

Summary
Pancreatic cancer has a dismal prognosis as cancer-specific symptoms occur only at an advanced stage. If the cancer is to be discovered early, it will have to be done in asymptomatic individuals. Since the incidence of pancreatic cancer is low, screening for asymptomatic cancer in the general population will not be feasible. Screening will have to be restricted to subjects at high risk for pancreatic cancer. The proportion of pancreatic cancer patients who also have hyperglycemia or diabetes has previously been under appreciated; new data show that up to 80% are either hyperglycemic or diabetic and this can be evident in the pre-symptomatic phase. Diabetes improves following pancreatic cancer resection suggesting that diabetes is caused by the cancer. Conversely, older subjects with new-onset diabetes have an approximately 8 fold higher risk of having pancreatic cancer compared to the general population. Recognition of new-onset diabetes as an early manifestation of pancreatic cancer could lead to diagnosis of asymptomatic, early stage pancreatic cancer. However, primary type 2 diabetes is common and pancreatic cancer is relatively uncommon in the general population and the two forms of diabetes are clinically indistinguishable. The success of the strategy to use new-onset hyperglycemia and diabetes as a screening tool to identify subjects with a high likelihood of having asymptomatic pancreatic cancer will depend largely on our ability to differentiate pancreatic cancer-associated diabetes from the more common type 2 diabetes using a (serologic) biomarker.
Keywords: Pancreatic cancer, diabetes, screening
Introduction
A screening strategy for sporadic pancreatic cancer has not been established; given that cancer-specific symptoms occur late, early detection will require screening of asymptomatic individuals. Pancreatic cancer is a markedly diabetogenic state and there is increasing evidence that diabetes is prevalent even in early stage, asymptomatic pancreatic cancer. The objective of this review is to present our current understanding of pancreatic cancer-associated diabetes and offer a perspective on the prospects and problems of using this strategy for early detection of pancreatic cancer.
Need for early detection of pancreatic cancer
Pancreatic cancer is the fourth leading cause of cancer death in the United States[1] and the fifth leading cause of cancer death in Europe.[2] It has a dismal five-year survival of ~5%,[3] primarily related to the fact that disease-specific symptoms occur late in the course of the disease; at the time of diagnosis, 50% have distant metastases, 29% have local and/or regional spread, and only 3% have tumors confined to the pancreas (19% remain unstaged/unknown).[4] By the time of diagnosis <15% of patients have surgically resectable disease (figure 1). The median survival of unresectable pancreatic cancer is 4–6 months. While the overall 5-year survival of resected pancreatic cancers (median size 32 mm) is 10–20%,[5, 6] it is 30–60% after resection of small pancreatic cancer (tumor size ≤20 mm)[7–9] and it exceeds 75% when minute pancreatic cancers (≤10 mm in size) are resected [9, 10]. While future treatment advances may improve survival, the above noted statistics imply that, in order to substantially impact long-term survival, we will need to detect pancreatic cancer earlier..
Figure 1.

Gross pathological specimen of resected pancreatic ductal adenocarcinoma split in half to show its typical appearance. Cancer-specific symptoms occur at an advanced stage and less than 15% of patients have resectable disease at presentation. For early detection of pancreatic cancer, we will need to identify a high-risk group among asymptomatic individuals.
Photograph courtesy of Dr Michael B. Farnell.
Opportunity to detect resectable pancreatic cancer
Invasive pancreatic cancer develops from precancerous non-invasive precursor lesions called pancreatic intraepithelial neoplasia (PanIN) which progress from PanIN1 through PanIN3 (carcinoma in situ).[11] The timeline of progression of pancreatic cancer is not well established. In a case series, Brat et al reported the presence of PanINs 17 months to 10 years prior to the clinical diagnosis of cancer.[12] To understand the timeline of progression of pancreatic cancer better, we retrospectively reviewed 114 CT scans in 45 patients performed at or prior to cancer diagnosis and found that pancreatic cancer was either undetectable or resectable on scans performed >6 months prior to clinical diagnosis.[13, 14] In these studies the onset of symptoms coincided with appearance of radiologic features of unresectability. These studies suggest that pancreatic cancer is resectable as little as 6 months prior to clinical diagnosis when it is still asymptomatic and would not normally come to clinical attention.
Challenges to screening for asymptomatic pancreatic cancer
Detecting early pancreatic cancer will require screening asymptomatic subjects for the disease. Two major obstacles limit our ability to screen for pancreatic cancer: lack of a high risk group and lack of sensitive and specific marker(s) of early pancreatic cancer. However, even if a biomarker with very high sensitivity and specificity is identified, screening the general population for asymptomatic pancreatic cancer is unlikely to be cost effective or practical. The age-adjusted incidence of pancreatic cancer in subjects ≥50 years of age is 38/100,000.[15] If a test with 99% sensitivity and 99% specificity for pancreatic cancer is used to screen 100,000 subjects ≥50 years of age, the test would identify nearly all pancreatic cancers in the population screened (n=37). However, the test would also falsely identify nearly a 1000 subjects as having pancreatic cancer. The test would have a positive predictive value of a mere 3.6%, which is unacceptably low to justify screening with invasive tests. Thus screening for asymptomatic pancreatic cancer will likely require at least two “sieves” to enrich the population to allow cost-effective screening.[16] The first sieve could be a high-risk group, i.e., a population of subjects at higher than average risk of pancreatic cancer and the second sieve could be a unique clinical phenotype, one or more biomarker(s) of early pancreatic cancer, or non-invasive imaging.[16] Currently, among genetic syndromes with a high incidence of pancreatic cancer (first sieve), those with ≥2 first degree relatives with pancreatic cancer (second sieve) are being screened using endoscopic ultrasonography (EUS).[17–19] However, such patients account for <5% of all pancreatic cancer. An entirely different approach will be needed to screen for sporadic pancreatic cancer. Here we propose that new-onset diabetes can be the first sieve for diagnosing asymptomatic early pancreatic cancer.
Diabetes and pancreatic cancer: what’s the relationship?
A review of studies examining the association between diabetes and pancreatic cancer suggests that while long-standing diabetes is an etiologic factor for pancreatic cancer, new-onset diabetes is its manifestation. There is a modestly elevated risk of pancreatic cancer among persons with long-standing diabetes.[20] A recent meta-analysis of 17 case–control and 19 cohort (or nested case–control) studies published between 1966 and 2005 found that the combined age- and sex-adjusted odds ratio (OR) for pancreatic cancer associated with diabetes was 1·8 (95% confidence interval (95% CI, 1·7–1·9) and it was still lower (OR 1.5) in subjects with diabetes duration of ≥5 years.[21] Given the modest association between long-standing diabetes and pancreatic cancer, the relatively low incidence of pancreatic cancer, and the fact that diabetes is quite prevalent in the general population, screening all subjects with long-standing diabetes for pancreatic cancer will not be cost-effective. However, new-onset diabetes caused by the cancer appears to be a clinically useful marker of asymptomatic pancreatic cancer.
Pancreatic cancer causes diabetes
Patients with pancreatic cancer often have new-onset diabetes which resolves with cancer resection as was observed in the case study presented (figure 2). Here we review the clinical and experimental evidence that support the notion that pancreatic cancer causes diabetes.
Figure 2.
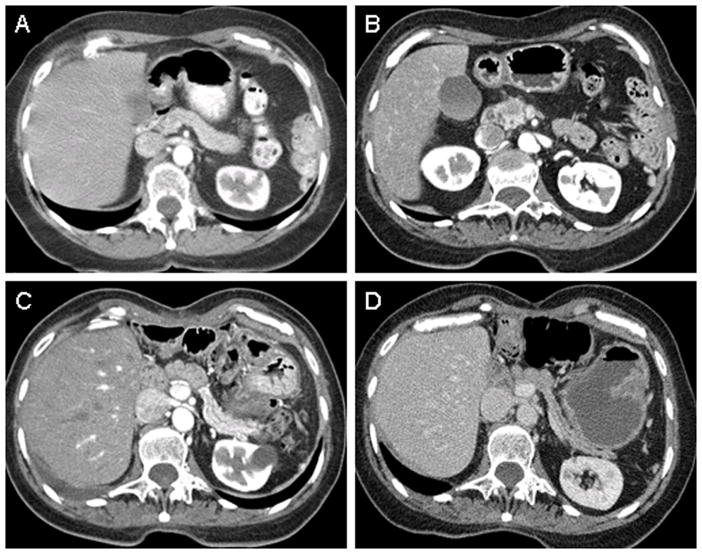
Representative case study of pancreatic cancer-associated diabetes. Patient is a 70-year old asymptomatic woman undergoing radiologic surveillance for recurrence of Hodgkin’s lymphoma treated with mantle radiation therapy in 1995. On yearly fasting glucose measurements, asymptomatic diabetes was noted in 2005, the same year her CT scan showed a pancreatic head mass (Table 1). She underwent an R0 surgical resection of a T3N0M0 ductal adenocarcimona and remains disease free to date.
a) Pancreatic cancer is a markedly diabetogenic state
The prevalence of diabetes in older studies that relied on medical records or proxy reports ranged from 4–20%. [22] However, studies that screened pancreatic cancer patients for diabetes using oral glucose tolerance tests reported diabetes prevalence of 45–65%.[23, 24] Using fasting blood glucose values and the diagnostic criteria for diabetes suggested by the American Diabetes Association [25], we collectively studied 642 newly diagnosed pancreatic cancer patients in two studies and noted that 303/642 (47%) met criteria for diabetes. [26, 27] Of the 512 pancreatic cancer patients in one of the studies,[26] 197 (38%) had impaired fasting glucose and only 72/512 patients (14%) had a normal fasting glucose.
b) Pancreatic cancer-associated diabetes is predominantly new-onset
The onset of diabetes is temporally associated with diagnosis of pancreatic cancer. Gullo et al reported that diabetes in pancreatic cancer was diagnosed either concomitantly with the cancer(in 40 %), or within two years before the diagnosisof cancer (16%).[28] In other studies, diabetes was new-onset, i.e., < 24 months in duration, in 74–88% of pancreatic cancer patients.[26, 27] In a recent population-based case-control study, we abstracted fasting blood glucose values from medical records for five years preceding cancer diagnosis in 736 pancreatic cancer patients and 1,875 age-and sex-matched controls.[29] Among the 736 pancreatic cancer cases, 296 cases (40·2%) had diabetes compared to 360/1875 (19.2%) controls; 50% of diabetes among cases was new-onset. In this study the proportion of diabetic cases and controls were similar >36 months before cancer diagnosis; however, a greater proportion of cases than controls met criteria for diabetes in the 36 to 24 (p=0·04), 24 to 12 (p<0·001) and 12 to 0 (p<0·001) month time periods before cancer diagnosis (figure 3). Collectively these studies indicate that the majority of pancreatic cancer-associated diabetes is of recent onset, beginning up to two years preceding the diagnosis of cancer.
Figure 3.

Observed and expected prevalence of diabetes in 60 months prior to index date in pancreatic cancer and controls. Reproduced with permission from [29]
Index date: Date of pancreatic cancer diagnosis
c) Pancreatic cancer resection ameliorates diabetes
Pancreatic cancer-associated diabetes improves following resection of tumor despite surgical removal of variable amounts of pancreas. In a study of 7 pancreatic cancer patients, Permert et al observed that after sub-total pancreatectomy, diabetic status and glucose tolerance improved in all 7 patients, 6 of whom had diabetes pre-operatively.[30] Fogar et al also reported improvement in oral glucose tolerance in 8/9 (89%) patients who underwent radical resection for pancreatic cancer.[31] In a study of 41 diabetic pancreatic cancer patients who underwent pancreaticoduodenectomy, we found that the diabetes resolved postoperatively in 57% of those with new-onset diabetes (n=30); none of the patients with long-standing diabetes (n=11) experienced any improvement in glycemic status [26]. These studies provide further evidence that pancreatic cancer induces glucose intolerance.
d) Experimental data that pancreatic cancer induces diabetes
Pancreatic cancer cell line conditioned media are metabolically active. They impair glucose metabolism in human myoblasts[32] and isolated and perfused rat hepatocytes.[33] Insulin release is reduced when isolated rat pancreatic islets are incubated with conditioned media of Panc-1 and HPAF cell lines or co-cultured with Panc-1 and HPAF cells.[34, 35] In a study by Basso et al,[36] daily intraperitoneal injection of supernatant from pancreatic cancer cell line (MIA PaCa2) into immunodeficient mice led to significant increase in blood glucose levels and significantly reduced glucose tolerance compared to control mice injected with saline. These studies suggest that pancreatic cancer cell lines produce soluble factor(s) that can impair glucose metabolism in vitro and cause hyperglycemia in vivo [36].
In summary, the very high prevalence of new-onset diabetes, which appears to improve after tumor resection coupled with the above experimental evidence, suggests that pancreatic cancer causes diabetes and this may have a unique pathogenic mechanism.
New-onset diabetes: a potential first sieve for pancreatic cancer screening
Patients with new-onset diabetes have a higher probability of subsequently being diagnosed with pancreatic cancer. In a population based cohort of 2,122 diabetic individuals in Olmsted County, MN, we determined that 18 (0·8%) new-onset diabetic individuals aged 50 or older were diagnosed with pancreatic cancer within 3 years of meeting criteria for diabetes and the observed-to-expected ratio of pancreatic cancer in this cohort of newly diagnosed diabetes subjects was 7·9 (95% confidence interval [CI] 4·7–12·5).[37]
For diabetes to be useful as a screening tool, it should be an early phenomenon that offers a window of opportunity to detect early stage pancreatic cancer in asymptomatic individuals. As noted previously, there is a marked and continuous increase in prevalence of diabetes from 24 to 36 months preceding the diagnosis of pancreatic cancer and leading up to the time of diagnosis.[29] In a recent retrospective study, we noted that the mean interval between onset of diabetes and diagnosis of pancreatic cancer was 10 months (range 5–29 months).[14] In addition, several studies have noted that diabetes is prevalent even in early stage pancreatic cancer.[23, 24, 27, 30, 31, 38, 39] In a recent large study we found that 115/232 (50%) patients with stage I and II pancreatic cancers had diabetes[26]. Permert et al conducted oral glucose tolerance tests in 44 consecutive resectable pancreatic cancer patients and noted that 28 (64%) had diabetes and an additional five patients (11%) had impaired glucose tolerance.[23] Small (≤20 mm) and minute pancreatic cancers (<10 mm) are often resectable. Tsuchiya et al reported that 61% of patients with small pancreatic cancer (≤2 cm) had an abnormal glucose tolerance test[39] and a study of minute pancreatic cancers (<10 mm) found 33% prevalence of diabetes[9]. Thus nearly half the patients with early stage, resectable tumors have diabetes.
In summary, new-onset diabetes not only defines a high-risk group for pancreatic cancer but is also a marker of early, asymptomatic cancer. Its occurrence in nearly half the patients with pancreatic cancer makes it an attractive screening target for early pancreatic cancer.
Proposed screening startegy based on new-onset diabetes
We propose that asymptomatic subjects be screened for diabetes and those with new-onset diabetes undergo “secondary sieving” to further enrich the group for pancreatic cancer. This cohort would then undergo noninvasive imaging or EUS to identify and confirm the diagnosis of pancreatic cancer (figure 4).
Figure 4.

Proposed screening strategy for pancreatic cancer using hyperglycemia as an indicator of underlying pancreatic cancer.
*A reliable biomarker for pancreatic cancer associated diabetes remains to be identified.
Approximately half the patients with sporadic pancreatic cancer have diabetes. In nearly 50% of diabetic pancreatic cancer patients, the diabetes is diagnosed concomitant with or shortly before cancer diagnosis [27, 28]. Hence, a strategy that relies only on physician diagnosed new-onset diabetes may capture at the most 25% of cancers. While this in itself would be a significant advance, it can certainly be improved upon. It is very likely that onset of diabetes precedes its symptomatic presentation by many months and sometimes a few years. Recent temporal association studies indicate that there is a substantial window of opportunity between the date subjects meet biochemical criteria for diabetes and diagnosis of cancer.[29] Therefore, to fully utilize the potential of recent hyperglycemia as a clue to early diagnosis of pancreatic cancer one would have to screen individuals for asymptomatic incident diabetes (for example, during annual physical examinations) rather than wait till symptomatic diabetes is diagnosed (figure 4). The case study presented (figure 2) illustrates this point. Presently the American Diabetes Association (ADA) recommends measurement of fasting blood glucose every 3 years beginning at age 45 in all individuals [40]. If a marker that can distinguish pancreatic cancer-associated diabetes from type 2 diabetes is identified, consideration could be given to shortening the interval of fasting blood glucose measurements to every year in individuals over the age of 50 years.
Whether subjects with new-onset diabetes should be directly screened for pancreatic cancer using CT and/or EUS or undergo “secondary sieving” to further enrich the population for pancreatic cancer is debatable (figure 4). The specific performance characteristics of CT and EUS in the setting of new-onset diabetes should be investigated in a clinical trial before advocating this approach clinically. We are currently enrolling patients at our center for such a study.
While in the case study presented (figure 2) CT scan revealed a resectable cancer at onset of diabetes, we believe that non-invasive imaging techniques, such as abdominal CT scans, are unlikely to consistently detect pancreatic cancer in asymptomatic individuals. This is based on retrospective reviews of CT scans done prior to pancreatic cancer diagnosis, which noted that pancreatic cancer was often undetectable >6 months prior to its diagnosis.[13, 14] Therefore, confirmation of the presence of pancreatic cancer in the screening population (i.e., in asymptomatic subjects) may require invasive tests such as EUS. Screening all new-onset diabetics for pancreatic cancer using invasive tests is unlikely to be cost-effective as the prevalence of pancreatic cancer in this population is <1%.[37] More importantly, it is becoming increasingly clear that abnormalities on EUS are seen even in the absence of clinical disease, especially in older subjects, smokers and alcohol users.[41] This “background noise” is likely to create doubt about the presence or absence of cancer possibly leading to unnecessary pancreatic resection. This indeed has been the experience of centers screening familial pancreatic cancer kindreds.[17] There are presently no studies that have screened asymptomatic new-onset diabetics for pancreatic cancer.
New-onset diabetes based screening strategy: search for the second sieve
The secondary sieve could be symptoms, clinical phenotype, or a unique biomarker of pancreatic cancer. Two studies have prospectively screened for pancreatic cancer in new-onset diabetes using symptoms and CA 19-9 elevation as secondary sieves.[42, 43] Though the prevalence of pancreatic cancer in the screened population was high (4.7% and 13%), most cancers identified were unresectable, again reiterating that screening for pancreatic cancer will have to be done in asymptomatic subjects.
Previous authors have suggested that new-onset diabetes in a lean patient or a patient without a family history of diabetes should raise suspicion for pancreatic cancer.[22] In a study of 240 pancreatic cancer patients with diabetes and 62 with type 2 diabetes, we noted that most patients with pancreatic cancer were over-weight or obese prior to onset of symptoms.[27] Indeed, obesity is a recognized risk factor for the development of pancreatic cancer.[44] We have also reported that the prevalence of conventional diabetes risk factors such as age, family history, and obesity were similar among patients with pancreatic cancer-associated diabetes and controls with type 2 diabetes.[26, 29] Therefore, a purely clinical distinction between pancreatic cancer-associated diabetes and type 2 diabetes is likely to be difficult.
In the absence of clinical features that distinguish pancreatic cancer-associated diabetes from type 2 diabetes, we will require a serologic or molecular biomarker to identify pancreatic cancer among patients with new-onset diabetes. This biomarker does not necessarily have to be metabolically active or even be the mediator of diabetes. For example, a marker in stool that detects early pancreatic cancer could serve as the secondary sieve. However, as new-onset glucose intolerance is a very frequent and characteristic feature of pancreatic cancer, it is reasonable to presume that the mediator(s) of diabetes will also be useful biomarker(s) of pancreatic cancer.
Previous candidate biomarkers of pancreatic cancer-associated diabetes have either failed in validation studies or remain to be validated. Though fasting levels of diabetogenic islet hormones, glucagon, somatostatin, and islet amyloid polypeptide (IAPP) are elevated in diabetic pancreatic cancer patients,[45] they have not proven to be useful discriminators between pancreatic cancer-associated diabetes and type 2 diabetes. Though initial reports of plasma islet amyloid peptide (IAPP) were promising,[46] this peptide has poor sensitivity for pancreatic cancer-associated diabetes or resectable pancreatic cancer.[27] Basso et al reported pancreatic cancer derived S100A8 N-terminal peptide in tumors from diabetics but not from non-diabetic pancreatic cancer or non-neoplastic tissues and this peptide had effects on glucose catabolism in myoblasts in vitro.[47] Recently Pfeffer et al reported that connexin26, a gap junction protein, is over-expressed in islets of pancreatic cancer and there is a correlation between connexin26 mRNA abundance and glucose level in glucose tolerance tests.[48] However, these preliminary findings remain to be confirmed.
In summary, we currently do not have a reliable marker of early pancreatic cancer or a marker specific for pancreatic cancer-associated diabetes. Understanding the pathogenesis of pancreatic cancer-associated diabetes and identifying unique defects in glucose turnover is likely to give important insights into its possible mediators.
Pathogenesis of pancreatic cancer-associated diabetes
Diabetes is caused by a relative or absolute impairment in insulin secretion (β-cell dysfunction) along with varying degrees of peripheral resistance to insulin action (insulin resistance).[49, 50] Though insulin resistance is frequently observed in a number of physiologic and pathologic states including puberty, pregnancy, ageing, and obesity, most insulin-resistant individuals do not develop hyperglycemia as normal pancreatic islet cells compensate for impaired insulin action by augmenting insulin release. In type 2 diabetes defective insulin secretion (β-cell failure) in the face of persistently impaired insulin action (insulin resistance), impaired glucose effectiveness (ability of glucose to stimulate its own uptake and suppress its own release in the absence of a dynamic insulin response), and defective suppression of glucagon secretion contribute to hyperglycemia (figure 5).
Figure 5.

Proposed pathogenesis of type 2 diabetes (left panel) and pancreatic cancer-associated diabetes (right panel)
The pathogenesis of pancreatic cancer-associated diabetes and its biochemical mediators are not known. This is unlikely to be simply due to destruction of the gland by the tumor or due to obstructive chronic pancreatitis induced by the tumor. The high prevalence of diabetes and glucose intolerance in small pancreatic cancer (tumors <2 cm)[10] and early stage tumors[26] and the onset of diabetes nearly 2 years before diagnosis[29] and prior to radiologically detectable tumor[14] tends to favor a humoral process rather than a local tumor effect. Additionally, insulin and C-peptide levels in pancreatic cancer, especially in those with diabetes have been reported to be higher than in healthy controls.[45, 46] If pancreatic destruction and resultant decrease in beta cell mass was the cause of diabetes in pancreatic cancer, one would have expected low levels of C-peptide and insulin, as are seen in diabetes associated with chronic pancreatitis. [51, 52]
In vitro studies
In vitro experiments reveal a combination of insulin resistance and β-cell dysfunction in pancreatic cancer. Skeletal muscle from pancreatic cancer patients, compared to controls, demonstrates impaired glucose transport and phosphatidylinositol 3-kinase (PI3-K) activity in the presence of physiologic concentrations of insulin.[53] Decreased glycogen synthase activity has also been reported in skeletal muscles of humans and rodents with pancreatic cancer.[32, 54] When human myoblasts are exposed to pancreatic cancer-conditioned media, enhanced glucose transformation into lactate is noted, a phenomenon which is more striking when myoblasts were exposed to tumor homogenates from pancreatic cancer with diabetes compared to pancreatic cancer without diabetes.[32] In isolated and perfused rat hepatocytes, pancreatic cancer-conditioned media reduces glycolysis and increases production of 1,2 diacyl-glycerol.[33] These data show that the insulin signaling cascade in skeletal muscle and liver is impaired by pancreatic cancer. Upon molecular weight fractionation of conditioned media, metabolic effects were retained by proteins or peptides in fractions with a molecular weight of <10,000 Da.[55]
Beta-cell dysfunction has also been observed in in vitro and animal experiments. As noted earlier, insulin release is reduced when isolated rat pancreatic islets are incubated with conditioned media of Panc-1 and HPAF cell lines or co-cultured with Panc-1 and HPAF cells.[34, 35] In the BOP-treated Syrian hamster model of pancreatic cancer, glucose stimulated insulin release was impaired in vivo.[56] A recent report noted a decrease in the size of islet cells and number of β-cells in pancreata from patients with pancreatic cancer-associated diabetes.[57] However, in these pancreata other characteristic morphometric alterations usually seen in type 2 diabetes were not observed suggesting that these changes are not chronic.
Whole body physiology studies
Whole body physiology studies in humans with pancreatic cancer have used hyperglycemic[23] or hyperinsulinemic[58] glucose clamp techniques to measure insulin action. In a series of studies Permert et al [23, 30, 45, 58] reported increased insulin resistance in pancreatic cancer subjects, more so in diabetic than in non-diabetic subjects. As noted earlier, these authors also demonstrated that insulin resistance improves after pancreatic cancer resection [30]. β-cell response, i.e., increase in insulin and C-peptide levels to oral glucose load [24, 59], hyperglycemic clamp[45], or glucagon stimulation[45, 60, 61] is also impaired in pancreatic cancer. Permert et al[45] found that while insulin-requiring pancreatic cancer patients did not elevate C-peptide levels in response to hyperglycemia or glucagon, non-insulin requiring diabetic pancreatic cancer patients raised C-peptide levels in response to glucagon but not hyperglycemia. Following sub-total pancreatectomy for tumor resection there was an expected marked decline in β-cell response to both stimuli.[45] Using Homeostasis Model Assessment (HOMA), we observed that β-cell function was markedly diminished in pancreatic cancer with impaired fasting glucose while insulin resistance was only modestly increased.[62]
In summary, in vitro and human studies show evidence for both insulin resistance and β-cell dysfunction in pancreatic cancer, likely caused by tumor secreted protein(s) or peptide(s) with a low molecular weight. As is evident from other insulin resistant states (e.g. obesity and polycystic ovarian syndrome), insulin resistance in the absence of β-cell failure is not sufficient to cause diabetes. Also, the prevalence of diabetes in pancreatic cancer (45 to 65%) is markedly higher than in other diabetogenic states. In order to explain the very high prevalence, we postulate that the β-cell is the primary target in pancreatic cancer-associated diabetes. β-cells are likely made susceptible to failure by humorally circulating factors and the modest increase in insulin resistance associated with pancreatic cancer is sufficient to cause β-cell decompensation and diabetes (figure 5). Also, the development of diabetes in pancreatic cancer is likely a result of interaction between host (age BMI, and family history of diabetes) and tumor factors (yet unknown). Subjects with predisposition to diabetes (older age, positive family history of diabetes or obesity) may decompensate and develop diabetes more often than those without risk factors for diabetes. How this interaction affects the development and progression of diabetes is unclear. Better understanding of the pathogenesis of pancreatic cancer-associated diabetes may shed light on this issue.
Conclusions
Early detection of pancreatic cancer appears to hold the greatest promise with regards to improving long-term survival. For this, screening will have to be targeted at asymptomatic individuals. New-onset diabetes is present in nearly half of all pancreatic cancer and various lines of evidence suggest that diabetes is caused by the cancer. Importantly, diabetes appears to be associated with early stage pancreatic cancer. However, further enrichment of the population of new-onset diabetics will be needed before screening becomes cost-effective. The pathogenesis of pancreatic cancer-associated diabetes remains to be elucidated. The very high prevalence of diabetes in pancreatic cancer suggests that β-cell dysfunction, likely related to tumor-secreted products, is the primary defect in pancreatic cancer-associated diabetes. Future studies should focus on understanding the pathogenesis of pancreatic cancer-associated diabetes and identifying biomarkers that can distinguish it from type 2 diabetes. In addition it would be important to investigate the prevalence, clinical profile, and temporal association between diabetes and pancreatic cancer in other patient populations.
Table 1.
FBG: fasting blood glucose. To convert mmol/L to mg/dl, multiply by 18. CT computed tomography, CA 19-9: Carbohydrate antigen 19-9, Body mass index (BMI) calculated as weight (kg)/height (m)2
(table 1 to accompany figure 2)
| A | B | C | D | ||
|---|---|---|---|---|---|
| Time Period | 1998–2003 | 2004 | September 2005 | November 2005 | 2007 |
| FBG (mmol/L) | 5.4 | 6.2 | 7.2 | 5.7 | 6.2 |
| BMI (kg/m2) | 25.5 | 25.7 | 25.9 | 24.3 | 22.5 |
| CT scan | Normal pancreas | - | 1.5 cm mass, head of pancreas | No recurrence | No recurrence |
| CA 19-9 (U/ml) | - | - | 47.8 | 7.7 | 8 |
Acknowledgments
Grant Support: Dr Chari’s research was funded by grants from NIH (R01 CA 100685) and the Mayo Clinic Pancreas Cancer SPORE (P50 CA 10270).
Footnotes
Financial Disclosures: None
Search Strategy: We searched Pubmed and Ovid databases using combinations of the following key words: “diabetes”, “pancreatic cancer”, and “screening” and retrieved articles from 1985 through present. Only English language studies were considered. All authors reviewed original studies and reviews for relevance and included all pertinent studies in the preparation of the manuscript. We also reviewed the bibliographies of the selected articles for other pertinent citations.
References
- 1.Jemal A, Seigel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007;18:581–592. doi: 10.1093/annonc/mdl498. [DOI] [PubMed] [Google Scholar]
- 3.Berrino F, De Angelis R, Sant M, Rosso S, Lasota MB, Coebergh JW, et al. Survival for eight major cancers and all cancers combined for European adults diagnosed in 1995–99: results of the EUROCARE-4 study. Lancet Oncol. 2007;8(9):773–83. doi: 10.1016/S1470-2045(07)70245-0. [DOI] [PubMed] [Google Scholar]
- 4.Key C. Cancer of the Pancreas. In: Ries LAG, Young JL, Keel GE, Eisner MP, Lin YD, Horner M-J, editors. SEER Survival Monograph: Cancer Survival Among Adults: U.S. SEER Program, 1988–2001, Patient and Tumor Characteristics. Bethesda: National Cancer Institute; 2007. SEER Program, NIH Pub. No. 07–6215. [Google Scholar]
- 5.Conlon KC, Klimstra DS, Brennan MF. Long-term survival after curative resection for pancreatic ductal adenocarcinoma. Clinicopathologic analysis of 5-year survivors. Ann Surg. 1996;223(3):273–9. doi: 10.1097/00000658-199603000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sohn TA, Yeo CJ, Cameron JL, Koniaris L, Kaushal S, Abrams RA, et al. Resected adenocarcinoma of the pancreas-616 patients: results, outcomes, and prognostic indicators. J Gastrointest Surg. 2000;4(6):567–79. doi: 10.1016/s1091-255x(00)80105-5. [DOI] [PubMed] [Google Scholar]
- 7.Furukawa H, Okada S, Saisho H, Ariyama J, Karasawa E, Nakaizumi A, et al. Clinicopathologic features of small pancreatic adenocarcinoma. A collective study Cancer. 1996;78(5):986–90. doi: 10.1002/(SICI)1097-0142(19960901)78:5<986::AID-CNCR7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu Y, Yasui K, Matsueda K, Yanagisawa A, Yamao K. Small carcinoma of the pancreas is curable: new computed tomography finding, pathological study and postoperative results from a single institute. J Gastroenterol Hepatol. 2005;20(10):1591–4. doi: 10.1111/j.1440-1746.2005.03895.x. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa O, Ohigashi H, Imaoka S, Nakaizumi A, Uehara H, Kitamura T, et al. Minute carcinoma of the pancreas measuring 1 cm or less in diameter-- collective review of Japanese case reports. Hepatogastroenterology. 1999;46(25):8–15. [PubMed] [Google Scholar]
- 10.Tsuchiya R, Noda T, Harada N, Miyamoto T, Tomioka T, Yamamoto K, et al. Collective review of small carcinomas of the pancreas. Ann Surg. 1986;203(1):77–81. doi: 10.1097/00000658-198601000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969–72. [PubMed] [Google Scholar]
- 12.Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol. 1998;22(2):163–9. doi: 10.1097/00000478-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Gangi S, Fletcher JG, Nathan MA, Christensen JA, Harmsen WS, Crownhart BS, et al. Time interval between abnormalities seen on CT and the clinical diagnosis of pancreatic cancer: retrospective review of CT scans obtained before diagnosis. Am J Roentgenol. 2004;182(4):897–903. doi: 10.2214/ajr.182.4.1820897. [DOI] [PubMed] [Google Scholar]
- 14.Pelaez-Luna M, Takahashi N, Fletcher JG, Chari ST. Resectability of presymptomatic pancreatic cancer and its relationship to onset of diabetes: a retrospective review of CT scans and fasting glucose values prior to diagnosis. Am J Gastroenterol. 2007;102(10):2157–63. doi: 10.1111/j.1572-0241.2007.01480.x. [DOI] [PubMed] [Google Scholar]
- 15.SEER Cancer Statistics Review, 1975–2004. National Cancer Institute; Bethesda, MD: [Accessed February 14, 2008]. http://seer.cancer.gov/csr/1975_2004/, based on November 2006 SEER data submission. [Google Scholar]
- 16.Chari ST. Detecting Pancreatic Cancer Early: Problems and Prospects. Semin Oncol. 2007;34:284–294. doi: 10.1053/j.seminoncol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canto MI, Goggins M, Yeo CJ, Griffin C, Axilbund JE, Brune K, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clin Gastroenterol Hepatol. 2004;2(7):606–21. doi: 10.1016/s1542-3565(04)00244-7. [DOI] [PubMed] [Google Scholar]
- 18.Brentnall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med. 1999;131(4):247–55. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- 19.Goggins M, Canto M, Hruban R. Can we screen high-risk individuals to detect early pancreatic carcinoma? J Surg Oncol. 2000;74(4):243–8. doi: 10.1002/1096-9098(200008)74:4<243::aid-jso1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 20.Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta- analysis JAMA. 1995;273(20):1605–9. [PubMed] [Google Scholar]
- 21.Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer. 2005;92(11):2076–83. doi: 10.1038/sj.bjc.6602619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noy A, Bilezikian JP. Clinical review 63: Diabetes and pancreatic cancer: clues to the early diagnosis of pancreatic malignancy. J Clin Endocrinol Metab. 1994;79(5):1223–31. doi: 10.1210/jcem.79.5.7962312. [DOI] [PubMed] [Google Scholar]
- 23.Permert J, Ihse I, Jorfeldt L, von Schenck H, Arnqvist HJ, Larsson J. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993;159(2):101–7. [PubMed] [Google Scholar]
- 24.Cersosimo E, Pisters PW, Pesola G, McDermott K, Bajorunas D, Brennan MF. Insulin secretion and action in patients with pancreatic cancer. Cancer. 1991;67(2):486–93. doi: 10.1002/1097-0142(19910115)67:2<486::aid-cncr2820670228>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 25.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2007;30:S42–47. doi: 10.2337/dc07-S042. [DOI] [PubMed] [Google Scholar]
- 26.Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM, Chari ST. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology. 2008;134 (4):981–987. doi: 10.1053/j.gastro.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chari ST, Klee GG, Miller LJ, Raimondo M, DiMagno EP. Islet amyloid polypeptide is not a satisfactory marker for detecting pancreatic cancer. Gastroenterology. 2001;121(3):640–5. doi: 10.1053/gast.2001.27210. [DOI] [PubMed] [Google Scholar]
- 28.Gullo L, Pezzilli R, Morselli-Labate AM. Diabetes and the risk of pancreatic cancer. Italian Pancreatic Cancer Study Group. N Engl J Med. 1994;331(2):81–4. doi: 10.1056/NEJM199407143310203. [DOI] [PubMed] [Google Scholar]
- 29.Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, de Andrade M, et al. Pancreatic Cancer-associated Diabetes Mellitus: Prevalence and Temporal Association with Diagnosis of Cancer. Gastroenterology. 2008;134(1):95–101. doi: 10.1053/j.gastro.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Permert J, Ihse I, Jorfeldt L, von Schenck H, Arnquist HJ, Larsson J. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br J Surg. 1993;80(8):1047–50. doi: 10.1002/bjs.1800800841. [DOI] [PubMed] [Google Scholar]
- 31.Fogar P, Pasquali C, Basso D, Sperti C, Panozzo MP, Tessari G, et al. Diabetes mellitus in pancreatic cancer follow-up. Anticancer Res. 1994;14(6B):2827–30. [PubMed] [Google Scholar]
- 32.Basso D, Millino C, Greco E, Romualdi C, Fogar P, Valerio A, et al. Altered glucose metabolism and proteolysis in pancreatic cancer cell conditioned myoblasts: searching for a gene expression pattern with a microarray analysis of 5000 skeletal muscle genes. Gut. 2004;53(8):1159–66. doi: 10.1136/gut.2003.024471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valerio A, Basso D, Brigato L, Ceolotto G, Baldo G, Tiengo A, et al. Glucose metabolic alterations in isolated and perfused rat hepatocytes induced by pancreatic cancer conditioned medium: a low molecular weight factor possibly involved. Biochem Biophys Res Commun. 1999;257(2):622–8. doi: 10.1006/bbrc.1999.0521. [DOI] [PubMed] [Google Scholar]
- 34.Wang F, Larsson J, Abdiu A, Gasslander T, Westermark P, Adrian TE, Permert J. Dissociated secretion of islet amyloid polypeptide and insulin in serum- free culture media conditioned by human pancreatic adenocarcinoma cell lines. Int J Pancreatol. 1997;21(2):157–64. doi: 10.1007/BF02822387. [DOI] [PubMed] [Google Scholar]
- 35.Wang F, Larsson J, Adrian TE, Gasslander T, Permert J. In vitro influences between pancreatic adenocarcinoma cells and pancreatic islets. Journal of Surgical Research. 1998;79(1):13–9. doi: 10.1006/jsre.1998.5393. [DOI] [PubMed] [Google Scholar]
- 36.Basso D, Brigato L, Veronesi A, Panozzo MP, Amadori A, Plebani M. The pancreatic cancer cell line MIA PaCa2 produces one or more factors able to induce hyperglycemia in SCID mice. Anticancer Res. 1995;15(6B):2585–8. [PubMed] [Google Scholar]
- 37.Chari ST, Liebson CL, de Andrade M, Rabe KG, Ransom JE, Petersen GM. Probability of pancreatic cancer following diabetes: A population-based study. Gastroenterology. 2005;129(2):504–511. doi: 10.1053/j.gastro.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green RC, Jr, Baggenstoss AH, Sprague RG. Diabetes mellitus in association with primary carcinoma of the pancreas. Diabetes. 1958;7(4):308–11. doi: 10.2337/diab.7.4.308. [DOI] [PubMed] [Google Scholar]
- 39.Tsuchiya R, Noda T, Harada N, Miyamoto T, Tomioka T, et al. Collective review of small carcinomas of the pancreas. Annals of Surgery. 1986;203(1):77–81. doi: 10.1097/00000658-198601000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.American Diabetes Association. Standards of Medical Care in Diabetes 2008. Diabetes Care. 2008;31:S12–S54. doi: 10.2337/dc08-S012. [DOI] [PubMed] [Google Scholar]
- 41.Yusoff IF, Sahai AV. A prospective, quantitative assessment of the effect of ethanol and other variables on the endosonographic appearance of the pancreas. Clin Gastroenterol Hepatol. 2004;2(5):405–9. doi: 10.1016/s1542-3565(04)00126-0. [DOI] [PubMed] [Google Scholar]
- 42.Ogawa Y, Tanaka M, Inoue K, Yamaguchi K, Chijiiwa K, Mizumoto K, et al. A prospective pancreatographic study of the prevalence of pancreatic carcinoma in patients with diabetes mellitus. Cancer. 2002;94(9):2344–9. doi: 10.1002/cncr.10493. [DOI] [PubMed] [Google Scholar]
- 43.Damiano J, Bordier L, Le Berre JP, Margery J, Dupuy O, Mayaudon H, et al. Should pancreas imaging be recommanded in patients over 50 years when diabetes is discovered because of acute symptoms? Diabetes & Metabolism. 2004;30(2):203–7. doi: 10.1016/s1262-3636(07)70111-8. [DOI] [PubMed] [Google Scholar]
- 44.Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. Bmj. 2007;335(7630):1134. doi: 10.1136/bmj.39367.495995.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Permert J, Larsson J, Fruin AB, Tatemoto K, Herrington MK, von Schenck H, et al. Islet hormone secretion in pancreatic cancer patients with diabetes. Pancreas. 1997;15(1):60–8. doi: 10.1097/00006676-199707000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Pour PM, et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med. 1994;330(5):313–8. doi: 10.1056/NEJM199402033300503. [DOI] [PubMed] [Google Scholar]
- 47.Basso D, Greco E, Fogar P, Pucci P, Flagiello A, Baldo G, et al. Pancreatic cancer-derived S-100A8 N-terminal peptide: A diabetes cause? Clin Chim Acta. 2006;372(1–2):120–8. doi: 10.1016/j.cca.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 48.Pfeffer F, Koczan D, Adam U, Benz S, von Dobschuetz E, Prall F, et al. Expression of connexin26 in islets of Langerhans is associated with impaired glucose tolerance in patients with pancreatic adenocarcinoma. Pancreas. 2004;29(4):284–90. doi: 10.1097/00006676-200411000-00007. [DOI] [PubMed] [Google Scholar]
- 49.Vella A, Camilleri M, Rizza RA. The gastrointestinal tract and glucose tolerance. Curr Opin Clin Nutr Metab Care. 2004;7(4):479–84. doi: 10.1097/01.mco.0000134375.01310.97. [DOI] [PubMed] [Google Scholar]
- 50.Dinneen S, Gerich J, Rizza R. Carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N Engl J Med. 1992;327(10):707–13. doi: 10.1056/NEJM199209033271007. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura T, Imamura K, Takebe K, Terada A, Arai Y, Tandoh Y, et al. Correlation between pancreatic endocrine and exocrine function and characteristics of pancreatic endocrine function in patients with diabetes mellitus owing to chronic pancreatitis. Int J Pancreatol. 1996;20(3):169–75. doi: 10.1007/BF02803765. [DOI] [PubMed] [Google Scholar]
- 52.Bank S, Marks IN, Vinik AI. Clinical and hormonal aspects of pancreatic diabetes. Am J Gastroenterol. 1975;64(1):13–22. [PubMed] [Google Scholar]
- 53.Isaksson B, Strommer L, Friess H, Buchler MW, Herrington MK, Wang F, et al. Impaired insulin action on phosphatidylinositol 3-kinase activity and glucose transport in skeletal muscle of pancreatic cancer patients. Pancreas. 2003;26(2):173–7. doi: 10.1097/00006676-200303000-00014. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Knezetic JA, Strommer L, Permert J, Larsson J, Adrian TE. The intracellular mechanism of insulin resistance in pancreatic cancer patients. J Clin Endocrinol Metab. 2000;85(3):1232–8. doi: 10.1210/jcem.85.3.6400. [DOI] [PubMed] [Google Scholar]
- 55.Basso D, Valerio A, Seraglia R, Mazza S, Piva MG, Greco E, et al. Putative pancreatic cancer-associated diabetogenic factor: 2030 MW peptide. Pancreas. 2002;24(1):8–14. doi: 10.1097/00006676-200201000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Ahren B, Andren-Sandberg A. Glucose tolerance and insulin secretion in experimental pancreatic cancer in the Syrian hamster. Res Exp Med. 1993;193(1):21–6. doi: 10.1007/BF02576207. [DOI] [PubMed] [Google Scholar]
- 57.Katsumichi I, Pour PM. Diabetes mellitus in pancreatic cancer: is it a causal relationship? Am J Surg. 2007;194:S71–S75. doi: 10.1016/j.amjsurg.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 58.Permert J, Adrian TE, Jacobsson P, Jorfelt L, Fruin AB, Larsson J. Is profound peripheral insulin resistance in patients with pancreatic cancer caused by a tumor-associated factor? Am J Surg. 1993;165(1):61–6. doi: 10.1016/s0002-9610(05)80405-2. [DOI] [PubMed] [Google Scholar]
- 59.Schwarts SS, Zeidler A, Moossa AR, Kuku SF, Rubenstein AH. A prospective study of glucose tolerance, insulin, C-peptide, and glucagon responses in patients with pancreatic carcinoma. Am J Dig Dis. 1978;23(12):1107–14. doi: 10.1007/BF01072886. [DOI] [PubMed] [Google Scholar]
- 60.Fox JN, Frier BM, Armitage M, Ashby JP. Abnormal insulin secretion in carcinoma of the pancreas: response to glucagon stimulation. Diabet Med. 1985;2(2):113–6. doi: 10.1111/j.1464-5491.1985.tb00612.x. [DOI] [PubMed] [Google Scholar]
- 61.Basso D, Plebani M, Fogar P, Del Favero G, Briani G, Meggiato T, et al. Beta- cell function in pancreatic adenocarcinoma. Pancreas. 1994;9(3):332–5. doi: 10.1097/00006676-199405000-00008. [DOI] [PubMed] [Google Scholar]
- 62.Chari ST, Zapiach M, Yadav D, Rizza RA. Beta-cell function and insulin resistance evaluated by HOMA in pancreatic cancer subjects with varying degrees of glucose intolerance. Pancreatology. 2005;5(2–3):229–33. doi: 10.1159/000085276. [DOI] [PubMed] [Google Scholar]