transporter (VGLUT1-3) have been identified. Of these, VGLUT1 is purportedly the major isoform in the cerebral cortex and hippocampus, where it is selectively located on synaptic vesicles of excitatory glutamatergic terminals. Variations in VGLUT1 expression levels have a major impact on the efficacy of glutamate synaptic transmission (reviewed in [26]).
Modification by antidepressants and mood stabilizers
Currently available antidepressants, including fluoxetine, paroxetine, and desipramine appear to increase VGLUT mRNA levels in the frontal, orbital, cingulate, and hippocampus in rodents; all of these areas have been implicated in emotional processes [39].
In addition, preliminary evidence suggests that lithium increases VGLUT1 mRNA expression in neurons of the cerebral cortex [40]. Another report found that valproate reduced VGLUT expression in the dentate gyrus of seizure-prone gerbils [41].
Antidepressant-like effects
Although existing data are sparse, a recent study found increased anxiety, depressive-like behaviors, and impaired recognition memory in mice with reduced VGLUT1 expression [42].
Glutamate release
The mechanism of glutamate release (described above) appears to be important to the mechanism of action of several anticonvulsant drugs with a predominantly antidepressant profile such as riluzole and lamotrigine; these drugs are reviewed below.
Modification by antidepressants and mood stabilizers
Chronic (but not acute) treatment with three antidepressants with different primary mechanisms (fluoxetine (an SSRI), reboxetine (an SNRI), and desipramine (a norepinephrine reuptake inhibitor)) was found to markedly reduce depolarization-evoked release of glutamate and the expression level of the three proteins forming the core SNARE [43].
Emerging data indicate that glutamate release appears to be important in the antidepressant-like effects of both lamotrigine and riluzole. Lamotrigine is currently approved by the U.S. Food and Drug Administration (FDA) for relapse prevention in BPD and a recent study reported that it acts primarily by inhibiting glutamate release through blockade of voltage-sensitivity sodium channels and stabilization of neuronal membrane [44]. In this study, veratrine, a sodium channel opener, reversed the antidepressant-like effect of lamotrigine in the forced swim test, while the antidepressant-like effects of imipramine, desipramine, and paroxetine were unchanged, thus indicating that the mechanism of action of lamotrigine differs from that of conventional antidepressants [44].
Antidepressant-like effects
As noted above, lamotrigine was found to have antidepressant-like effects in the forced swim test in mice. However, it has been suggested that lamotrigine’s antidepressant-like effects may also be related to the noradrenergic system [45]. In addition, preclinical studies support the antidepressant properties of riluzole, as evidenced by its antidepressant-like effects in the unpredictable stress paradigm [46].
Studies in patients with mood disorders
Riluzole
Riluzole (2-amino-6-(trifluoromethoxy) benzothiazole) is indicated for the treatment of amyotrophic lateral sclerosis (ALS). It has diverse effects within different components of the glutamatergic system, including glutamate inhibitory release, AMPA receptor trafficking, and glutamate reuptake properties. However, its best-known mechanism of action involves the inhibition of glutamate release. The evidence indicates that riluzole reduces potassium-evoked release from slices of hippocampal area CA1 [47] and rat cortical synaptosomes [48].
Other putative mechanisms involved in the antidepressant properties of riluzole include its capacity to stimulate nerve growth factor, brain derived growth factor (BDNF), and glia cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes [49]. Another report found that repeated, but not single, injections of riluzole resulted in prolonged elevation of hippocampal BDNF, and were associated with increased numbers of recently created cells in the granule cell layer [50].
Clinical use and clinical investigations have examined the antidepressant profile of riluzole. These reports involved either case series or open-label studies (summarized in Table 3). A six-week open label monotherapy study in 19 patients with treatment-resistant MDD (mean daily dose of 169 mg) found that riluzole had significant antidepressant effects [51]. In bipolar depression, augmentation of lithium with riluzole (mean daily dose 171 mg) for eight weeks significantly improved depressive symptoms [52] with no evidence of hypomania or mania. More recently, riluzole (mean dose 95 mg/day) was added to ongoing antidepressant therapy in 10 patients with treatment-resistant MDD; a significant improvement in depressive symptoms was noted after six to 12 weeks of treatment [53]. The most common side effects reported with riluzole were fatigue, nausea, and weight loss. These preliminary findings need to be confirmed in a controlled study.
Table 3.
Clinical and preclinical evidence for effects of riluzole on mood disorders
| Type of Study | Findings | Reference |
|---|---|---|
| Case study | In a patient diagnosed with OCD and MDD, significant antiobsessional and antidepressant effects were noted with eight weeks of riluzole (50 mg bid) treatment when added to the ongoing medication regimen | [152] |
| Case study | Successful augmentation therapy with riluzole in a patient with treatment-resistant bipolar depression who had developed a severe rash from lamotrigine | [153] |
| Clinical trial | Significant antidepressant effects in a 6-week open label monotherapy study in 19 patients with treatment-resistant depression | [51] |
| Clinical trial | In 14 patients with bipolar depression of moderate severity who had failed to respond to a prospective trial of therapeutic levels of lithium, augmentation of lithium with riluzole for 8 weeks significantly improved depressive symptoms | [52] |
| Clinical trial | When added to ongoing antidepressant therapy in 10 patients with treatment-resistant unipolar depression, led to significant improvements in depressive symptoms after 6–12 weeks | [53] |
To date, there is no indication that the glutamatergic modulator riluzole acts more rapidly that existing antidepressants. Riluzole has not yet been studied in the manic phase of BPD, although it purportedly reduced hyperactivity caused by amphetamine in an animal model of mania [54]; these data suggest that riluzole should be considered for further testing in other phases of BPD.
N-acetyl cysteine (NAC)
Evidence of NAC’s antidepressant effects in patients with BPD was recently reported [55]. These have been hypothesized to result from its ability to increase glutathione, which is a potent antioxidant. Glutathione is the one of the principal antioxidant substrates in tissue, and its production is rate-limited by its precursor, cysteine. Glutathione alterations have been reported in BPD [56, 57]. Consequently, NAC would increase glutathione levels, leading to postulated benefits in BPD because of its antioxidant properties. Its antidepressant effects might also be due to its actions on glutamatergic neurotransmission. NAC increases cysteine levels in glia. The uptake of cysteine by glia is mediated by the cysteine-glutamate antiporter, a protein that liberates a molecule of glutamate into the extrasynaptic space in exchange for each molecule of cysteine or cysteine transported into glia. A corollary is that raising glutamate levels in the extrasynaptic space could result in stimulation of mGluR-2 receptors on glutamate nerve terminals and, subsequently, the reduction of synaptic release of glutamate [58].
Berk and colleagues conducted a randomized, double-blind, multicenter, placebo-controlled study involving 75 patients with BPD who were treated with NAC (1g twice daily) added on to treatment as usual over 24 months, followed by a four-week washout phase. The investigators found that NAC significantly improved Montgomery Asberg Depression Rating Scale (MADRS) scores and most secondary scale scores by endpoint. Improvement was seen in the Global Assessment of Functioning (GAF) scale and the Social and Occupational Functioning Assessment Scale (SOFAS) at eight weeks, and the MADRS at 20 weeks. Notably, all interested patients could participate in the study; that is, patients were not necessarily selected for having a major depressive episode. In addition, there was a considerable lag in the benefits obtained; effects occurred mainly towards the end of the trial. NAC was reported to be well-tolerated. The side effects numerically greater than placebo were headaches, heartburn, and increased joint pain.
Ionotropic Receptors
Glutamate receptor findings pertinent to mood disorders are summarized in Table 2.
NMDA Receptors
Over a decade ago, Nowak and colleagues [59] and Paul and colleagues [60] conducted a series of elegant studies with tricyclic antidepressants—the prototypic antidepressants—in order to decipher whether glutamate contributed to the mechanism of action of antidepressants. They concluded that the NMDA receptor serves as a final common pathway of antidepressant action, and that directly targeting NMDA receptors could possibly bring about a rapid onset of antidepressant effects. This important work led to a reconceptualization of the original theory of antidepressants’ mechanism of action.
Physiology
The NMDA receptor is activated by glutamate and requires the presence of a co-agonist glycine or D-serine to be activated. The receptor is permanently blocked by extracellular magnesium when the membrane is in a resting state. Only depolarization mediated by the fast activation of AMPA receptor releases magnesium plug blockade from the NMDA receptor pore, thus permitting the flow of other electrolytes (e.g., calcium) (reviewed in [33, 61]; Figure 1).
The channel is composed of a combination of NR1, NR2, NR2B, NR2C, NR2D, NR3A, and NR3B subunits. The genes encoding for these subunits are GRIN1, GRIN2A, GRIN2B, GRIN2C, GRIN2D, GRIN3A, and GRIN3B, respectively. The binding site for glutamate has been found in the NR2 subunit and the site for the co-agonist glycine has been localized on the NR1 subunit. Two molecules of glutamate and two of glycine are believed to be mandatory to activate the ion channel. Within the ion channel, two other sites have been identified: the “s” site and the phencyclidine site.
Several drugs bind to the PCP site within the channel and are considered noncompetitive receptor antagonists that inhibit NMDA receptor channel function, including the hallucinogenic drug phencyclidine (PCP), ketamine, and dizocilpine (MK-801). A series of intracellular proteins associated with the postsynaptic density that have a specific association with the NMDA receptor complex have been identified. NR1 appears to interact with NL-L and Yotiao, while NR2 interacts with PSD95, PSD93, SAP102, CIPP, and Densin-180 [62–64]. Extrasynaptic NMDA receptors are also hypothesized to be involved in the mechanisms of antidepressant action.
Modification by antidepressants and mood stabilizers
One of the most consistent and replicated finding in the glutamate literature as it pertains to conventional antidepressants is that the tricyclic antidepressants, SSRIs, and SNRIs all downregulate NMDA and potentiate AMPA [59, 60, 65]. It should be emphasized that these actions are often subtle and delayed after administration. The considerable lag in antidepressant efficacy, which is often several weeks in duration, is perhaps due to the fact that a series of intracellular signaling pathways effects have to first transpire before the involvement of the end targets are affected (in this case, NMDA and AMPA receptors). Skolnick [30] and others hypothesized that directly targeting these end targets would probably result in a more rapid onset of antidepressant action than existing antidepressants. Below, we discuss how this question was directly addressed in a proof-of-principle study using the NMDA antagonist ketamine.
Compared to antidepressants, fewer experiments have examined the effects of mood stabilizers on the NMDA receptor complex; in fact only lithium has been studied. In one study, lithium was found to abrogate NMDA function [66, 67], a property subsequently demonstrated to be important in neuroprotection models.
Antidepressant-like effects
In animal models, a series of NMDA receptor antagonists have been found to have antidepressant-like effects (see review [33, 61]). For example, administration of ketamine and related NMDA antagonists has been shown to have anxiolytic and antidepressant effects in animal models of anxiety and depression[68–71]. However, these non-subunit NMDA receptor type compounds were not clinically developed because they produce psychotomimetic effects when used acutely; more subunit selective NMDA antagonists should be better tolerated. A study from our laboratory did indeed find antidepressant-like effects with a subunit selective NR2B antagonist in rodents [72]. Other previously reported issues that will have to be addressed with non-selective NMDA antagonists include problems with cognition, neurotoxicity (i.e., vacuolation in rodents in retrosplenial cortex), and psychotomimetic effects [73].
NMDA receptor binding and postsynaptic density changes in post-mortem studies of patients with mood disorders
Several studies have reported intracellular proteins changes associated with the postsynaptic density of the NMDA receptor complex (see above). One study found that reduced expression of SAP102 (a synapse-associated protein that primarily interacts with the NR2B subunit) corresponded with a reduction in expression of subunits NR1 and NR2A in the hippocampus of patients with BPD [74]; this finding has also been observed in the striatum [75] and thalamus [76] of patients with BPD and MDD. Decreases in NF-L and PSD95 transcripts were observed only in subjects with BPD. PSD95 immunoreactivity was decreased in the hippocampal dentate in patients with BPD, but not in patients with MDD or in the orbital frontal cortex or hippocampal hilus in patients with either MDD or BPD [77]. In a more recent study, NR2A and PSD95 protein levels in the lateral amygdala were significantly increased in depressed subjects compared to controls [78]. Finally, Beneyto and colleagues reported lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex (PFC) in patients with BPD [79].
Studies in patients with mood disorders
Cycloserine
Cycloserine (4-amino-isoxazolidin-3) is a broad-spectrum antibiotic now being widely studied in anxiety disorders, and it has also been tested in MDD. Initial cases reports from patients with tuberculosis suggested that cycloserine had antidepressant or euphorogenic properties, particularly at higher doses. However, the only controlled study that has been conducted to date found no significant antidepressant effects for cycloserine in MDD. In this double-blind, placebo-controlled, six-week crossover study involving 22 patients with treatment-resistant MDD, D-cycloserine at 250 mg/day was added to existing antidepressants and found to be ineffective [80]. Some methodological limitations, including the dose used, may in part explain the negative findings. At present, this drug is being tested in BPD depression. However, the lack of efficacy of D-cycloserine in MDD diminishes enthusiasm for further pursuing this partial agonist at the NMDA receptor in the treatment of mood disorders.
Memantine
Memantine (-amino-3, 5-dimethyladamantane) is derived from amantadine, and is a low-affinity, uncompetitive, open-channel NMDA blocker. Memantine is very well-tolerated [81] and is approved for use in minimizing the cognitive decline of patients with Alzheimer’s Disease [82].
Memantine evinces antidepressant-like properties in animal models [83, 84]. However, evidence from clinical studies of this low-affinity antidepressant is mixed. One recent paper reported that two patients with treatment-resistant BPD had improved cognition and mood stabilization with memantine [85]. In another recent report, memantine was successfully used to treat an 80 year old woman with MDD with catatonic features [86]; a confounder in this case was that the patient was receiving multiple medications in addition to 5 mg of memantine, including lorazepam, which has been reported to help with catatonia.
To date, there have been two controlled studies of memantine in MDD; the first was placebo controlled [87], and the second used an active comparator [88]. The first study, a double-blind, placebo-controlled trial of memantine in patients with MDD, found no significant antidepressant or antianxiety effects at the dose studied (20 mg/day). In the second study, 80 subjects with MDD and comorbid alcohol dependence were randomly assigned 1:1 to receive either memantine 20 mg/day or escitalopram 20 mg/day for up to 26 weeks. Twenty-nine patients in each group completed the study. Both treatments significantly reduced the baseline level of depression and anxiety according to MADRS and the Hamilton Anxiety Rating Scale (HAM-A), which were the primary measures (p < .0001). Memantine and escitalopram were equally effective [88], however, the lack of a placebo control limits the interpretation that can be drawn from this study. Further controlled studies are necessary to determine whether memantine does have antidepressant properties; if so, it is possible that these might occur primarily in a subgroup of patients (i.e., MDD with comorbid alcohol dependence). Another possibility is that more potent NMDA antagonists than memantine are required to induce antidepressant effects.
Ketamine
Ketamine (dl2-(o-chlorophenyl)-2-(methylamino) cyclohexanone hydrochloride) is an NMDA antagonist [89] and a derivative of phencyclidine (PCP). In humans, psychotomimetic effects and cognitive deficits become apparent with its use, most likely because ketamine binds to the PCP site within the ionotropic channel. Clinically, ketamine is a popular agent for emergency department procedural sedation in children, with a long safety record in both clinical and research settings [90, 91]. In patients with MDD, a preliminary study found that ketamine had significant, rapid-onset antidepressant effects [32].
Another double-blind, placebo-controlled, crossover study found that a single intravenous dose of ketamine (0.5 mg/kg over 40 minutes) resulted in rapid and significant antidepressant effects in patients with treatment-resistant MDD [92]; significant improvement was noted as soon as 110 minutes after ketamine administration. Seventy-one percent of subjects met response criteria, and 29% achieved remission at 24 hours following the infusion of ketamine. Thirty-five percent of subjects maintained response to ketamine for at least one week; two of these maintained response at least two weeks. By contrast, no subject on placebo responded at one or seven days. Mild perceptual disturbances occurred in most patients, and in all cases lasted less than one hour. No serious adverse events occurred.
Subsequent reports of ketamine’s antidepressant effects concur with the previous controlled studies. Such reports include: a case noting that ketamine had rapid and significant antidepressant effects in a patient with treatment-resistant MDD and concurrent alcohol dependence [93]; a patient with severe recurrent MDD who demonstrated marked improvement within eight hours of receiving a pre-operative dose of ketamine and one treatment of electroconvulsive therapy (ECT) [94]; a patient with treatment-resistant MDD who improved after ketamine use during ECT [95]; two cases of MDD patients with concomitant pain syndrome who improved after treatment with ketamine [96]; and 35 cases of medical-surgical patients with some symptoms of depression who underwent orthopedic surgery, and whose depressive symptoms improved after being anesthetized with propofol, fentanyl, and ketamine [97].
Although ketamine is an NMDA antagonist, it is likely that antagonism of the NMDA receptor is only one aspect of its mechanism of antidepressant action. Indeed, evidence exists that the rapid onset of ketamine is due to a sudden and robust increase in AMPA relative to NMDA glutamatergic throughput, which results in increased synaptic potentiation [72]. In addition to NMDA antagonism, ketamine also induces presynaptic glutamate release [98], a property believed to be essential to its antidepressant action. A corollary is that this increase in extracellular glutamate concentration preferentially favors AMPA receptors over NMDA receptors because the latter are antagonized by ketamine; thus the resultant effect on a molecular level is enhanced AMPA throughput. In an animal study, we found that in the forced swim test— a test with fairly high predictive validity in identifying antidepressant compounds—ketamine significantly decreased immobility time. In the same experiment, NBQX, an AMPA/KA antagonist, did not affect forced swim test results when given alone; however when NBQX was given immediately prior to ketamine and imipramine, it abolished the decrease in immobility time with ketamine but not imipramine [72]. This finding suggests that, at least in animal models, the antidepressant-like properties of ketamine are mediated in part by AMPA receptor throughput.
NR2B antagonists
As discussed above, it is possible that a more selective subunit NMDA antagonist could be better tolerated than a non-selective one. A study by our group found antidepressant-like effects with a subunit selective NR2B antagonist in rodents [72]. A proof-of-principle study is currently underway with such compounds in patients with MDD in order to determine whether acute antidepressant effects could indeed be obtained with a lower propensity for psychotomimetic adverse events. There are, however, no currently available subunit selective NR2B antagonists for clinical use. Brain-penetrant NR2B antagonists currently being developed include indole-2-carboxamides, benzimidazole-2-carboxamides, and HON0001 [99–101].
Other NMDA subunits
It will also be important to determine whether antagonizing other NMDA subunits would lead to antidepressant effects. Evidence is mounting that upregulation and/or overstimulation of the NR2A receptor subtype of the NMDA receptor plays a pivotal role in the etiology of MDD (see, for instance, [26]). An animal model reported that NR2A knockout mice had a highly robust anxiolytic- and antidepressant-like phenotype [102]. These data provide the first evidence linking the NR2A subunit of the NMDA receptor to the regulation of emotional behavior.
AMPA Receptors
Physiology
Long-lasting, activity-dependent changes in synaptic strength at excitatory synapses are believed to be crucial for many aspects of experience-dependent plasticity, including learning and memory. The most extensively studied and accepted models of synaptic plasticity in the mammalian brain are long-term depression (LTD) and long-term potentiation (LTP). There is now persuasive evidence that changes in trafficking of AMPA receptors both to and from synapses significantly contributes to changes in synaptic strength during LTP and LTD [103]. The AMPA receptor is stimulated by the presence of glutamate, characteristically produces a fast excitatory synaptic signal, and is responsible for the initial reaction to glutamate in the synapse. In fact, its activation opens the pore permitting the inward flow of sodium resulting in the depolarization of the neuronal membrane. This change in the intracellular charge liberates the magnesium cation from the NMDA receptor, permitting the entrance of calcium through that pore. The channel is composed of the combination of glutamate receptor GluR1, GluR2, GluR3, and GluR4 subunits (Figure 1). The genes encoding for these subunits are GRIA1, GRIA2, GRIA3, and GRIA4, respectively. AMPA receptors have a lower affinity for glutamate than the NMDA receptor, allowing for more rapid dissociation of glutamate and therefore a rapid deactivation of the AMPA receptor (for review, see [33, 61]).
Many of the known protein-protein interactions of AMPA receptors involve intracellular proteins so-called postsynaptic density (PSD) domains [104]. GluR1 appears to interact specifically with the PDZ-containing protein SAP97, while GluR2 and GluR3 interact with GRIP, ABP, and PICK1 through a different class of PDZ domain [105, 106]. Another protein that plays an important role in AMPA receptor trafficking and hence synaptic plasticity is N-ethylmaleimide-sensitive fusion protein (NSF) [107], which interacts with the intracellular C-terminus of GluR2.
Modification by antidepressants
Svenningson and colleagues reported that acute treatment with fluoxetine increased phosphorylation of GluR1 at Ser831 and Ser845 while chronic treatment with fluoxetine increased phosphorylation only of GluR1 Ser845 [108]. This result is supported by a recent study from our group that also found that chronic imipramine increased p845 GluR1 (associated with increased GluR1 insertion), and also found that NMDA receptor phosphorylation at NR2B 1303 was increased (thus reducing NMDA receptor function)[72].With regards to GluR2, several tricyclic antidepressants were found to bind to the S1 S2 domain [109]. The interaction of GluR1 subunit with Stargazin and the interaction of GluR2/3 with NSF was recently examined with desipramine and paroxetine [110]. Paroxetine increased the interaction of GluR1 with Rab4A, while desipramine markedly increased the interaction of GluR1 with SAP97 [110].Chronic fluoxetine treatment affected transcription levels in prefrontal cortex, and variously affected GluR2, GluR5, and GluR6 in the hippocampus (GluR5 and GluR6 are Kainate subunits). Chronic desipramine treatment selectively decreased GluR3 expression both in pre-frontal/frontal cortex and hippocampus of rats [111].
In recent years, there has been considerable enthusiasm regarding the use of the atypical antidepressant tianeptine. Tianeptine prevents or reverses stress-associated structural and cellular changes in the brain and normalizes altered glutamatergic neurotransmission. In the hippocampus, it purportedly averts stress-induced dendritic atrophy, induces neurogenesis, abrogates apoptosis, and normalizes metabolite concentrations and hippocampal volume (reviewed in [112]). A recent study found that tianeptine increased P-Ser831-GluR1 in the frontal cortex and the CA3 region of hippocampus, as well as P-Ser845-GluR1 in the CA3 region of hippocampus. In the same study, behavioral analyses showed that mice bearing point mutations at both Ser831- and Ser845-GluR1 exhibited increased immobility in the tail-suspension test compared to their wild-type counterparts. Tianeptine has no affinity for any of the monoaminergic receptors or transporters, confirming its atypical profile [113].
With regards to non-pharmacological treatments for MDD, in situ hybridization studies [114, 115] have shown that repeated electroconvulsive shock in rats increased expression of mRNA for GLUR1 in the dentate gyrus, as well as the CA1 and CA3 cell fields of the hippocampus.
Finally, there is increasing evidence that riluzole, noted above as a glutamate release inhibitor, also affects AMPA receptors by increasing cell surface expression of the AMPA subunits GLUR1 and GLUR2, and induces changes involved in AMPA trafficking; the latter is a component of synaptic plasticity [116]. Riluzole has no known direct effects on NMDA or kainate receptors [117].
Modification by mood stabilizers
Recent studies show that lithium decreases synaptic expression of AMPA receptor subunits GluR1 and GluR2 in rat hippocampus, a property shared with valproate and believed to be important to the “antimanic” drug class; such effects are believed to be a distinctive property of effective antimanic therapies [118]. The mood stabilizer lamotrigine, which has a predominantly antidepressant profile, was recently found to increase expression of GluR1 and GluR2 subunits in the hippocampus of rats in a manner similar to riluzole [116].
Antidepressant-like effects
Further support for the role of the AMPA subunit (GluR1) in MDD comes from a recent study examining the consequences of GluR1 knockout. GluR1 knockout mice displayed increased learned helplessness, glutamate levels, and NMDA receptor (NR1) expression, coincidentally with decreased serotonin and norepinephrine levels. This knockout could be a useful means of studying the biological mechanisms of AMPA receptor modulators and the efficacy of NMDA antagonists in reducing behavioral or biochemical changes that correlate with increased helplessness [119].
There are now several AMPA receptor positive modulators or AMPA receptor potentiators (ARPs) compounds under development. Unlike agonists, they do not activate AMPA receptors themselves but slow the rate of receptor desensitization and/or deactivation in the presence of an agonist (see [62, 120] for review). A series of modulators from several structural classes have been synthesized, including benzothiazides (e.g., cyclothiazide), the benzoyliperidines (e.g., CX-516), and the birylpropylsulfonamides (e.g., LY392098) [27, 121]. These compounds are classified based on their effects on the biophysical processes of desensitization and deactivation.
AMPA and KA receptor binding changes
Neuropathological studies indicate that there is increased AMPA binding coupled with a decreased GluR1 subunit expression in the striatum in BPD [122]. Decreases of GluR2 levels have also been found in the dorsolateral prefrontal cortex (DLPFC) of patients with BPD, and GluR2 and GluR3 in individuals with MDD [123].With regards to AMPA receptor trafficking and signaling molecules (NSF, PICK1, Stargazin, and syntenin), non-significant changes in expression were found in the DLPFC of patients with mood disorders [123].
Kainate Receptors
Physiology
KA receptors have pre- and post-synaptic roles and share some of the same properties as AMPA receptors. They are composed of the combination of GluR5, GluR6, GluR7, KA1, and KA2 subunits. The genes encoding for KA1 and KA2 are GRIK4 and GRIK5, respectively.
Although data on the effects of antidepressants on KA receptors are sparse, chronic fluoxetine treatment was found to affect transcription levels in prefrontal cortex, and GluR5 and GluR6 levels in the hippocampus[111]. Notably, in the second wave of a genotype study of patients enrolled in the STAR*D study, an association between treatment response to citalopram and the marker (rs1954787) in the GRIK4 gene, which codes for the kainic acid-type glutamate receptor KA1, was found [124]. Beyond these data, there is little other information on the role of kainate receptors in mood disorders.
Metabotropic Glutamate Receptors
In addition to ionotropic receptors, metabotropic glutamate receptors (mGlu receptors) have also been implicated in the mechanism of action of effective antidepressant treatments. Convergent biochemical, pharmacological, behavioral, and clinical data imply that glutamatergic neurotransmission via mGlu receptors is biologically relevant to the regulation of mood, and that these receptors may serve as novel targets for the discovery of small molecule modulators with unique antidepressant properties.
Physiology
The mGlu receptors are involved in the early phase of memory formation and the mechanism of LTD [125–127]. The mGlu group of receptors was identified after the ionotropic group and is believed to be involved in modulating synaptic transmission and cell excitability through the activation of guanosine triphosphate (GTP)-binding protein (or G-protein) linked receptors. Currently, eight types of receptors have been cloned. They are organized into three different subgroups (I, II, and III) that have been characterized based on the signaling transduction pathway that they activate. The glutamate metabotropic receptor Group I includes the mGlu1 (a, b, c, d), and mGlu5 (a, b) receptors. Group I subunit, mGlu1 and mGlu4 interact with proteins Homer 1, 2, and 3. They preferentially interact with the Gq/11 subunit G-proteins, resulting in activation of the enzyme phospholipase C (PLC) and, through phosphoinositide hydrolysis, activate the inositide triphosphate/calcium and diacylglycerol/protein kinase C (PKC) cascades. The receptors are located in both the pre- and post-synaptic membranes. Group II includes mGlu2 and mGlu3 receptors. mGluR3 interacts with proteins GRIP and PICK-1. These receptors are well positioned both anatomically and physiologically to regulate glutamate with the CNS. Group II mGluR2and mGluR2/3 are negatively linked to the adenylyl cyclase signal transduction pathway and decrease glutamate release, especially under conditions of glutamate excess in the synapse; moreover, they regulate glutamate transmission by post-synaptic mechanisms. Group III mGlu receptors represent the largest family of mGlu receptors and are classified into four receptor subtypes (mGlu4, mGlu6, mGlu7 and mGlu8 receptors) (see [27] for a review). mGlu4, 6, and 7 interact with proteins GRIP and Sytenin.
Notably, two recent studies, the STAR*D and the Munich Antidepressant Response Signature (MARS) project, found a relationship between treatment-emergent suicidal ideation and the glutamate system, specifically identifying the involvement of the genes GRIA3 and GRIK2 [128, 129].
Modification by antidepressants
Evidence exists that chronic imipramine treatment reduces the inhibitory properties of group II mGlu receptors [130]. The same study found no changes in the density or affinity of the mGlu2 and mGlu3 receptor ligand for group II mGlu receptors, as determined by the receptor radioligand [3H]-LY341495.
Antidepressant-like effects
The first types of compounds that were antagonists for mGlu5 were SIB1757 and SIB1893. Unlike previous mGlu ligands, these compounds do not bind to the glutamate binding site but act at an allosteric site to inhibit mGlu receptor activation of G proteins. SIB1757 and SIB1893 subsequently led to the development of the mGlu5 receptor antagonists MPEP (2-methyl-6-[phenylethynyl]-pyridine) and MTEP ([(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine), both of which induce antidepressant-like activity in animal models of depression [131, 132]. MPEP exhibited a 100-fold greater potency than its predecessors. MTEP has fewer off-target activities and more favorable pharmacokinetic properties than MPEP [133]. In addition, the mGlu1 receptor antagonist EMQMCM ([3-ethyl-2-methyl-quinolin-6-yl]-(4-methyoxy-cyclohexyl)-methanone methanesulfonate) showed antidepressant-like properties in mice [134].
Group II mGlu receptor antagonists have similarly been shown to induce antidepressant-like effects in rodents. For instance, MGS-0039, a mGlu2/3 receptor antagonist, appears to be effective in the learned helplessness model of depression [135]. In addition, activation of AMPA receptors appears to be at least partially responsible for the antidepressant-like activity of group II mGlu receptor antagonists; the AMPA antagonist NBQX blocked the antidepressant-like activity of MGS-0039 in the tail suspension test in mice [136].
Selective group III mGlu receptor agonists (ACPT-I, [1S,3R,4S]-1-aminocyclo-pentane-1,3,4-tricarboxylic acid) and a mGlu8 receptor agonist (RS-PPG, [RS]-4-phosphonophenylglycine) also showed antidepressant-like effects in the forced swim test in rats [137]. While the nonselective group III mGlu (mGlu 4–8) receptor agonist ACPT-I had antidepressant properties, a positive allosteric modulator of mGlu4 receptor did not when given alone in the forced swim test [138]. Additional corroboration for the involvement of group III mGlu receptors in the mechanism of antidepressant action comes from the work of Cryan and colleagues, who showed that mGlu7 receptor knockout in mice produces antidepressant-like effects in the forced swim and tail suspension tests in mice [139]. Group III mGlu receptor agonists also have antidepressant-like properties in the behavioral despair test [140]. Finally, AMN082 (N,N'-dibenzyhydryl-ethane-1,2-diamine dihydrochloride), a selective mGlu7 receptor agonist, was recently found to have antidepressant-like properties in animal models [141].
In summary, many agents that modulate the metabotropic receptors are under development. Ultimately, these are likely to result in new therapeutics for mood, anxiety, psychotic, and addictive disorders, Parkinson’s Disease and pain. Studies conducted over the next few years should provide insight into the effectiveness and safety of these types of compounds.
Studies in patients with mood disorders
No studies have yet been published investigating mGlu receptor compounds in patients with mood disorders. The only available data from which results could be extrapolated come from a study of individuals with generalized anxiety disorder (GAD). In this study, a selective mGlu 2/3 agonist (LY544344) was found to improve anxiety symptoms. Although depression ratings were obtained, there were no significant changes compared to placebo. Notably, however, baseline depression scores were quite low, as patients were selected primarily for an anxiety disorder, and the study subjects may not have displayed sufficient severity of depressive symptoms to assess the antidepressant effects of the compound [142].
Glia Reuptake Transporters
The anatomy and physiology of the glial transporter system was reviewed above. Reduced glia cell number and density over several brain regions have been reported in individuals with mood disorders. Reduced expression of EAAT1, EAAT2, and glutamine synthetase have been found in the frontal brain regions in postmortem brain tissue from individuals with MDD [143] (see Table 1). Decreases in EAAT3 and EAAT4 mRNA expression in the striatum of individuals with mood disorders have also been reported [144].
Modification by antidepressants or drugs with antidepressant effects
As noted above, riluzole is an inhibitor of glutamate release. Emerging data also indicate that it might affect the activity of the glutamate transporters GLAST, GLT1, and EAAC1 [145]. Concentrations of riluzole ranging from 0.01 to 100 µM increased specific glutamate uptake in HEKGLT1, HEKEAAC1, and HEKGLAST in a dose-dependent fashion; at the highest concentrations the increase was about 30% in the three cell lines [145]. Another report found that that 0.1 and 1uM riluzole significantly increased glutamate uptake both in vitro and after in vivo treatment in animals [146]. Finally, Frizzo and colleagues (2004) reported that riluzole had a biphasic effect, with significant increases in uptake at 1 and 10 µM (15%); higher concentrations were found to be ineffective or even toxic in astrocyte cultures [147].
Modification by mood stabilizers
In addition to the mechanisms of action discussed earlier, the beneficial mood effects of valproate could be due to its ability to clear excess synaptic glutamate by increasing expression of glutamate transporters. Chronic treatment with valproate was found to increase EAAT1 levels and decrease EAAT2 levels in the hippocampus [148, 149]. Drugs that selectively regulate EAAT1 and EAAT2 would help clarify the importance of this mechanism.
Antidepressant-like effects
It has been hypothesized that increased GLT1 expression could result in antidepressant effects. An animal model of depression found that increasing GLT1 expression resulted in antidepressant-like effects [150]. One candidate drug (ceftriaxone, a β-lactam antibiotic) was recently reported to increase expression of the gene encoding for the GLT1 receptor within a few days of its administration; the expression for this receptor continues well beyond drug discontinuation [151]. As noted above, the GLT1 receptor is one of the glial glutamate transporters involved in regulating glutamate synaptic concentrations. Thus, enhancing GLT1 expression may be another putative target for modulating the glutamatergic neurotransmitter system. Notably, this is one of the mechanisms by which riluzole is believed to regulate excess synaptic glutamate [147].
CONCLUSIONS
The study of the glutamatergic system provides a rich target for developing novel therapeutics for mood disorders. Furthermore, developing this next generation of treatments is likely to result in significant gains in our understanding of the cellular and molecular underpinnings of these devastating disorders. In this review we discussed evidence that existing effective antidepressants and mood stabilizers modulate different components of the glutamate neurotransmission system, effects that might be relevant to drug development. Furthermore, directly focusing on these targets in animal studies has yielded contributory information that supports the relevance of some of the glutamate targets. Other data reviewed here (i.e., brain imaging, MRS, postmortem) argue that the excitatory amino acid system may play a role in the pathogenesis of mood disorders.
More recently, an increasing number of proof-of-principle studies have attempted to hone in on what is or is not relevant about the glutamate system as it pertains to mood therapeutics. Most notably, drugs such as riluzole and ketamine, which enhance AMPA throughput and acutely increase synaptic plasticity, are worthy of further study. Continuing to explore the mechanism of action of glutamatergic modulators, in particular those that exert full antidepressant effects in hours such as ketamine, holds considerable promise for developing new treatments for mood disorders, and for reducing at least some of the morbidity and mortality associated with the delayed onset of action of traditional antidepressants. Refining relevant aspects of their therapeutic action is likely to yield more refined and better treatments than existing ones. Furthermore, a host of other benefits could possibly emerge from these next generation compounds. For example, the neuroprotective effects of glutamatergic modulators could possibly exert long-term salutary benefits. Such properties become increasingly important as individuals are living longer, and mood disorders are typically chronic diseases often accompanied by psychiatric and medical comorbidity.
Acknowledgments
We would like to acknowledge the support of the Intramural Research Program of the National Institute of Mental Health and NARSAD. Dr. Zarate is listed among the inventors on a patent application submitted for the use of ketamine in depression. Ioline Henter provided invaluable editorial assistance.
REFERENCES
- 1.Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- 2.Dunn V, Goodyer IM. Longitudinal investigation into childhood-and adolescence-onset depression: psychiatric outcome in early adulthood. Br J Psychiatry. 2006;188:216–222. doi: 10.1192/bjp.188.3.216. [DOI] [PubMed] [Google Scholar]
- 3.Tohen M, Strakowski SM, Zarate C, Jr, Hennen J, Stoll AL, Suppes T, et al. The McLean-Harvard first-episode project: 6-month symptomatic and functional outcome in affective and nonaffective psychosis. Biol Psychiatry. 2000;48:467–476. doi: 10.1016/s0006-3223(00)00915-x. [DOI] [PubMed] [Google Scholar]
- 4.Murray CJ, Lopez AD. Evidence-based health policy--lessons from the Global Burden of Disease Study. Science. 1996;274:740–743. doi: 10.1126/science.274.5288.740. [DOI] [PubMed] [Google Scholar]
- 5.Tohen M, Zhang F, Taylor CC, Burns P, Zarate C, Sanger T, et al. A meta-analysis of the use of typical antipsychotic agents in bipolar disorder. J Affect Disord. 2001;65:85–93. doi: 10.1016/s0165-0327(00)00162-2. [DOI] [PubMed] [Google Scholar]
- 6.Zarate CA, Jr, Tohen M. Antipsychotic drug treatment in first-episode mania: a 6-month longitudinal study. J Clin Psychiatry. 2000;61:33–38. doi: 10.4088/jcp.v61n0109. [DOI] [PubMed] [Google Scholar]
- 7.Baldessarini RJ, Hennen J, Wilson M, Calabrese J, Chengappa R, Keck PE, Jr, et al. Olanzapine versus placebo in acute mania: treatment responses in subgroups. J Clin Psychopharmacol. 2003;23:370–376. doi: 10.1097/01.jcp.0000085410.08426.9a. [DOI] [PubMed] [Google Scholar]
- 8.Zarate CA, Jr, Quiroz JA. Combination treatment in bipolar disorder: a review of controlled trials. Bipolar Disord. 2003;5:217–225. doi: 10.1034/j.1399-5618.2003.00034.x. [DOI] [PubMed] [Google Scholar]
- 9.McElroy SL, Zarate CA, Cookson J, Suppes T, Huffman RF, Greene P, et al. A 52-week, open-label continuation study of lamotrigine in the treatment of bipolar depression. J Clin Psychiatry. 2004;65:204–210. doi: 10.4088/jcp.v65n0210. [DOI] [PubMed] [Google Scholar]
- 10.Zarate CA, Jr, Tohen M. Double-blind comparison of the continued use of antipsychotic treatment versus its discontinuation in remitted manic patients. Am J Psychiatry. 2004;161:169–171. doi: 10.1176/appi.ajp.161.1.169. [DOI] [PubMed] [Google Scholar]
- 11.Sajatovic M, Valenstein M, Blow FC, Ganoczy D, Ignacio RV. Treatment adherence with antipsychotic medications in bipolar disorder. Bipolar Disord. 2006;8:232–241. doi: 10.1111/j.1399-5618.2006.00314.x. [DOI] [PubMed] [Google Scholar]
- 12.Zarate CA, Jr, Tohen M, Narendran R, Tomassini EC, McDonald J, Sederer M, et al. The adverse effect profile and efficacy of divalproex sodium compared with valproic acid: a pharmacoepidemiology study. J Clin Psychiatry. 1999;60:232–236. doi: 10.4088/jcp.v60n0405. [DOI] [PubMed] [Google Scholar]
- 13.Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 14.Manji HK, Zarate CA. Molecular and cellular mechanisms underlying mood stabilization in bipolar disorder: implications for the development of improved therapeutics. Mol Psychiatry. 2002;7 Suppl 1:S1–S7. doi: 10.1038/sj.mp.4001068. [DOI] [PubMed] [Google Scholar]
- 15.Insel TR, Scolnick EM. Cure therapeutics and strategic prevention: raising the bar for mental health research. Mol Psychiatry. 2006;11:11–17. doi: 10.1038/sj.mp.4001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agid Y, Buzsaki G, Diamond DM, Frackowiak R, Giedd J, Girault JA, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6:189–201. doi: 10.1038/nrd2217. [DOI] [PubMed] [Google Scholar]
- 17.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 18.Jick H, Kaye JA, Jick SS. Antidepressants and the risk of suicidal behaviors. Jama. 2004;292:338–343. doi: 10.1001/jama.292.3.338. [DOI] [PubMed] [Google Scholar]
- 19.Zarate CA, Jr, Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry. 2006;59:1006–1020. doi: 10.1016/j.biopsych.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 20.Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, N AG, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry. 2003;53:707–742. doi: 10.1016/s0006-3223(03)00117-3. [DOI] [PubMed] [Google Scholar]
- 21.Carlson PJ, Singh JB, Zarate CA, Jr, Drevets WC, Manji HK. Neural circuitry and neuroplasticity in mood disorders: insights for novel therapeutic targets. NeuroRx. 2006;3:22–41. doi: 10.1016/j.nurx.2005.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- 23.Rajkowska G. Cell pathology in bipolar disorder. Bipolar Disord. 2002;4:105–116. doi: 10.1034/j.1399-5618.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol. 2007;18:391–418. doi: 10.1097/FBP.0b013e3282ee2aa8. [DOI] [PubMed] [Google Scholar]
- 25.Quiroz JA, Gray NA, Kato T, Manji HK. Mitochondrially Mediated Plasticity in the Pathophysiology and Treatment of Bipolar Disorder. Neuropsychopharmacology. 2008 doi: 10.1038/sj.npp.1301671. [DOI] [PubMed] [Google Scholar]
- 26.Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quiroz JA, Singh J, Gould TD, Denicoff KD, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: clues from the molecular pathophysiology. Mol Psychiatry. 2004;9:756–776. doi: 10.1038/sj.mp.4001521. [DOI] [PubMed] [Google Scholar]
- 28.Javitt DC, Zukin SR. The role of excitatory amino acids in neuropsychiatric illness. J Neuropsychiatry Clin Neurosci. 1990;2:44–52. doi: 10.1176/jnp.2.1.44. [DOI] [PubMed] [Google Scholar]
- 29.Rajkowska G. Histopathology of the prefrontal cortex in major depression: what does it tell us about dysfunctional monoaminergic circuits? Prog Brain Res. 2000;126:397–412. doi: 10.1016/S0079-6123(00)26026-3. [DOI] [PubMed] [Google Scholar]
- 30.Skolnick P. Antidepressants for the new millennium. Eur J Pharmacol. 1999;375:31–40. doi: 10.1016/s0014-2999(99)00330-1. [DOI] [PubMed] [Google Scholar]
- 31.Skolnick P, Legutko B, Li X, Bymaster FP. Current perspectives on the development of non-biogenic amine-based antidepressants. Pharmacol Res. 2001;43:411–423. doi: 10.1006/phrs.2000.0806. [DOI] [PubMed] [Google Scholar]
- 32.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 33.Zarate CA, Quiroz J, Payne J, Manji HK. Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacol Bull. 2002;36:35–83. [PubMed] [Google Scholar]
- 34.Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- 35.Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 36.Danbolt NC. Glutamate uptake. Progress in Neurobiology. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 37.Masson J, Sagne C, Hamon M, El Mestikawy S. Neurotransmitter transporters in the central nervous system. Pharmacol Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- 38.Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM. Inhibitor of the glutamate vesicular transporter (VGLUT) Curr Med Chem. 2005;12:2041–2056. doi: 10.2174/0929867054637635. [DOI] [PubMed] [Google Scholar]
- 39.Tordera RM, Pei Q, Sharp T. Evidence for increased expression of the vesicular glutamate transporter, VGLUT1, by a course of antidepressant treatment. J Neurochem. 2005;94:875–883. doi: 10.1111/j.1471-4159.2005.03192.x. [DOI] [PubMed] [Google Scholar]
- 40.Moutsimilli L, Farley S, Dumas S, El Mestikawy S, Giros B, Tzavara ET. Selective cortical VGLUT1 increase as a marker for antidepressant activity. Neuropharmacology. 2005;49:890–900. doi: 10.1016/j.neuropharm.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 41.Kang TC, Kim DS, Kwak SE, Kim JE, Kim DW, Kang JH, et al. Valproic acid reduces enhanced vesicular glutamate transporter immunoreactivities in the dentate gyrus of the seizure prone gerbil. Neuropharmacology. 2005;49:912–921. doi: 10.1016/j.neuropharm.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Tordera RM, Totterdell S, Wojcik SM, Brose N, Elizalde N, Lasheras B, et al. Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (VGLUT1) Eur J Neurosci. 2007;25:281–290. doi: 10.1111/j.1460-9568.2006.05259.x. [DOI] [PubMed] [Google Scholar]
- 43.Bonanno G, Giambelli R, Raiteri L, Tiraboschi E, Zappettini S, Musazzi L, et al. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J Neurosci. 2005;25:3270–3279. doi: 10.1523/JNEUROSCI.5033-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prica C, Hascoet M, Bourin M. Antidepressant-like effect of lamotrigine is reversed by veratrine: a possible role of sodium channels in bipolar depression. Behav Brain Res. 2008;191:49–54. doi: 10.1016/j.bbr.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Kaster MP, Raupp I, Binfare RW, Andreatini R, Rodrigues AL. Antidepressant-like effect of lamotrigine in the mouse forced swimming test: evidence for the involvement of the noradrenergic system. Eur J Pharmacol. 2007;565:119–124. doi: 10.1016/j.ejphar.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Banasr M, Chowdhury G, Duman RS, Behar K, Sanacora G. Antidepressant-like effects in the unpredictable stress paradigm. Neuropsychopharmacology. 2006;31 S159-S-160. [Google Scholar]
- 47.Martin D, Thompson MA, Nadler JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- 48.Lingamaneni R, Hemmings HC., Jr Effects of anticonvulsants on veratridine-and KCl-evoked glutamate release from rat cortical synaptosomes. Neurosci Lett. 1999;276:127–130. doi: 10.1016/s0304-3940(99)00810-1. [DOI] [PubMed] [Google Scholar]
- 49.Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Kuno S. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. 2001;310:117–120. doi: 10.1016/s0304-3940(01)02098-5. [DOI] [PubMed] [Google Scholar]
- 50.Katoh-Semba R, Asano T, Ueda H, Morishita R, Takeuchi IK, Inaguma Y, et al. Riluzole enhances expression of brain-derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. Faseb J. 2002;16:1328–1330. doi: 10.1096/fj.02-0143fje. [DOI] [PubMed] [Google Scholar]
- 51.Zarate CA, Jr, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161:171–174. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- 52.Zarate CA, Jr, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57:430–432. doi: 10.1016/j.biopsych.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 53.Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, et al. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry. 2007;61:822–825. doi: 10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lourenco Da Silva A, Hoffmann A, Dietrich MO, Dall'Igna OP, Souza DO, Lara DR. Effect of riluzole on MK-801 and amphetamine-induced hyperlocomotion. Neuropsychobiology. 2003;48:27–30. doi: 10.1159/000071825. [DOI] [PubMed] [Google Scholar]
- 55.Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder--a double-blind randomized placebo-controlled trial. Biol Psychiatry. 2008;64:468–475. doi: 10.1016/j.biopsych.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 56.Kuloglu M, Ustundag B, Atmaca M, Canatan H, Tezcan AE, Cinkilinc N. Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct. 2002;20:171–175. doi: 10.1002/cbf.940. [DOI] [PubMed] [Google Scholar]
- 57.Andreazza AC, Cassini C, Rosa AR, Leite MC, de Almeida LM, Nardin P, et al. Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res. 2007;41:523–529. doi: 10.1016/j.jpsychires.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 58.Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nowak G, Trullas R, Layer RT, Skolnick P, Paul IA. Adaptive changes in the N-methyl-D-aspartate receptor complex after chronic treatment with imipramine and 1-aminocyclopropanecarboxylic acid. J Pharmacol Exp Ther. 1993;265:1380–1386. [PubMed] [Google Scholar]
- 60.Paul IA, Nowak G, Layer RT, Popik P, Skolnick P. Adaptation of the N-methyl-D-aspartate receptor complex following chronic antidepressant treatments. J Pharmacol Exp Ther. 1994;269:95–102. [PubMed] [Google Scholar]
- 61.Zarate CA, Jr, Du J, Quiroz J, Gray NA, Denicoff KD, Singh J, et al. Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann N Y Acad Sci. 2003;1003:273–291. doi: 10.1196/annals.1300.017. [DOI] [PubMed] [Google Scholar]
- 62.Bleakman D, Lodge D. Neuropharmacology of AMPA and kainate receptors. Neuropharmacology. 1998;37:1187–1204. doi: 10.1016/s0028-3908(98)00139-7. [DOI] [PubMed] [Google Scholar]
- 63.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 64.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 65.Skolnick P, Layer RT, Popik P, Nowak G, Paul IA, Trullas R. Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry. 1996;29:23–26. doi: 10.1055/s-2007-979537. [DOI] [PubMed] [Google Scholar]
- 66.Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9:2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- 67.Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80:589–597. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- 68.Aguado L, San Antonio A, Perez L, del Valle R, Gomez J. Effects of the NMDA receptor antagonist ketamine on flavor memory: conditioned aversion, latent inhibition, and habituation of neophobia. Behav Neural Biol. 1994;61:271–281. doi: 10.1016/s0163-1047(05)80010-x. [DOI] [PubMed] [Google Scholar]
- 69.Silvestre JS, Nadal R, Pallares M, Ferre N. Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in Wistar rats. Depress Anxiety. 1997;5:29–33. [PubMed] [Google Scholar]
- 70.Mickley GA, Schaldach MA, Snyder KJ, Balogh SA, Len T, Neimanis K, et al. Ketamine blocks a conditioned taste aversion (CTA) in neonatal rats. Physiol Behav. 1998;64:381–390. doi: 10.1016/s0031-9384(98)00097-3. [DOI] [PubMed] [Google Scholar]
- 71.Adamec RE, Burton P, Shallow T, Budgell J. Unilateral block of NMDA receptors in the amygdala prevents predator stress-induced lasting increases in anxiety-like behavior and unconditioned startle--effective hemisphere depends on the behavior. Physiol Behav. 1999;65:739–751. doi: 10.1016/s0031-9384(98)00225-x. [DOI] [PubMed] [Google Scholar]
- 72.Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 73.Zarate CA, Jr, Charney DS, Manji HK. Searching for rational anti-N-methyl-D-aspartate treatment for depression. Arch Gen Psychiatry. 2007;64:1100–1101. doi: 10.1001/archpsyc.64.9.1099. [DOI] [PubMed] [Google Scholar]
- 74.McCullumsmith RE, Kristiansen LV, Beneyto M, Scarr E, Dean B, Meador-Woodruff JH. Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Research. 2007;1127:108–118. doi: 10.1016/j.brainres.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kristiansen LV, Meador-Woodruff JH. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res. 2005;78:87–93. doi: 10.1016/j.schres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 76.Clinton SM, Meador-Woodruff JH. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2004;29:1353–1362. doi: 10.1038/sj.npp.1300451. [DOI] [PubMed] [Google Scholar]
- 77.Toro C, Deakin JF. NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr Res. 2005;80:323–330. doi: 10.1016/j.schres.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 78.Karolewicz B, Szebeni K, Gilmore T, Maciag D, Stockmeier CA, Ordway GA. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int J Neuropsychopharmacol. 2008:1–11. doi: 10.1017/S1461145708008985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60:585–598. doi: 10.1002/syn.20329. [DOI] [PubMed] [Google Scholar]
- 80.Heresco-Levy U, Javitt DC, Gelfin Y, Gorelik E, Bar M, Blanaru M, et al. Controlled trial of D-cycloserine adjuvant therapy for treatment-resistant major depressive disorder. J Affect Disord. 2006;93:239–243. doi: 10.1016/j.jad.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 81.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease. Arch Neurol. 2006;63:49–54. doi: 10.1001/archneur.63.1.49. [DOI] [PubMed] [Google Scholar]
- 82.Areosa SA, Sherriff F. Memantine for dementia. Cochrane Database Syst Rev. 2003 doi: 10.1002/14651858.CD003154. CD003154. [DOI] [PubMed] [Google Scholar]
- 83.Moryl E, Danysz W, Quack G. Potential antidepressive properties of amantadine, memantine and bifemelane. Pharmacol Toxicol. 1993;72:394–397. doi: 10.1111/j.1600-0773.1993.tb01351.x. [DOI] [PubMed] [Google Scholar]
- 84.Rogoz Z, Skuza G, Maj J, Danysz W. Synergistic effect of uncompetitive NMDA receptor antagonists and antidepressant drugs in the forced swimming test in rats. Neuropharmacology. 2002;42:1024–1030. doi: 10.1016/s0028-3908(02)00055-2. [DOI] [PubMed] [Google Scholar]
- 85.Teng CT, Demetrio FN. Memantine may acutely improve cognition and have a mood stabilizing effect in treatment-resistant bipolar disorder. Rev Bras Psiquiatr. 2006;28:252–254. doi: 10.1590/s1516-44462006000300020. [DOI] [PubMed] [Google Scholar]
- 86.Munoz C, Yulan N, Achaval V, Appiani F, Carroll BT. Memantine in major depression with catatonic features. J Neuropsychiatry Clin Neurosci. 2008;20:119–120. doi: 10.1176/jnp.2008.20.1.119. [DOI] [PubMed] [Google Scholar]
- 87.Zarate CA, Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163:153–155. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- 88.Muhonen LH, Lonnqvist J, Juva K, Alho H. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69:392–399. doi: 10.4088/jcp.v69n0308. [DOI] [PubMed] [Google Scholar]
- 89.Harrison NL, Simmonds MA. Quantitative studies on some antagonists of N-methyl D-aspartate in slices of rat cerebral cortex. Br J Pharmacol. 1985;84:381–391. doi: 10.1111/j.1476-5381.1985.tb12922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Green SM, Rothrock SG, Harris T, Hopkins GA, Garrett W, Sherwin T. Intravenous ketamine for pediatric sedation in the emergency department: safety profile with 156 cases. Acad Emerg Med. 1998;5:971–976. doi: 10.1111/j.1553-2712.1998.tb02773.x. [DOI] [PubMed] [Google Scholar]
- 91.Perry EB, Jr, Cramer JA, Cho HS, Petrakis IL, Karper LP, Genovese A, et al. Psychiatric safety of ketamine in psychopharmacology research. Psychopharmacology (Berl) 2007;192:253–260. doi: 10.1007/s00213-007-0706-2. [DOI] [PubMed] [Google Scholar]
- 92.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 93.Liebrenz M, Borgeat A, Leisinger R, Stohler R. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137:234–236. doi: 10.4414/smw.2007.11852. [DOI] [PubMed] [Google Scholar]
- 94.Goforth HW, Holsinger T. Rapid relief of severe major depressive disorder by use of preoperative ketamine and electroconvulsive therapy. J Ect. 2007;23:23–25. doi: 10.1097/01.yct.0000263257.44539.23. [DOI] [PubMed] [Google Scholar]
- 95.Ostroff R, Gonzales M, Sanacora G. Antidepressant effect of ketamine during ECT. Am J Psychiatry. 2005;162:1385–1386. doi: 10.1176/appi.ajp.162.7.1385. [DOI] [PubMed] [Google Scholar]
- 96.Correll GE, Futter GE. Two case studies of patients with major depressive disorder given low-dose (subanesthetic) ketamine infusions. Pain Med. 2006;7:92–95. doi: 10.1111/j.1526-4637.2006.00101.x. [DOI] [PubMed] [Google Scholar]
- 97.Kudoh A, Takahira Y, Katagai H, Takazawa T. Small-dose ketamine improves the postoperative state of depressed patients. Anesth Analg. 2002;95:114–118. doi: 10.1097/00000539-200207000-00020. table of contents. [DOI] [PubMed] [Google Scholar]
- 98.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suetake-Koga S, Shimazaki T, Takamori K, Chaki S, Kanuma K, Sekiguchi Y, et al. In vitro and antinociceptive profile of HON0001, an orally active NMDA receptor NR2B subunit antagonist. Pharmacol Biochem Behav. 2006;84:134–141. doi: 10.1016/j.pbb.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 100.Borza I, Bozo E, Barta-Szalai G, Kiss C, Tarkanyi G, Demeter A, et al. Selective NR1/2B N-methyl-D-aspartate receptor antagonists among indole-2-carboxamides and benzimidazole-2-carboxamides. J Med Chem. 2007;50:901–914. doi: 10.1021/jm060420k. [DOI] [PubMed] [Google Scholar]
- 101.Liverton NJ, Bednar RA, Bednar B, Butcher JW, Claiborne CF, Claremon DA, et al. Identification and characterization of 4-methylbenzyl 4-[(pyrimidin-2-ylamino)methyl]piperidine-1-carboxylate, an orally bioavailable, brain penetrant NR2B selective N-methyl-D-aspartate receptor antagonist. J Med Chem. 2007;50:807–819. doi: 10.1021/jm060983w. [DOI] [PubMed] [Google Scholar]
- 102.Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic-and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–2414. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- 103.Zarate CA, Jr, Manji HK. The role of AMPA receptor modulation in the treatment of neuropsychiatric diseases. Exp Neurol. 2008;211:7–10. doi: 10.1016/j.expneurol.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 105.Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 106.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 107.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 108.Svenningsson P, Tzavara ET, Witkin JM, Fienberg AA, Nomikos GG, Greengard P. Involvement of striatal and extrastriatal DARPP-32 in biochemical and behavioral effects of fluoxetine (Prozac) Proc Natl Acad Sci U S A. 2002;99:3182–3187. doi: 10.1073/pnas.052712799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stoll L, Seguin S, Gentile L. Tricyclic antidepressants, but not the selective serotonin reuptake inhibitor fluoxetine, bind to the S1S2 domain of AMPA receptors. Arch Biochem Biophys. 2007;458:213–219. doi: 10.1016/j.abb.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 110.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 111.Barbon A, Popoli M, La Via L, Moraschi S, Vallini I, Tardito D, et al. Regulation of editing and expression of glutamate alpha-amino-propionic-acid (AMPA)/kainate receptors by antidepressant drugs. Biol Psychiatry. 2006;59:713–720. doi: 10.1016/j.biopsych.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 112.Kasper S, McEwen BS. Neurobiological and clinical effects of the antidepressant tianeptine. CNS Drugs. 2008;22:15–26. doi: 10.2165/00023210-200822010-00002. [DOI] [PubMed] [Google Scholar]
- 113.Svenningsson P, Bateup H, Qi H, Takamiya K, Huganir RL, Spedding M, et al. Involvement of AMPA receptor phosphorylation in antidepressant actions with special reference to tianeptine. Eur J Neurosci. 2007;26:3509–3517. doi: 10.1111/j.1460-9568.2007.05952.x. [DOI] [PubMed] [Google Scholar]
- 114.Wong ML, Smith MA, Licinio J, Doi SQ, Weiss SR, Post RM, et al. Differential effects of kindled and electrically induced seizures on a glutamate receptor (GluR1) gene expression. Epilepsy Res. 1993;14:221–227. doi: 10.1016/0920-1211(93)90046-a. [DOI] [PubMed] [Google Scholar]
- 115.Naylor P, Stewart CA, Wright SR, Pearson RC, Reid IC. Repeated ECS induces GluR1 mRNA but not NMDAR1A-G mRNA in the rat hippocampus. Brain Res Mol Brain Res. 1996;35:349–353. doi: 10.1016/0169-328x(95)00264-s. [DOI] [PubMed] [Google Scholar]
- 116.Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z, et al. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32:793–802. doi: 10.1038/sj.npp.1301178. [DOI] [PubMed] [Google Scholar]
- 117.Debono MW, Le Guern J, Canton T, Doble A, Pradier L. Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur J Pharmacol. 1993;235:283–289. doi: 10.1016/0014-2999(93)90147-a. [DOI] [PubMed] [Google Scholar]
- 118.Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST, et al. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004;24:6578–6589. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chourbaji S, Vogt MA, Fumagalli F, Sohr R, Frasca A, Brandwein C, et al. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. Faseb J. 2008;22:3129–3134. doi: 10.1096/fj.08-106450. [DOI] [PubMed] [Google Scholar]
- 120.Borges K, Dingledine R. AMPA receptors: molecular and functional diversity. Prog Brain Res. 1998;116:153–170. doi: 10.1016/s0079-6123(08)60436-7. [DOI] [PubMed] [Google Scholar]
- 121.Miu P, Jarvie KR, Radhakrishnan V, Gates MR, Ogden A, Ornstein PL, et al. Novel AMPA receptor potentiators LY392098 and LY404187: effects on recombinant human AMPA receptors in vitro. Neuropharmacology. 2001;40:976–983. doi: 10.1016/s0028-3908(01)00027-2. [DOI] [PubMed] [Google Scholar]
- 122.Meador-Woodruff JH, Hogg AJ, Jr, Smith RE. Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Research Bulletin. 2001;55:631–640. doi: 10.1016/s0361-9230(01)00523-8. [DOI] [PubMed] [Google Scholar]
- 123.Scarr E, Pavey G, Sundram S, MacKinnon A, Dean B. Decreased hippocampal NMDA, but not kainate or AMPA receptors in bipolar disorder. Bipolar Disord. 2003;5:257–264. doi: 10.1034/j.1399-5618.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- 124.Paddock S, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, et al. Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. Am J Psychiatry. 2007;164:1181–1188. doi: 10.1176/appi.ajp.2007.06111790. [DOI] [PubMed] [Google Scholar]
- 125.Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behav Brain Res. 2003;140:1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 126.Salinska E, Stafiej A. Metabotropic glutamate receptors (mGluRs) are involved in early phase of memory formation: possible role of modulation of glutamate release. Neurochem Int. 2003;43:469–474. doi: 10.1016/s0197-0186(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 127.Tan Y, Hori N, Carpenter DO. The mechanism of presynaptic long-term depression mediated by group I metabotropic glutamate receptors. Cell Mol Neurobiol. 2003;23:187–203. doi: 10.1023/A:1022949922364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Laje G, Paddock S, Manji H, Rush AJ, Wilson AF, Charney D, et al. Genetic markers of suicidal ideation emerging during citalopram treatment of major depression. Am J Psychiatry. 2007;164:1530–1538. doi: 10.1176/appi.ajp.2007.06122018. [DOI] [PubMed] [Google Scholar]
- 129.Menke A, Lucae S, Kloiber S, Horstmann S, Bettecken T, Uhr M, et al. Genetic markers within glutamate receptors associated with antidepressant treatment-emergent suicidal ideation. Am J Psychiatry. 2008;165:917–918. doi: 10.1176/appi.ajp.2008.08020274. [DOI] [PubMed] [Google Scholar]
- 130.Palucha A, Branski P, Klak K, Sowa M. Chronic imipramine treatment reduces inhibitory properties of group II mGlu receptors without affecting their density or affinity. Pharmacol Rep. 2007;59:525–530. [PubMed] [Google Scholar]
- 131.Li X, Need AB, Baez M, Witkin JM. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther. 2006;319:254–259. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- 132.Wieronska JM, Szewczyk B, Branski P, Palucha A, Pilc A. Antidepressant-like effect of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist in the olfactory bulbectomized rats. Amino Acids. 2002;23:213–216. doi: 10.1007/s00726-001-0131-5. [DOI] [PubMed] [Google Scholar]
- 133.Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, et al. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 134.Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur Neuropsychopharmacol. 2007;17:172–179. doi: 10.1016/j.euroneuro.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 135.Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 2006;186:587–593. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
- 136.Karasawa J, Shimazaki T, Kawashima N, Chaki S. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 2005;1042:92–98. doi: 10.1016/j.brainres.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 137.Gasparini F, Bruno V, Battaglia G, Lukic S, Leonhardt T, Inderbitzin W, et al. (R,S)-4-phosphonophenylglycine, a potent and selective group III metabotropic glutamate receptor agonist, is anticonvulsive and neuroprotective in vivo. J Pharmacol Exp Ther. 1999;289:1678–1687. [PubMed] [Google Scholar]
- 138.Klak K, Palucha A, Branski P, Sowa M, Pilc A. Combined administration of PHCCC, a positive allosteric modulator of mGlu4 receptors and ACPT-I, mGlu III receptor agonist evokes antidepressant-like effects in rats. Amino Acids. 2007;32:169–172. doi: 10.1007/s00726-006-0316-z. [DOI] [PubMed] [Google Scholar]
- 139.Cryan JF, Kelly PH, Neijt HC, Sansig G, Flor PJ, van Der Putten H. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci. 2003;17:2409–2417. doi: 10.1046/j.1460-9568.2003.02667.x. [DOI] [PubMed] [Google Scholar]
- 140.Palucha A, Tatarczynska E, Branski P, Szewczyk B, Wieronska JM, Klak K, et al. Group III mGlu receptor agonists produce anxiolytic-and antidepressant-like effects after central administration in rats. Neuropharmacology. 2004;46:151–159. doi: 10.1016/j.neuropharm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 141.Palucha A, Klak K, Branski P, van der Putten H, Flor PJ, Pilc A. Activation of the mGlu7 receptor elicits antidepressant-like effects in mice. Psychopharmacology (Berl) 2007;194:555–562. doi: 10.1007/s00213-007-0856-2. [DOI] [PubMed] [Google Scholar]
- 142.Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology. 2008;33:1603–1610. doi: 10.1038/sj.npp.1301531. [DOI] [PubMed] [Google Scholar]
- 143.Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. Epub 2005 Oct 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McCullumsmith RE, Meador-Woodruff JH. Striatal excitatory amino acid transporter transcript expression in schizophrenia, bipolar disorder, and major depressive disorder. Neuropsychopharmacology. 2002;26:368–375. doi: 10.1016/S0893-133X(01)00370-0. [DOI] [PubMed] [Google Scholar]
- 145.Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–176. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 146.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 147.Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ueda Y, Willmore LJ. Molecular regulation of glutamate and GABA transporter proteins by valproic acid in rat hippocampus during epileptogenesis. Exp Brain Res. 2000;133:334–339. doi: 10.1007/s002210000443. [DOI] [PubMed] [Google Scholar]
- 149.Hassel B, Iversen EG, Gjerstad L, Tauboll E. Up-regulation of hippocampal glutamate transport during chronic treatment with sodium valproate. J Neurochem. 2001;77:1285–1292. doi: 10.1046/j.1471-4159.2001.00349.x. [DOI] [PubMed] [Google Scholar]
- 150.Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61:250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 151.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 152.Coric V, Milanovic S, Wasylink S, Patel P, Malison R, Krystal JH. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology (Berl) 2003;167:219–220. doi: 10.1007/s00213-003-1396-z. [DOI] [PubMed] [Google Scholar]
- 153.Singh J, Zarate CA, Jr, Krystal AD. Case report: Successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology (Berl) 2004;173:227–228. doi: 10.1007/s00213-003-1756-8. [DOI] [PubMed] [Google Scholar]