

Abstract
OBJECTIVES
Our aim was to determine the frequency of genomic imbalances in neonates with birth defects by using targeted array-based comparative genomic hybridization, also known as chromosomal microarray analysis.
METHODS
Between March 2006 and September 2007, 638 neonates with various birth defects were referred for chromosomal microarray analysis. Three consecutive chromosomal microarray analysis versions were used: bacterial artificial chromosome-based versions V5 and V6 and bacterial artificial chromosome emulated oligonucleotide-based version V6 Oligo. Each version had targeted but increasingly extensive genomic coverage and interrogated >150 disease loci with enhanced coverage in genomic rearrangement-prone pericentromeric and subtelomeric regions.
RESULTS
Overall, 109 (17.1%) patients were identified with clinically significant abnormalities with detection rates of 13.7%, 16.6%, and 19.9% on V5, V6, and V6 Oligo, respectively. The majority of these abnormalities would not be defined by using karyotype analysis. The clinically significant detection rates by use of chromosomal microarray analysis for various clinical indications were 66.7% for “possible chromosomal abnormality” ± “others” (other clinical indications), 33.3% for ambiguous genitalia ± others, 27.1% for dysmorphic features + multiple congenital anomalies ± others, 24.6% for dysmorphic features ± others, 21.8% for congenital heart disease ± others, 17.9% for multiple congenital anomalies ± others, and 9.5% for the patients referred for others that were different from the groups defined. In all, 16 (2.5%) patients had chromosomal aneuploidies, and 81 (12.7%) patients had segmental aneusomies including common microdeletion or microduplication syndromes and other genomic disorders. Chromosomal mosaicism was found in 12 (1.9%) neonates.
CONCLUSIONS
Chromosomal microarray analysis is a valuable clinical diagnostic tool that allows precise and rapid identification of genomic imbalances and mosaic abnormalities as the cause of birth defects in neonates. Chromosomal microarray analysis allows for timely molecular diagnoses and detects many more clinically relevant genomic abnormalities than conventional cytogenetic studies, enabling more informed decision-making and management and appropriate assessment of recurrence risk.
Keywords: sporadic birth defects, array comparative genomic hybridization, copy-number variation, molecular cytogenetic analysis, mosaicism
In the united States, birth defects are the leading cause of neonatal morbidity and mortality1 and occur in ~3% of all newborns.2 Genetic factors have been long recognized to be an important cause for syndromic and nonsyndromic congenital anomalies.3 Previous cytogenetic investigations demonstrated that constitutional chromosomal abnormalities affect ~0.5% of live-born infants4,5 and contribute to a significant percentage of birth defects.6 Chromosomal abnormalities are found in ~25% of deaths in newborns with congenital anomalies.7 Common chromosomal aneuploidies, such as monosomy X and trisomy for chromosomes 21, 18, and 13, have traditionally been diagnosed during the neonatal period by chromosome studies by using conventional cytogenetic methods, trypsin-Giemsa staining (GTG banding) analysis. Improved cytogenetic methods, such as fluorescence in situ hybridization (FISH) by using chromosome- or locus-specific probes, enable the diagnosis of the common microdeletion syndromes such as DiGeorge/velocardiofacial syndrome (DG/VCFS),8 Wolf-Hirschhorn syndrome (4p-),9 and Cri-du-chat syndrome (5p-)10 in patients with dysmorphic features and/or congenital anomalies; however, FISH assays using genomic locus-specific probes are applicable only for patients with a strong clinical suspicion of a specific genetic defect. This is often challenging in neonates with birth defects because their clinical presentations may be atypical, they may have nonspecific phenotypic features shared by several different genetic disorders, or they may lack specific syndromic features that appear only at a later age. Syndrome recognition can also be challenging in premature children because some clinical features may not yet be evident or may be obscured by other pathologies seen in these newborns.
Despite advances in prenatal diagnosis and perinatal management, birth defects remain a major cause of neonatal morbidity and mortality,11 and the genetic cause of the majority of birth defects remains unknown.12 Submicroscopic genomic rearrangements may be the cause in a significant proportion of birth defects, and many such cryptic genomic imbalances are not identifiable by using conventional cytogenetic methods because of their limited coverage (eg, FISH) and resolution (eg, GTG banding) for genomic analysis.13 Precise determinations of a diagnosis for the child’s pattern of birth defects is critical because a diagnosis may determine medical and surgical management as well as help to provide accurate information to families regarding prognosis and recurrence risks. Thus, newer molecular cytogenetic diagnostic tools with improved genomic coverage and resolution are needed to increase the detection rate for genomic imbalances, especially in neonates with major birth defects and seemingly normal constitutional chromosomes.
Array comparative genomic hybridization (aCGH)14 has proved to be such a powerful innovative tool for high-resolution genome analysis and for the detection of gains or losses (ie, genomic imbalances) resulting in copy-number variations (CNVs). CNVs are segments of deletion or duplication in the genome that may represent rare or common benign variants (benign CNVs) or pathologic variants (eg, deletions in DG/VCFS or Williams-Beuren syndrome). The use of aCGH for genomic analysis detects both benign and pathologic CNVs, and there is an important need to better understand benign CNVs in the normal population15,16 or in the various ethnic populations17 and to distinguish the pathologic CNV that can be responsible for the human constitutional disorders18 and autism.19 Two well-established aCGH platforms have been developed and clinically implemented: the large insert bacterial artificial chromosome (BAC)-based array20 and the oligonucleotide-based array.21-23 Both whole-genome arrays that interrogate the entire genome and targeted arrays focused on specific genomic regions of clinical interest24 have had a major impact on postnatal evaluation25-29 and have the potential for use in prenatal diagnosis.30-32 aCGH also has enabled the identification of novel disease genes,33 discovery of new microdeletion syndromes,34,35 and detection of low-level mosaicism that is undetected by conventional chromosome analysis.36,37 This has significantly increased the identification of genomic imbalances (eg, pathogenic CNV) as the potential cause in patients with dysmorphic features (DFs), multiple congenital anomalies (MCA), or mental retardation.13,27,38-41
To our knowledge, to date, no studies have specifically addressed CNVs by using aCGH in the neonatal population. We used a targeted aCGH platform, termed chromosomal microarray analysis (CMA), for 638 consecutive neonates who presented with various birth defects.
METHODS
Neonatal Patient Samples
Peripheral blood samples from a total of 638 neonates, aged ≤28 days, with a wide range of birth defects were submitted for CMA to the Clinical Cytogenetics Laboratory at Baylor College of Medicine between March 2006 and September 2007. The clinical indications at the time of the referral were taken from clinical comments on laboratory requisitions and are therefore relatively crude, but they are summarized in Table 1. Patients who had ≥2 indications were included in ≥2 categories. A total of 21 patients were suspected for possible chromosomal abnormality ± “others” (eg, cleft palate, club foot, polydactyly), and 12 patients were referred for ambiguous genitalia ± others. A total of 179 patients were referred for MCA ± others, including 59 for both MCA and DFs. For 138 patients, the clinical indications were DFs ± others (excluding MCA). A total of 101 patients’ samples were submitted because of congenital heart disease (CHD) including atrial septal defect, ventricular septal defect, tetralogy of Fallot, or CHD ± others. A total of 295 patients were referred for other indications, including a variety of anomalies involving neuromuscular, craniofacial, skeletal, gastrointestinal, and renal defects; cleft lip and/or palate; respiratory distress syndrome; and congenital diaphragmatic hernia, summarized in a large group referred as others (see Table 1). A total of 24 samples from neonatal patients had no clinical information provided at the time of submission of a sample for clinical CMA testing.
TABLE 1. Clinical Indications at the Time of Referral and Detection Rates for Clinically Significant CNV.
| Clinical Indications at the Time of the Referrala |
Cases (N = 638), n |
Clinically Significant CNVs (N = 109), n |
Overall Detection Rate (17.1%), % |
|---|---|---|---|
| Possible chromosomal abnormalities ± othersb |
21 | 14 | 66.7 |
| Ambiguous genitalia ± others | 12 | 4 | 33.3 |
| Ambiguous genitalia alone | 7 | 3 | 42.9 |
| Ambiguous genitalia ± others | 5 | 1 | 20.0 |
| MCA ± others | 179 | 32 | 17.9 |
| MCA + CHD | 7 | 2 | 28.6 |
| MCA + DFs ± others | 59 | 16 | 27.1 |
| MCA alone | 97 | 14 | 14.4 |
| MCA + others (excluding CHD or DFs) |
16 | 0 | 0.0 |
| DFs ± others (excluding MCA) | 138 | 34 | 24.6 |
| DFs alone | 81 | 19 | 23.5 |
| DFs + others | 57 | 15 | 26.3 |
| CHD ± others | 101 | 22 | 21.8 |
| Others also including following subgroups |
295 | 28 | 9.5 |
| Respiratory distress disorder | 9 | 2 | 22.2 |
| Possible syndrome | 24 | 5 | 20.8 |
| Heterotaxy | 6 | 1 | 16.7 |
| Failure to thrive, gastrointestinal or renal anomalies |
44 | 6 | 13.6 |
| Neuromuscular | 85 | 9 | 10.6 |
| Intrauterine growth retardation or premature |
21 | 2 | 9.5 |
| Hydrops | 13 | 1 | 7.7 |
| Cleft lip/palate | 21 | 1 | 4.8 |
| No indications | 24 | 1 | 4.2 |
| Seizure disorder | 19 | 0 | 0.0 |
| Congenital diaphragmatic hernia |
11 | 0 | 0.0 |
| Skeletal | 13 | 0 | 0.0 |
| Family history of chromosomal abnormality or DFs |
5 | 0 | 0.0 |
Patients who had ≥2 clinical indications were included in ≥2 groups in this table.
Broad spectrum of birth defects.
aCGH
Genomic DNA isolated from peripheral blood obtained from the 638 neonatal patients was analyzed on 1 of the 3 different CMA versions that interrogated >150 genomic disorder loci, with expanded coverage in pericentromeric and subtelomeric regions, depending on the time of sample submission (Table 2). The first 197 samples were analyzed on BAC V5, which consisted of 853 BAC clones.27 There were 175 samples in the second set analyzed on BAC V6, which consisted of 1475 BAC clones and with greater backbone coverage of the genome with inclusion of 1 clone per band at 650 cytogenetic banding resolution.22 The most recent group of the patients consisted of 266 neonates analyzed using V6 Oligo, which consisted of 42 640 oligonucleotides, with the average of 20 to 40 oligonucleotides corresponding to each V6 BAC clone genomic locus.22 CMA procedures, data analyses, and confirmation of abnormalities by using partial karyotype analysis and/or FISH were performed as previously described.22,25,27
TABLE 2. CNVs Detected by CMA in 638 Neonates With Birth Defects.
| Parameter | March 2006 BAC V5 |
November 2006 BAC V6a |
March 2007 V6 Oligo |
|---|---|---|---|
| Coverage of BACs/Oligos, n | 853 | 1475 | 1475; 44 K |
| Genomic disorders, n | >75 | >150 | >150 |
| Subtelomeric coverage, Mb | 10 | 10 | 10 |
| Pericentric coverage | Yes | Yes | Yes |
| Backbone coverage | Yes | Expanded | Expanded |
| Neonates studied, n | 197 | 175 | 266 |
| Clinically significant CNVs, n | 27 | 29 | 53 |
| Clinically significant CMA detection rate, % |
13.7 | 16.6 | 19.9 |
| Benign or unknown CNVs, n | 18 | 20 | 24 |
| Benign or unknown CNVs detection rate, % |
9.1 | 11.4 | 9.0 |
V6 has coverage of 1 clone per band at the 650-band level.
RESULTS
CMA Resolution and Clinically Significant CNV-Detection Rates
CNVs were classified as clinically significant, likely benign, or of uncertain significance as follows. Clinically significant abnormalities included aneuploidy, detection of well-characterized deletion/duplication syndromes, deletion/duplication >3 Mb in size or cytogenetically visible, and de novo deletions or duplications <1 Mb of which most were >0.5 Mb in size. Parental studies were performed or recommended for the patients for whom we reported clinically significant CMA findings (except for the patients with common trisomies or monosomy X) to determine whether the CMA findings represent de novo or familial events. Deletions or duplications that were not diagnostic of well-characterized syndromes were classified as “likely benign” when the variant was well documented to occur in the normal population on the basis of public databases (http://projects.tcag.ca/variation) or internal experience or when the variant was also present in a healthy parent. In cases in which parental samples were unavailable and the variants were <1 Mb in size and not cytogenetically visible, deletions were considered to be CNVs of uncertain significance. That parental samples were unavailable in such cases leads to an underestimation of the clinically significant abnormalities.27 The expectation is that parental studies will eventually be performed in most of these cases.
The detection rates of clinically significant CNVs or genetic aberrations in this study were 13.7% (27 of 197) for BAC V5, 16.6% (29 of 175) for BAC V6, and 19.9% (53 of 266) for V6 Oligo (see Table 2). A consistent increase in detection rate was observed for each advanced version of the arrays; especially in the 266 neonates who were studied by use of the most recent oligonucleotide array with expanded genomic coverage. In addition, 62 (9.6%) patients were found to have CNVs (see Table 2), including 2 likely benign groups of 13 (2.0%) with CNVs that were also reported to be present in the normal population and 22 (3.4%) with familial variants present in phenotypically normal parents; 27 (4.2%) were classified as being of uncertain significance and awaiting parental studies (data not shown). The detection rates of the “likely benign” group decreased from 9.1% (18 of 197) and 11.4% (20 of 175) to 9.0% (24 of 266) on the 3 sequential versions of arrays (see Table 2), through systematic removal of interrogating clones or oligonucleotides from genomic regions that consistently detected seemingly benign variants. This restricts or reduces the need to perform parental studies on variants that ultimately prove to be benign. These results also reflect our learning curve and experience in designing high-resolution targeted clinical diagnostic arrays through better BAC and oligonucleotide probe selection.
Clinically Significant Abnormality-Detection Rate and Clinical Indications
Of a total of 638 neonates, CMA revealed 109 (17.1%) patients with clinically significant chromosomal aberrations (Fig 1, Table 1, and Supplemental Table 3, which is published as supporting information on www.pediatrics.org/content/full/122/6/1310); all of these abnormalities were verified by a secondary independent laboratory test: FISH and/or high-resolution retrospective partial karyotype analyses. The clinical information available for each individual neonatal patient is limited. As a general measure of clinical presentation, we used the clinical indication as determined by the clinician referring to our diagnostic laboratory.
FIGURE 1.
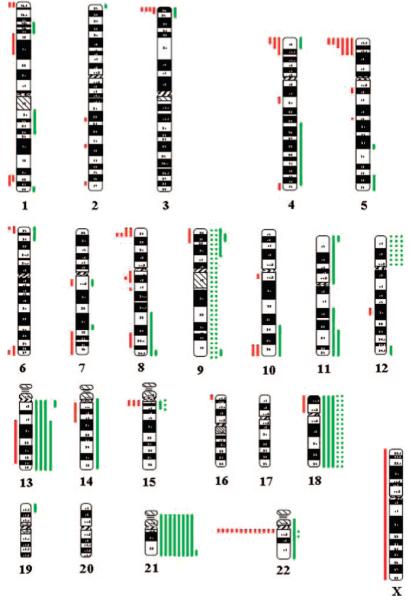
Ideogram summarizing the clinically significant CNVs in 109 neonates with birth defects. Red and green indicate loss and gain in copy numbers, respectively. The lengths of the vertical bars indicate the size of the CNVs corresponding to the cytogenetic bands; the vertical dotted lines represent mosaicism for chromosomal aneuploidy. The Y chromosome is not shown in this ideogram.
In the group of 21 patients who were suspected clinically to have chromosomal abnormalities ± others (eg, DFs, cleft palate, MCA), the CMA clinically significant detection rate was the highest at 66.7% (n = 14; see Table 1). CMA identified trisomies 21 (6), 18 (2), and 13 (1); all of these were concordant with the clinical indication for the concurrent karyotype analysis. In addition, a derivative chromosome 6 was identified in a patient who was suspected to have trisomy 18; 2 patients were found to have a deletion in Prader-Willi/Angelman syndrome (PWS/AS) critical region, 1 with a deletion in chromosome 8q and 1 with 45,X, which was consistent with a diagnosis of Turner Syndrome.
In 12 patients with ambiguous genitalia ± other indications (see Table 1), CMA detected abnormalities in 4 (33.3%) patients, including 2 with a mosaic 45,X karyotype with the presence of the Y material, 1 with a complex chromosome 5 rearrangement, and 1 with an interstitial deletion in the subtelomeric region of the long arm of chromosome 10.
A total of 179 patients were referred for MCA ± others (eg, DFs, CHD, club feet, broad thumbs), and CMA identified significant chromosomal aberrations in 32 cases. All of the CMA findings were summarized in 4 subgroups (see Table 1). For 7 patients who were referred for MCA and CHD, CMA revealed that 2 (28.6%) patients had clinically significant CNVs involving subtelomeric imbalances in chromosome 1q (1) and 3p (1). A total of 59 patients were referred for MCA + DFs ± others (eg, polydactyly, failure to thrive; see Table 2), CMA detected 16 (27.1%) with clinically significant genomic imbalance including 2 mosaic tetrasomy 12p, 1 trisomy 21, 1 trisomy 22, 1 deletion in PWS/AS critical region, a complex chromosome 8 rearrangement, and other genomic imbalances involving chromosome 1 (4), chromosome 4 (3), chromosome 11 (2), and 10qter (1).The identified nonmosaic trisomy 22 in 1 patient was later verified by metaphase FISH analysis. This infant was admitted to the NICU secondary to DFs and MCA, where she died at the age of 35 days. She received a diagnosis of CHD with major abnormalities involving double-outlet right ventricle and pulmonary vein stenosis. No autopsy was performed, thereby limiting the clinical information available. The identified aneuploidy and early death are clearly compatible with a severe phenotype often seen in this rarely reported live-born trisomy. In 113 patients who were referred for MCA alone or MCA + others (eg, hydrocephalus, limb anomalies, but excluding CHD or DFs), CMA detected significant CNVs in 14 patients (see Table 1). Most of these aberrations involved subtelomeric (6) and pericentromeric (3) imbalances; 3 had microdeletions in DG/VCFS critical region, and 2 had mosaicism for inv dup (15) (1) and inv dup (22) (1).
CMA detected 34 (24.6%) patients with clinically significant abnormalities in 138 patients who were referred for DFs alone or DFs + others (eg, cleft palate, hypotonia, but excluding MCA; see Table 1). Of these, 14 were found to have subtelomeric imbalances, 4 were revealed to have microdeletions involving DG/VCFS (2), Williams-Beuren syndrome (1), and PWS/AS (1) critical regions; 11 were observed to have aneuploidies including trisomy 21 (4), trisomy 13 (2), trisomy 18 (1), 45,X (1), mosaic trisomy 9 (2) and mosaic trisomy 18 (1); the remaining 5 had genomic imbalances that involved other rare disease regions. It is interesting that in the mosaic trisomy 18, the CMA ratio plot was suggestive of copy-number differences between the p and q arms of chromosome 18; this result was concordant with the concurrent karyotype analysis of 2 cell lines that consisted of 47,XX,+18[27]/47,XX,+i(18)(p10)[3] (Fig 2).
FIGURE 2.
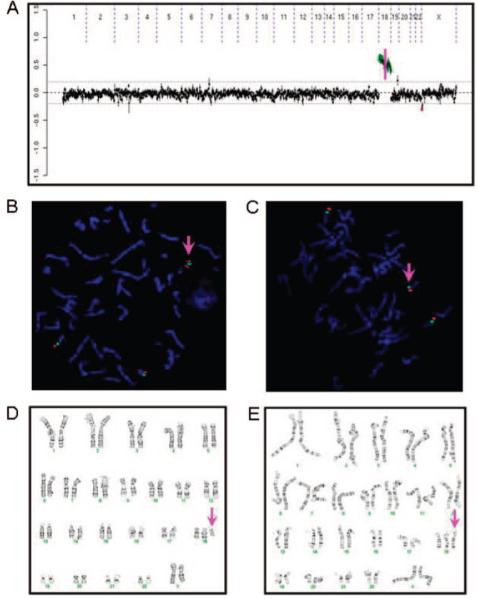
CMA ratio plot, FISH, and karyotype analyses for mosaic trisomy 18 in a female neonate with DFs and failure to thrive. A, Different copy-number gains for chromosome 18p and 18q (separated by a pink vertical line) detected by CMA. B and C, FISH using chromosome 18 centromere probe (green) and a probe for 18p (red) showing i(18p) (B) and trisomy 18 (C). D and E, Karyotype analysis showing i(18p) (D) and trisomy 18 (E).
Among 101 patients who were referred for CHD ± others (see Table 1), CMA identified 22 (21.8%) patients with significant chromosomal aberrations. Eight patients with a common 3-Mb deletion in 22q11.2 had clinical features compatible with a diagnosis of DG/VCFS; 1 patient had an interstitial deletion at 1p36, and the remaining patients had subtelomeric imbalances in chromosomes 3p, 6p, 8p, and 11q.
In 295 patients who were referred for other birth defects (see Table 1), the average clinically significant detection rate was 9.5% (28 of 295). In a small group of 9 patients who were referred for respiratory distress syndrome, CMA detected 2 (22.2%) of them with genomic imbalance, including 1 mosaic tetrasomy 12p, that can be associated with the Pallister-Killian syndrome, and another 1 involving an unbalanced translocation between chromosomes 14 and 18. In 24 patients who were suspected for a possible syndrome, CMA clinically significant detection rate was 20.8%.
In summary, of the 109 (17.1% of the 638 neonates referred for testing) patients in which clinically significant genomic imbalances or pathogenic CNVs were detected by CMA (see Fig 1), 16 (14.7%) had numerical anomalies including trisomy 21 (8), trisomy 18 (3), trisomy 13 (3), trisomy 22 (1), and monosomy X (1). The remaining 93 (85.3%) patients had genomic imbalances that may not be detected by standard cytogenetic studies, including 37 (33.9%) with common microdeletions or microduplications involving 22q11.2 (13), 5p15.2 (6), 3p26.3 (4), 4p16.3 (4), and other chromosomal regions (10); 44 (40.4%) with genomic imbalances at relatively rare disease loci; and 12 (11.0%) with chromosomal mosaicism.
DISCUSSION
Constitutional chromosomal aberrations often result in birth defects and are frequently reported as the underlying cause of DFs, MCA, CHD, and developmental delay or mental retardation by aCGH.42-45 The aim of this study was to investigate the frequencies of submicroscopic chromosomal aberrations including pathogenic CNVs in neonates with birth defects by using targeted aCGH or CMA that enables higher resolution genomic analysis than standard clinical GTG-banding chromosome analysis. In 638 neonates with various birth defects, clinically significant genomic imbalances or pathogenic CNVs were detected in 17.1% of the patients, with an observation of the highest clinically significant detection rate of 19.9% in the 266 neonates analyzed on the most recent CMA V6 Oligo version with expanded human genome coverage. In patients with abnormal CMA results, not surprising, the majority of the abnormalities could not be defined by GTG-banded karyotype (see Supplemental Table 3). In fact, the 85.3% (93 of 109) of genomic imbalances detected by CMA were not simply aneuploidies and might have been missed by conventional clinical cytogenetic analysis. Although many could have been diagnosed by locus-specific FISH tests or subtelomeric FISH, ordering locus-specific FISH requires a presumptive clinical diagnosis. CMA provides a more efficient and reliable method for detecting all of these cases with a single test that enables a “genomic” screen.
More than half of the patients (see Table 1) were referred for MCA ± others, DFs ± others, or CHD ± others. CMA detected the most abnormalities (28.6% and 27.1%) in neonates with MCA + CHD and MCA + DFs, respectively. CMA detection rates for DFs ± others and CHD ± others referrals were 24.6% and 21.8%, respectively. These data suggest that the neonates with these phenotypes are mostly likely to have higher frequencies of genomic imbalances or pathogenic CNVs. In the neonates who were referred with MCA alone, the detection rate was relatively lower at 14.4% (see Table 1).
CMA detected the highest abnormal rate for neonates who were referred with a clinical indication of suspected chromosomal abnormalities. Previous cytogenetic studies showed that the average chromosomal aberration incidence was 21.6% in a total of 6183 pediatric patients who were suspected to have chromosomal abnormalities.46-49 Remarkably, by using CMA, ~66.7% (see Table 1) of the neonates who were referred for possible chromosomal aberrations, a threefold increase from previous cytogenetic studies, were found to have genomic imbalances. The significantly improved detection of chromosome aberrations substantiates the power of CMA as a high-resolution molecular cytogenetic diagnostic tool.
Many submicroscopic genomic rearrangements have been associated with well-defined clinical syndromes, such as contiguous gene microdeletion/microduplication disorders.50-52 CMA tests hundreds or thousands of genomic loci for CNVs (gains or losses) simultaneously and thus greatly improves the detection rate for identifying microdeletion and microduplication syndromes.27,53 At least 37 (5.8%) patients were identified as having microdeletion or microduplication syndromes that involved the common chromosomal regions in 22q11.2, 5p15.4, and 4p16.3. Among these, the common 3-Mb deletion in DG/VCFS critical region at 22q11.2 (Online Mendelian Inheritance in Man [OMIM] No. 188400 and 192430) was the most prevalent, found in 13 (2.0%) of the referred neonates. In 1 neonate with an apparently balanced inversion of chromosome 2 (not included in this series), CMA revealed a reciprocal microduplication in DG/VCFS critical region at 22q11.2 that was consistent with the clinical diagnosis of 22q11.2 microduplication syndrome (OMIM No. 608363). Both syndromes present with CHD and have other overlapping clinical features.54 Often, a significant number of duplication patients were first referred for a FISH test for suspected DG/VCFS. Many patients with FISH-negative results were later found to have the 22q11.2 microduplication syndromes by CMA27 or interphase FISH analysis.55 Recent work that applied clinical arrays documented a new genomic disorder that is associated with deletion just distal to the common DG/VCFS locus.56 Common subtelomeric rearrangements were identified in 3 chromosomal regions in the distal 3p, 4p, and 5p. Monosomy 4p and 5p have been well defined to be associated with Wolf-Hirschhorn syndrome (OMIM No. 194190) and Cri-du-chat syndrome (OMIM No. 123450); however, the clinical manifestation of microduplications in the distal 4p as well as the deletions in the distal region of 3p57,58 are not very well defined, and they could represent new genomic syndromes.
CMA in this study also demonstrated an increased detection rate for identifying chromosomal mosaicism. CMA for 12 patients, mostly referred for DFs and MCA, were suggestive for mosaicism. Five (38.5%) patients had low-level mosaicism with aberrations that were not detected in retrospective chromosomal metaphase analyses, including tetrasomy 12p (3) and trisomy 9 (2). Tetrasomy 12p has been associated with Pallister-Killian syndrome, and skin biopsy for conventional cytogenetic studies is generally recommended when clinically indicated; however, none of these 3 neonates whom we reported with mosaic tetrasomy 12p in this study was suspected for this diagnosis, including 1 patient who was reported to have a 45,X karyotype before CMA and was known to manifest clinically DFs and MCA findings that were not consistent with a Turner syndrome phenotype. Interphase FISH analysis on blood smears was needed to verify mosaicism in these cases. As elucidated in previous studies,36,37 CMA on DNA from multiple cell lineages in whole blood improves the ability to detect mosaicism compared with conventional karyotype analysis, which examines only the stimulated T lymphocytes that respond to mitogens. Chromosomal low-level mosaicism has been shown in this study to exist in neonates with unexplained birth detects such as MCA, DFs, and CHD and seemingly normal chromosomes by karyotype analysis, and, therefore aCGH/CMA is highly recommended for these neonates for additional evaluation of the possible chromosomal mosaicism.
To our knowledge, this is the largest study to date to use aCGH to detect the frequencies of genomic imbalances in a large series of neonates with birth defects and has important implications for the evaluation of congenital anomalies with unknown genetic cause. Limitations of this study include the relatively modest sample size (638) and that neonates with birth defects in this study were not characterized on the basis of the coding of birth defects because of limited clinical information.12,59 We propose that additional investigations use CMA in large neonatal sample cohorts, such as the DNA sample collection of the National Birth Defects Prevention Study (NBDPS),60 to identify the potential genomic imbalances and disease gene(s) that underlie specific birth defects.
What recommendations might be made for the use of cytogenetic studies in the neonatal setting? First, infants who are suspected for Down syndrome should have a karyotype and not CMA because chromosome analysis would be more informative than CMA when the additional chromosome 21 is part of a Robertsonian translocation [eg, rob(14,21)(q10;q10), rob(21,21)(q10;q10)]. Experienced clinicians should recognize the great majority of such infants. In 8 patients with trisomy 21 in this study, only 2 of them were referred for DFs or DFs + MCA instead of “possible trisomy 21”; in the majority, a diagnosis of Down syndrome was made by clinical examination. Second, infants with possible or suspected trisomy 13 or trisomy 18 could also be analyzed initially by karyotype or STAT FISH if findings are classical, but concurrent CMA is desirable in most instances. Finally, for most other circumstances, we believe that CMA is already or soon will be the preferred first-line test. It is more rapid (3–5 days) than karyotype (7–14 days) and has a far higher detection rate for clinically significant abnormalities, and this would be beneficial in timely clinical management. Very few clinically significant abnormalities are detected by karyotype but not by targeted aCGH or CMA. A balanced rearrangement that disrupts a dosage-sensitive gene is one such circumstance. Recent studies that used high-resolution aCGH analysis revealed that many seemingly balanced translocations were in fact not balanced.61,62
CONCLUSIONS
This study demonstrates that CMA can effectively diagnose birth defects that are caused by submicroscopic genomic imbalances in the neonatal period and, therefore, assist the clinician in diagnosis, early neonatal intervention, and genetic counseling. Importantly, the CMA was ~3 times as likely to detect an abnormality than routine clinical chromosome analysis in a neonate who was referred with a clinical suspicion of a chromosomal syndrome. Better understanding of the causes of birth defects can potentially enable effective prenatal detection and potential avenues for therapy on the basis of mitigating the clinical consequences of the dosage-sensitive gene(s) whose CNV is responsible for disease.63 We recommend the consideration of using CMA in lieu of routine cytogenetic testing to evaluate neonates with birth defects of unknown cause.
What’s Known on This Subject.
Birth defects are the leading cause of neonatal morbidity and mortality. Genomic imbalances are believed to be the cause in a significant number of birth defects; however, submicroscopic genomic imbalances are not identifiable using the conventional clinical cytogenetic analysis.
What This Study Adds.
Molecular cytogenetic studies using chromosomal microarray analysis (CMA) in neonates with birth defects detects many more genomic imbalances than conventional cytogenetics. Furthermore, the improved resolution provided by CMA enables more informed decision making and medical management in a timely manner.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported in part by Baylor College of Medicine Mental Retardation Developmental Disabilities Research Center grant HD024064-20.
We thank all of the referring physicians, patients, and families for cooperation. We also thank all of our colleagues and co-workers from the CMA laboratory, Array Production Laboratory, Cytogenetic/FISH Laboratory, and DNA Diagnostic Laboratory at Baylor College of Medicine for contributions and support.
Abbreviations
- GTG banding
trypsin-Giemsa banding
- FISH
fluorescence in situ hybridization
- DG/VCFS
DiGeorge/velocardiofacial syndrome
- aCGH
array comparative genomic hybridization
- CNV
copy-number variation
- BAC
bacterial artificial chromosome
- CMA
chromosomal microarray analysis
- DF
dysmorphic feature
- MCA
multiple congenital anomalies
- CHD
congenital heart disease
- PWS/AS
Prader-Willi/Angelman syndrome
- OMIM
Online Mendelian Inheritance in Man
Footnotes
The authors have indicated they have no financial relationships relevant to this article to disclose.
REFERENCES
- 1.Mathews TJ, MacDorman MF. Infant mortality statistics from the 2003 period linked birth/infant death data set. Natl Vital Stat Rep. 2006;54(16):1–29. [PubMed] [Google Scholar]
- 2.Canfield MA, Honein MA, Yuskiv N, et al. National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999–2001. Birth Defects Res A Clin Mol Teratol. 2006;76(11):747–756. doi: 10.1002/bdra.20294. [DOI] [PubMed] [Google Scholar]
- 3.Yoon PW, Rasmussen SA, Lynberg MC, et al. The National Birth Defects Prevention Study. Public Health Rep. 2001;116(suppl 1):32–40. doi: 10.1093/phr/116.S1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamerton JL, Canning N, Ray M, Smith S. A cytogenetic survey of 14,069 newborn infants: I—incidence of chromosome abnormalities. Clin Genet. 1975;8(4):223–243. doi: 10.1111/j.1399-0004.1975.tb01498.x. [DOI] [PubMed] [Google Scholar]
- 5.Evans JA, de von FR, Greenberg C, Ramsay S, Hamerton JL. A cytogenetic survey of 14,069 newborn infants: IV—further follow-up on the children with sex chromosome anomalies. Birth Defects Orig Artic Ser. 1982;18(4):169–184. [PubMed] [Google Scholar]
- 6.Seashore MR. Chromosomal abnormalities in the newborn period. Semin Perinatol. 1993;17(5):312–317. [PubMed] [Google Scholar]
- 7.Smith A, Bannatyne P, Russell P, Ellwood D, den Dulk G. Cytogenetic studies in perinatal death. Aust N Z J Obstet Gynaecol. 1990;30(3):206–210. doi: 10.1111/j.1479-828x.1990.tb03214.x. [DOI] [PubMed] [Google Scholar]
- 8.Larson RS, Butler MG. Use of fluorescence in situ hybridization (FISH) in the diagnosis of DiGeorge sequence and related diseases. Diagn Mol Pathol. 1995;4(4):274–278. doi: 10.1097/00019606-199512000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandelman KY, Gibson L, Meyn MS, Yang-Feng TL. Molecular definition of the smallest region of deletion overlap in the Wolf-Hirschhorn syndrome. Am J Hum Genet. 1992;51(3):571–578. [PMC free article] [PubMed] [Google Scholar]
- 10.Gersh M, Grady D, Rojas K, et al. Development of diagnostic tools for the analysis of 5p deletions using interphase FISH. Cytogenet Cell Genet. 1997;77(3–4):246–251. doi: 10.1159/000134586. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Joseph KS, Wen SW. Trends in fetal and infant deaths caused by congenital anomalies. Semin Perinatol. 2002;26(4):268–276. doi: 10.1053/sper.2002.34776. [DOI] [PubMed] [Google Scholar]
- 12.Rasmussen SA, Moore CA. Effective coding in birth defects surveillance. Teratology. 2001;64(suppl 1):S3–S7. doi: 10.1002/tera.1077. [DOI] [PubMed] [Google Scholar]
- 13.Ming JE, Geiger E, James AC, et al. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum Mutat. 2006;27(5):467–473. doi: 10.1002/humu.20322. [DOI] [PubMed] [Google Scholar]
- 14.Pinkel D, Segraves R, Sudar D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 15.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 16.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 17.White SJ, Vissers LE, Geurts van KA, et al. Variation of CNV distribution in five different ethnic populations. Cytogenet Genome Res. 2007;118(1):19–30. doi: 10.1159/000106437. [DOI] [PubMed] [Google Scholar]
- 18.Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39(7 suppl):S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- 19.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai WW, Mao JH, Chow CW, et al. Genome-wide detection of chromosomal imbalances in tumors using BAC microarrays. Nat Biotechnol. 2002;20(4):393–396. doi: 10.1038/nbt0402-393. [DOI] [PubMed] [Google Scholar]
- 21.Rouillard JM, Herbert CJ, Zuker M. OligoArray: genome-scale oligonucleotide design for microarrays. Bioinformatics. 2002;18(3):486–487. doi: 10.1093/bioinformatics/18.3.486. [DOI] [PubMed] [Google Scholar]
- 22.Ou Z, Kang SH, Shaw CA, et al. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med. 2008;10(4):278–289. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman JM, Baross A, Delaney AD, et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet. 2006;79(3):500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aradhya S, Cherry AM. Array-based comparative genomic hybridization: clinical contexts for targeted and whole-genome designs. Genet Med. 2007;9(9):553–559. doi: 10.1097/gim.0b013e318149e354. [DOI] [PubMed] [Google Scholar]
- 25.Cheung SW, Shaw CA, Yu W, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7(6):422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 26.Shaffer LG, Bejjani BA. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet Genome Res. 2006;115(3–4):303–309. doi: 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- 27.Lu X, Shaw CA, Patel A, et al. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS ONE. 2007;2(3):e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38(9):1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 29.Menten B, Maas N, Thienpont B, et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet. 2006;43(8):625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahoo T, Cheung SW, Ward P, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med. 2006;8(11):719–727. doi: 10.1097/01.gim.0000245576.47154.63. [DOI] [PubMed] [Google Scholar]
- 31.Van den Veyver I, Beaudet AL. Comparative genomic hybridization and prenatal diagnosis. Curr Opin Obstet Gynecol. 2006;18(2):185–191. doi: 10.1097/01.gco.0000192986.22718.cc. [DOI] [PubMed] [Google Scholar]
- 32.Rickman L, Fiegler H, Shaw-Smith C, et al. Prenatal detectionof unbalanced chromosomal rearrangements by array CGH. J Med Genet. 2006;43(4):353–361. doi: 10.1136/jmg.2005.037648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vissers LE, Veltman JA, van Kessel AG, Brunner HG. Identification of disease genes by whole genome CGH arrays. Hum Mol Genet. 2005;14(Spec No 2):R215–R223. doi: 10.1093/hmg/ddi268. [DOI] [PubMed] [Google Scholar]
- 34.Lupski JR. Genome structural variation and sporadic disease traits. Nat Genet. 2006;38(9):974–976. doi: 10.1038/ng0906-974. [DOI] [PubMed] [Google Scholar]
- 35.Ballif BC, Hornor SA, Jenkins E, et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2–p12.2. Nat Genet. 2007;39(9):1071–1073. doi: 10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- 36.Ballif BC, Rorem EA, Sundin K, et al. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am J Med Genet A. 2006;140(24):2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- 37.Cheung SW, Shaw CA, Scott DA, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143A(15):1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- 38.Bar-Shira A, Rosner G, Rosner S, Goldstein M, Orr-Urtreger A. Array-based comparative genome hybridization in clinical genetics. Pediatr Res. 2006;60(3):353–358. doi: 10.1203/01.pdr.0000233012.00447.68. [DOI] [PubMed] [Google Scholar]
- 39.Caselli R, Mencarelli MA, Papa FT, et al. A 2.6 Mb deletion of 6q24.3–25.1 in a patient with growth failure, cardiac septal defect, thin upperlip and asymmetric dysmorphic ears. Eur J Med Genet. 2007;50(4):315–321. doi: 10.1016/j.ejmg.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17(3):182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 41.Shaw-Smith C, Redon R, Rickman L, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41(4):241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoumans J, Ruivenkamp C, Holmberg E, et al. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH) J Med Genet. 2005;42(9):699–705. doi: 10.1136/jmg.2004.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aradhya S, Manning MA, Splendore A, Cherry AM. Whole-genome array-CGH identifies novel contiguous gene deletions and duplications associated with developmental delay, mental retardation, and dysmorphic features. Am J Med Genet A. 2007;143A(13):1431–1441. doi: 10.1002/ajmg.a.31773. [DOI] [PubMed] [Google Scholar]
- 44.de Vries BB, Pfundt R, Leisink M, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77(4):606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krepischi-Santos AC, Vianna-Morgante AM, Jehee FS, et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisited and new alterations. Cytogenet Genome Res. 2006;115(3–4):254–261. doi: 10.1159/000095922. [DOI] [PubMed] [Google Scholar]
- 46.Shah VC, Murthy DS, Murthy SK. Cytogenetic studies in a population suspected to have chromosomal abnormalities. Indian J Pediatr. 1990;57(2):235–243. doi: 10.1007/BF02722094. [DOI] [PubMed] [Google Scholar]
- 47.Kenue RK, Raj AK, Harris PF, el-Bualy MS. Cytogenetic analysis of children suspected of chromosomal abnormalities. J Trop Pediatr. 1995;41(2):77–80. doi: 10.1093/tropej/41.2.77. [DOI] [PubMed] [Google Scholar]
- 48.Kim SS, Jung SC, Kim HJ, Moon HR, Lee JS. Chromosome abnormalities in a referred population for suspected chromosomal aberrations: a report of 4117 cases. J Korean Med Sci. 1999;14(4):373–376. doi: 10.3346/jkms.1999.14.4.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goud MT, Al-Harassi SM, Al-Khalili SA, et al. Incidence of chromosome abnormalities in the Sultanate of Oman. Saudi Med J. 2005;26(12):1951–1957. [PubMed] [Google Scholar]
- 50.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14(10):417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 51.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18(2):74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 52.Lupski JR, Stankiewicz P, editors. Genomic Disorders: The Genomic Basis of Disease. Humana Press; Totowa, NJ: 2006. pp. 1–427. [DOI] [PubMed] [Google Scholar]
- 53.Berg JS, Brunetti-Pierri N, Peters SU, et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9(7):427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- 54.Ou Z, Berg JS, Yonath H, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med. 2008;10(4):267–277. doi: 10.1097/GIM.0b013e31816b64c2. [DOI] [PubMed] [Google Scholar]
- 55.Ensenauer RE, Adeyinka A, Flynn HC, et al. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am J Hum Genet. 2003;73(5):1027–1040. doi: 10.1086/378818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-Shachar S, Ou Z, Shaw CA, et al. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet. 2008;82(1):214–221. doi: 10.1016/j.ajhg.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dijkhuizen T, van ET, van der Vlies P, et al. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am J Med Genet A. 2006;140(22):2482–2487. doi: 10.1002/ajmg.a.31487. [DOI] [PubMed] [Google Scholar]
- 58.McCullough BJ, Adams JC, Shilling DJ, et al. 3p-syndrome defines a hearing loss locus in 3p25.3. Hear Res. 2007;224(1–2):51–60. doi: 10.1016/j.heares.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rasmussen SA, Olney RS, Holmes LB, et al. Guidelines for case classification for the National Birth Defects Prevention Study. Birth Defects Res A Clin Mol Teratol. 2003;67(3):193–201. doi: 10.1002/bdra.10012. [DOI] [PubMed] [Google Scholar]
- 60.Rasmussen SA, Lammer EJ, Shaw GM, et al. Integration of DNA sample collection into a multi-site birth defects case-control study. Teratology. 2002;66(4):177–184. doi: 10.1002/tera.10086. [DOI] [PubMed] [Google Scholar]
- 61.De Gregori M, Ciccone R, Magini P, et al. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: a study of 59 patients. J Med Genet. 2007;44(12):750–762. doi: 10.1136/jmg.2007.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higgins AW, Alkuraya FS, Bosco AF, et al. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am J Hum Genet. 2008;82(3):712–722. doi: 10.1016/j.ajhg.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lupski JR. Structural variation in the human genome. N Engl J Med. 2007;356(11):1169–1171. doi: 10.1056/NEJMcibr067658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.