

Abstract
Solid-contact ion-selective electrodes (SC-ISEs) can exhibit very low detection limits and, in contrast to conventional ISEs, do not require an optimization of the inner filling solution. This work shows that subnanomolar detection limits can also be achieved with SC-ISEs with three-dimensionally ordered macroporous (3DOM) carbon contacts, which have been shown recently to exhibit excellent long-term stabilities and good resistance to the interferences from oxygen and light. The detection limit of 3DOM carbon-contacted electrodes with plasticized poly(vinyl chloride) as membrane matrix can be improved with a high polymer content of the sensing membrane, a large ratio of ionophore and ionic sites, and conditioning with a low concentration of analyte ions. This permits detection limits as low as 1.6×10−7 M for K+ and 4.0×10−11 M for Ag+.
Keywords: Solid-contact ion-selective electrode, Three-dimensionally ordered macroporous (3DOM) carbon, Detection limit, Subnanomolar, Silver, Potassium
Introduction
Over the last four decades, ionophore-based ion-selective electrodes (ISEs) have been extensively studied [1–5]. However, while ISEs have been designed for more than 60 analytes and are used for billions of measurements each year, their practical use has been limited to a selected number of application fields, most importantly in clinical chemistry. One of the limitations that has hindered their wider use has been the frequent observation of insufficient detection limits. Until a decade ago, most potentiometric sensors could only detect sample concentrations down to the micromolar level, disqualifying them for trace-level measurements. Fortunately, the recently obtained understanding of ion fluxes through ISE membranes allowed the lowering of detection limits drastically [6, 7]. The successful minimization of such fluxes was first achieved by an appropriate choice of the internal solution of conventional ISEs, improving detection limits to the nanomolar and even picomolar level [8–15]. However, the optimization of the inner filling solution of a conventional ISE depends on the membrane selectivity, diffusion coefficients and—most importantly—the anticipated sample, which can make the procedure somewhat cumbersome to perform under real-life conditions [16–18].
As an alternative approach to ISEs with low detection limits, solid contact ISEs (SC-ISEs) have attracted a lot of attention. Initially, SC-ISEs were prepared by direct contact of the ionophore-doped polymeric membrane with a metallic conductor, resulting in membrane/metal interfaces with poorly defined and unstable phase boundary potentials [19]. This problem has been addressed by the use of intermediate layers with localized redox-active groups separating the sensing membrane and the metallic conductor [20, 21]. Also, many conducting polymers such as polypyrrole [22–26], polyaniline [27, 28], polythiophene [29, 30] and derivatives thereof have been used for the intermediate layer. Indeed, this has become the most popular approach to prepare SC-ISEs.
Our group developed SC-ISEs based on three-dimensionally ordered macroporous (3DOM) carbon as the intermediate layer (see Figure 1) [31]. 3DOM carbon consists of a skeleton of glassy carbon surrounding a periodic array of uniform spherical pores that are interconnected in three dimensions [32, 33]. Typical pore sizes in these materials are on the order of a few hundred nanometers, and skeletal walls are tens of nanometers thick. Due to the well-interconnected pore and wall structure of 3DOM carbon, filling of the 3DOM pores with an electrolyte solution results in a nanostructured material that exhibits good ionic and electronic conductivity. Very recently, a similar system that relies on the high capacitance of a high surface area carbon nanotube interface has been described [34].
Figure 1.
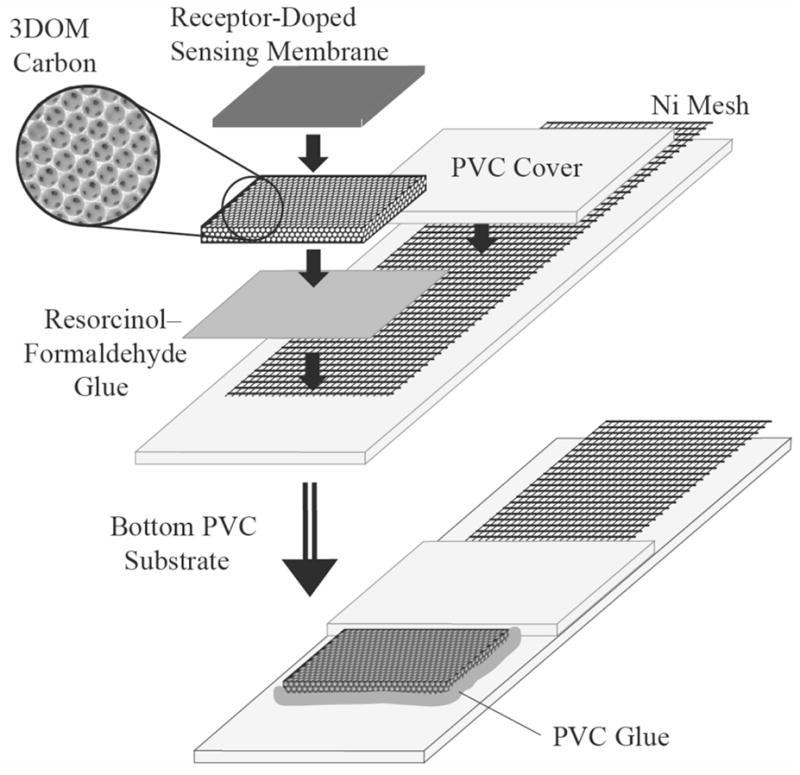
Schematic setup of a 3DOM carbon-contacted ion-selective electrode.
First efforts to use SC-ISEs for measurements with low detection limits were reported by Michalska and co-workers [35]. They incorporated the complexing agent ethylenediaminetetraacetate (EDTA) in electropolymerized poly(3-methylthiophene) intermediate layers. More recently, they presented an impressive nanomolar detection limit for calcium ions by using a polypyrrole solid contact doped with the Ca2+ ligand Tiron (4,5-dihydroxy-m-benzenedisulfonate) [36]. As could be expected, it has been recently demonstrated that the detection limit of SC-ISEs is much worse when a water-layer is present because ion fluxes between the sample and the water layer arise [37, 38]. Indeed, SC-ISEs with electropolymerized polypyrrole that do not show any evidence of a water layer reached a nanomolar detection limit for Pb2+ [26]. Even better detection limits as low as 5.0×10−10 M were achieved with sensing membranes based on the plasticizer-free methyl methacrylate-decyl methacrylate copolymer and poly(3-octylthiophene) intermediate layers that were not doped with the potentially leaching hexacyanoferrate [39]. The improvement in detection limit was probably not only due to the absence of hexacyanoferrate but at least partly due to the use of copolymer, in which diffusion is much slower than in PVC membranes modified with a high concentration of plasticizer [40]. A nanomolar detection limit for Ag+ was reported using this approach [41]. Importantly, whatever type of intermediate layer is being used for SC-ISEs, the conditioning procedure prior to measurements has a crucial effect on the observed detection [41, 42].
The objective of the present work was to determine whether 3DOM carbon-contacted ISEs offer the same advantages of low detection limits as the SC-ISEs with conducting polymers as intermediate electron- and ion-conducting layer. With this goal in view, the effect of membrane composition and the conditioning procedure on the detection limits of these electrodes for K+ and Ag+ were determined.
Experimental
Reagents
High molecular weight poly(vinyl chloride) (PVC), 2-nitrophenyl octyl ether (NPOE) and o-xylylenebis(N,N-diisobutyldithiocarbamate)—despite its Ag+ selectivity sometimes referred to as Copper(II) Ionophore (I)—were purchased from Fluka (Buchs, Switzerland), valinomycin from Sigma (St. Louis, MO, USA), and sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaTFPB) from Dojindo (Kumamoto, Japan). Deionized and charcoal-treated water (18.2 MΩ·cm specific resistance) obtained with a Milli-Q PLUS reagent-grade water system (Millipore, Bedford, MA, USA) was used for all sample solutions. All chemicals were used as received.
Membranes
Valinomycin-doped K+-ISE membranes were prepared according to a standard procedure by pouring a tetrahydrofuran (THF) solution of 200 mg of the membrane components into a glass dish (31 mm i.d.) and letting the THF evaporate slowly at room temperature over 24 h. The same type of membrane containing 32.8% PVC as polymer matrix, 65.6% NPOE as plasticizer, 0.6% NaTFPB to provide for ionic sites, and 1% valinomycin as ionophore was used for all K+ measurements. Membranes doped with the ionophore o-xylylenebis(N,N-diisobutyldithiocarbamate) were prepared in the same way, but the amount of PVC and the ratio of ionophore and ionic sites were varied. The thickness of the resulting membranes was approximately 100 μm.
Electrodes
3DOM carbon monoliths were prepared as reported by colloidal crystal templating with monodisperse PMMA spheres [33, 43–45]. SC-ISEs (Figure 1) were assembled in the same way as reported in earlier work [31], i.e., by positioning a small piece of 3DOM carbon between a Ni mesh and a PVC membrane. The 3DOM carbon was glued onto the Ni mesh with a resorcinol–formaldehyde (RF) adhesive (60:120:1 molar ratio of resorcinol, formaldehyde, and sodium carbonate; hardened at 85° C), and a section of the Ni mesh was embedded between two PVC sheets (Goodfellow, Oakdale, PA, USA), which served as the substrate for the whole setup. The RF glue is not conductive. Finally, the 3DOM carbon was covered with the ionophore-doped, plasticized PVC membrane. A solution of PVC was used to tightly glue the two PVC sheets together and to cover all still exposed Ni mesh and 3DOM carbon, ensuring that there was no direct contact between the ionophore-doped PVC membrane and the Ni mesh, and between the 3DOM carbon and the sample solution. A fairly concentrated THF solution of the same PVC used in the preparation of the membranes could be used for this purpose, but for convenience commercially available PVC cement (a solution of PVC in a mixture of THF, methyl ethyl acetone and cyclohexane; ACE Hardware Corp., Oak Brook, IL, USA) was used in this study.
EMF Measurements
Potentials were monitored at room temperature in stirred solutions with an EMF 16 potentiometer (Lawson Labs, Malvern, PA, USA) controlled with EMF Suite 1.02 software (Fluorous Innovations, Arden Hills, MN, USA). The external reference electrode consisted of a double-junction Ag/AgCl electrode with a 1 M LiOAc bridge electrolyte and AgCl-saturated 3 M KCl as reference electrolyte. All EMF values were corrected for liquid-junction potentials according to the Henderson equation [46]. Activity coefficients were calculated with a two-parameter Debye–Hückel approximation [47]. All K+ and Ag+ measurements were performed with polypropylene beakers to minimize ion leaching from the sample container into the sample. In the case of the Ag+ measurements, the beakers were cleaned overnight in 0.1 M HNO3 before use.
Results and Discussion
As reported earlier, K+ selective 3DOM carbon-contacted ISEs exhibited excellent long-term stability with potential drifts of only 11.7 μV/h and a very good resistance to the interference from oxygen and light [31]. Moreover, the hydrophobic surface of 3DOM carbon suppressed the formation of an aqueous layer. While a detection limit of 6.3×10−7 M for K+ was reported, it was suspected that the detection limit could be improved by more carefully preventing direct exposure of the electrode membrane to solutions of high K+ concentration [31]. This is indeed the case, as shown in the following.
When freshly prepared electrodes were first conditioned in 1 μM KCl solution for 1 day and then transferred to 1 mM KCl solutions for the measurement of calibration curves by successive sample dilution, a detection limit for K+ of 1.0×10−6 M was observed. Similarly, conditioning in 1 nM KCl solution for 2 days and calibration curve measurements by dilution of 1 mM KCl gave the nearly identical detection limit of 1.2×10−6 M. This suggests that any effect from the low concentration of the conditioning solution is overpowered by the 1 mM KCl concentration that the electrode is first exposed to when the calibration curve is measured by successive dilution. Therefore, calibration curves were determined with the successive sample dilution method, starting from different KCl concentrations (see Figure 2 and Table 1). Clearly, lower detection limits were obtained for lower concentrations of the conditioning and sample solutions. The best detection limit, 1.6×10−7 M, was obtained with a 1 μM conditioning solution and a calibration curve obtained by successive dilution of a 1 μM K+ starting solution. The use of high concentrations of the analyte ion in the conditioning solution is apparently followed by a release of analyte ions from the membrane phase back into the sample when the membrane is exposed to more dilute solutions.
Figure 2.
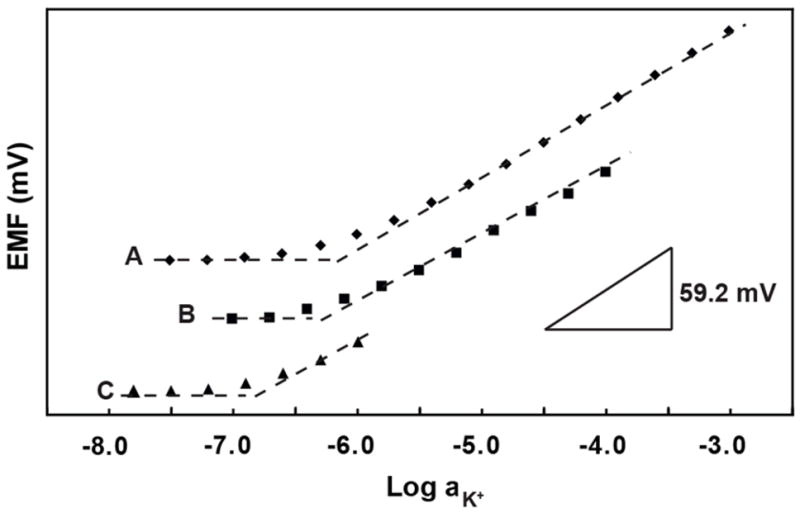
Potassium ion calibration curves for K+-selective ISEs recorded:Conditioning in (A) 1 mM KCl for 1 d, (B) 0.1 mM KCl for 1 d, and (C) 1 μM KCl for 1 d. For clarity, response curves have been shifted vertically relative to one another.
Table 1.
Detection limits achieved for K+ with different concentrations of conditioning and starting solutions.
| Conditioning solution (M) | Starting solution (M) | Detection limit (M) |
|---|---|---|
| 10−6 | 10−3 | 1.0×10−6 |
| 10−9 | 10−3 | 1.2×10−6 |
| 10−3 | 10−3 | 6.3×10−7 |
| 10−4 | 10−4 | 4.0×10−7 |
| 10−6 | 10−6 | 1.6×10−7 |
The K+ SC-ISEs presented here compare well to previously reported ones. For example, solid contact K+ ISEs with polypyrrole intermediate layers, electropolymerized in the presence of the water-soluble hexacyanoferrate, exhibited a 1.2×10−7 M detection limit [38]. Chumbimuni-Torres and co-workers reported a detection limit for K+ at 1×10−7 M for electrodes with a plasticizer-free copolymer as the membrane matrix and poly(3-octylthiophene) as the intermediate layer [41]. In comparison, the lowest detection limit reported thus far for any ISE was ~5.0×10−9 M for a K+ ISE with an inner filling solution in equilibrium with an ion-exchange resin buffering the activity of the primary ion [13]. It appears that, independent of the approach, very similar detection limits are observed for all SC ISEs. This may be the result of sample contamination with K+ from ambient sources. Only clean room conditions are likely to exclude the possibility of ambient K+ contamination. Therefore, further tests of the low detection limits of 3DOM carbon-contacted ISEs were performed with Ag+ ISEs. Silver is not only a ubiquitous contaminant, but its determination is also of practical interest. Due to their antibacterial properties, silver salts are used in the disinfection of drinking water, as well as in implanted prostheses. The amount of silver entering the aquatic system and the atmosphere each year is estimated to be as high as 450,000 kg. Although very small portions of the total silver is biologically available in water, a concentration higher than 0.17 μg/L is toxic to fish and microorganisms, making it crucial to monitor the concentration of silver in the environment [48].
SC-ISEs for Ag+ have been used previously [49, 50], however, it was not until 2006 that Bakker and co-workers reported on the first SC-ISE for Ag+ with a detection limit in the nanomolar range. In a report on trace level measurements of five different ions, they described detection limits for Ag+ of 2.0×10−9 M for a SC-ISE with o-xylylenebis(N,N-diisobutyldithiocarbamate) as ionophore [41]. In the work with 3DOM carbon-contacted ISEs reported here, the same ionophore has been used, and different factors that affect the detection limit were studied. These include the polymer content, the molar ratio of ionophore and ionic sites, the concentration of the conditioning solution, and the concentration range of the calibration curves.
Polymer Content
The choice of the polymer content of a PVC-based membrane is a trade-off. On one hand, a higher polymer content decreases the diffusion of ions through the membrane, which is helpful for lowering ion fluxes and, thereby, detection limits. The diffusion coefficients in the membrane can be strongly varied by changing the concentration ratio of polymer to plasticizer [51, 52]. An increase of the polymer content of the membrane from the usual value of 30% to 50% (w/w) has been reported to improve the detection limit for calcium from the micromolar down to the nanomolar range [53]. On the other hand, increasing the polymer content of the sensing membrane increases its electrical resistance, and eventually makes it impossible to carry out potentiometric experiments. In this study, the polymer content of NPOE/PVC membranes was varied from 33% to 75%. All electrodes were conditioned in 1 nM AgNO3 for 2 days, and calibration curves were measured by dilution of 0.1 mM AgNO3 solutions. The thus obtained detection limits are listed in Table 2. Clearly, membranes with a higher polymer content provided lower detection limits. With 75% PVC, the electrodes responded to Ag+ down to the nanomolar level, which was two orders of magnitude lower than for the membranes with 33% PVC. This is consistent with the observation that the resistance of a 33% PVC membrane (5.1×105 Ω) was approximately two orders of magnitude lower than the resistance of a 75% PVC membranes (1.7×108 Ω).
Table 2.
Detection limits for Ag+ upon optimization of the sensing membranes and the experimental protocol.
| Polymer content (wt %)a | 33 | 43 | 66 | 70 | 75 |
|---|---|---|---|---|---|
| Detection limit (M) | 1.0×10−7 | 3.1×10−8 | 1.8×10−8 | 7.2×10−9 | 3.7×10−9 |
| Molar ratio of ionophore and ionic sitesb | 3.2:1.0 | 2.1:1.0 | |||
| Detection limit (M) | 1.6×10−8 | 3.7×10−8 | |||
| Conditioning solution (M)c | 1 mM AgNO3 (1 d) | 1 nM AgNO3 (1 d) | 1 nM AgNO3 (2 d) | ||
| Detection limit (M) | 1.1×10−7 | 4.3×10−8 | 4.0×10−8 | ||
| Starting concentration of AgNO3 (M)d | 1.0×10−4 | 1.0×10−6 | 1.0×10−8 | ||
| Detection limit (M) | 4.0×10−8 | 8.0×10−9 | 7.9×10−11 | ||
Molar ratio of ionophore and ionic sites 2.1:1.0. Electrodes conditioned in 1 nM AgNO3 solution for 2 d. Calibration curves measured by dilution of 0.1 mM AgNO3 solution.
Polymer content 43%. Conditioning and calibration curve procedure as for a.
Polymer content 43%. Molar ratio of ionophore and ionic sites 3.2:1.0. After conditioning in different solutions, calibration curves measured by dilution of 0.1 mM AgNO3 solutions.
Membrane components as for c. All electrodes conditioned in 1 nM AgNO3 solution for 1 d.
Molar Ratio of Ionophore and Ionic Sites
The molar ratio of ionophore and ionic sites is well known to affect the selectivities [54, 55] and, thereby, the detection limits of ISEs. For a given concentration of interfering ions, electrodes with better selectivities can detect the primary ion at lower concentrations. Moreover, the ionophore and ionic sites are buffering the primary ion activity in the sensing membrane, stronger binding of the primary ion to the ionophore resulting in a lower primary ion activity in the membrane. Under circumstances where the detection limit is not determined by interfering ions but by the release of primary ions from the membrane into the sample, a lower primary ion activity in the membrane is also expected to result in a lower detection limit. Therefore, if the concentration of ionic sites is kept constant while the ionophore concentration in the sensing membrane is increased and, thereby the concentration of free analyte ions in the sensing membrane decreases, the detection limit is expected to improve. Experimentally, the molar ratio of the Ag+ ionophore o-xylylenebis(N,N-diisobutyldithiocarbamate) and the ionic sites in the sensing membrane was varied, while the weight percentages of PVC, NPOE and NaTFPB were kept constant at 43%, 56% and 1%, respectively. Two molar ratios of ionophore and ionic sites, i.e., 3.2:1.0 and 2.1:1.0, were tested. All electrodes were conditioned in 1 nM AgNO3 for 2 days, and calibration curves were obtained by successive dilution of 0.1 mM AgNO3. With the ratio of ionophore to ionic site ratio of 3.2:1.0, the detection limit was 1.6×10−8 M, which was approximately two times better than for electrodes with the ionophore:ionic site ratio of 2.1:1.0 (Table 2). This seems to be consistent with the fact that the activity of Ag+ in the membranes with the 3.2:1.0 ionophore to site ratio is calculated from the known 1:1 binding constants to be two times lower than in the membranes with the 2.1:1.0 ratio [56].
Conditioning Solutions and Starting Solutions
As the above mentioned results for K+ show, different conditioning solutions and starting solutions strongly affect the detection limits. To investigate these effects, membranes containing 43% PVC and a 3.2:1.0 molar ratio of ionophore and ionic sites were used, and calibration curves were measured by successive dilution, starting with 0.1 mM AgNO3 solutions. Conditioning for one day in 1 nM solutions gave a detection limit of 4.0×10−8 M, which was approximately four times lower than when conditioning was performed with a 1 mM AgNO3 solution. Conditioning in 1 nM AgNO3 for one additional day did not further improve the detection limit. For this lower concentration of the conditioning solution of 1 nM, different concentrations of starting solutions were subsequently investigated, i.e., 0.1 mM, 1 μM and 10 nM. As expected, the lower the concentration of the starting solution, the lower the detection limit that could be achieved. With a starting solution containing 10 nM AgNO3, the detection limit was 7.9×10−11 M, which is about 3 and 2 orders of magnitude lower than the results for the 0.1 mM and 1 μM starting solutions, respectively.
The best conditions for low detection limit detection of Ag+ were selected based on a combination of the relevant experimental parameters. In this experiment, the sensing membrane was composed of 75% PVC, and the molar ratio of ionophore and ionic sites was 3.2:1.0. The electrode was conditioned in 1 L of 1 μM AgNO3 for one day to ensure complete Na+/Ag+ exchange between the sample and the sensing membrane. Subsequently, the membrane was conditioned in 1 nM AgNO3 for 2 days. Then the electrode was immersed into 500 mL of a 10 nM AgNO3 solution for the measurement of a calibration curve by successive dilution (see Figure 3). The thus obtained detection limit was 4.0×10−11 M, or 4.3 ppt for Ag+, which is more than two orders of magnitude lower than for the previously reported SC-ISEs based on a plasticizer-free methyl methacrylate-decyl methacrylate copolymer matrix [41]. As shown by others previously, this two-step conditioning procedure starting with a relatively high concentration of analyte in the first step ensures that even a limited volume of conditioning solution contains enough analyte ions to fill the sensing membrane by ion-exchange with primary ions, while the second step with the solution of much lower primary ion activity removes excess primary ion from the sensing membrane that may have entered in there due to co-extraction. Once the ion exchange is performed this way, the electrodes can be stored in the conditioning solution of lower primary ion activity and used without further preparatory steps.
Figure 3.
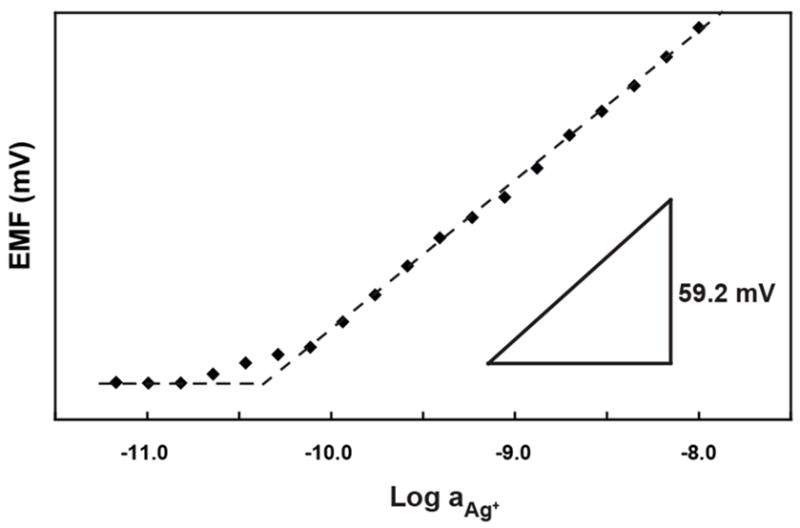
Silver ion calibration curve for Ag+-selective electrode, with the lowest detection limit achieved in this work, recorded after conditioning in 1 mM AgNO3 for 1 d and 1 nM AgNO3 for 2 days.
Conclusions
An improved detection limit of 3DOM carbon-contacted ISEs for K+ was obtained with conditioning at low K+ concentration. The resulting detection limit is comparable to that of other K+ selective SC-ISEs operated under optimized conditions. This paper also presents the first application of 3DOM carbon-contacted ISEs for the trace-level detection of Ag+. With optimizations of the polymer content, the molar ratio of ionophore and ionic sites, and the concentrations of the conditioning and starting solutions, the detection limit for Ag+ could be lowered into the subnanomolar concentration range. The successful detection of Ag+ at these low concentrations and the excellent stability and resistance to interferences reported previously make 3DOM carbon-contacted ISEs interesting for trace-level measurements in real life samples.
Acknowledgments
This work was supported by the National Science Foundation (CTS-0428046), the National Institute of Health (R01 EB005225-01), the Office of Naval Research (ONR, Grant N00014-07-1-0608), and the MRSEC program of the NSF (DMR-0212302), which supports the University of Minnesota Characterization Facility. MAF thanks 3M for a Science & Technology Fellowship. MMJ and NDP acknowledge the NSF/Lando summer undergraduate research fellowship program of the University of Minnesota (CHE-0552622).
References
- 1.Johnson RD, Bachas LG. Anal Bioanal Chem. 2003;376:328. doi: 10.1007/s00216-003-1931-0. [DOI] [PubMed] [Google Scholar]
- 2.Diamond D. Anal Chem. 2004;76:278A. doi: 10.1021/ac041598m. [DOI] [PubMed] [Google Scholar]
- 3.Bakker E, Bühlmann P, Pretsch E. Chem Rev. 1997;97:3083. doi: 10.1021/cr940394a. [DOI] [PubMed] [Google Scholar]
- 4.Bühlmann P, Pretsch E, Bakker E. Chem Rev. 1998;98:1593. doi: 10.1021/cr970113+. [DOI] [PubMed] [Google Scholar]
- 5.Buck RP, Linder E. Anal Chem. 2001;73:88A. doi: 10.1021/ac012390t. [DOI] [PubMed] [Google Scholar]
- 6.Sokalski T, Ceresa A, Zwickl T, Pretsch E. J Am Chem Soc. 1997;119:11347. [Google Scholar]
- 7.Mathison S, Bakker E. Anal Chem. 1998;70:303. doi: 10.1021/ac990387s. [DOI] [PubMed] [Google Scholar]
- 8.Ion AC, Bakker E, Pretsch E. Anal Chim Acta. 2001;440:71. [Google Scholar]
- 9.Ceresa A, Bakker E, Hattendorf B, Gunther D, Pretsch E. Anal Chem. 2001;73:343. doi: 10.1021/ac001034s. [DOI] [PubMed] [Google Scholar]
- 10.Ceresa A, Radu A, Peper S, Bakker E, Pretsch E. Anal Chem. 2002;74:4027. doi: 10.1021/ac025548y. [DOI] [PubMed] [Google Scholar]
- 11.Wang C-Y, Hu X-Y, Leng Z-Z, Jin G-D. Electroanalysis. 2003;15:709. [Google Scholar]
- 12.Malon A, Radu A, Qin W, Qin Y, Ceresa A, Maj-Zurawska M, Bakker E, Pretsch E. Anal Chem. 2003;75:3865. doi: 10.1021/ac026454r. [DOI] [PubMed] [Google Scholar]
- 13.Qin W, Zwickl T, Pretsch E. Anal Chem. 2000;72:3236. doi: 10.1021/ac000155p. [DOI] [PubMed] [Google Scholar]
- 14.Linder E, Gyurcsanyi RE, Buck RP. Electroanalysis. 1999;11:695. [Google Scholar]
- 15.Pergel E, Gyurcsanyi RE, Toth K, Linder E. Anal Chem. 2001;73:4249. doi: 10.1021/ac010094a. [DOI] [PubMed] [Google Scholar]
- 16.Zwickl T, Sokalski T, Pretsch E. Electroanalysis. 1999;11:673. [Google Scholar]
- 17.Sokalski T, Zwickl T, Bakker E, Pretsch E. Anal Chem. 1999;71:1204. [Google Scholar]
- 18.Sokalski T, Ceresa A, Fibbioli M, Zwickl T, Bakker E, Pretsch E. Anal Chem. 1999;71:1210. [Google Scholar]
- 19.Buck RP. In: Ion-Selective Electrodes in Analytical Chemistry. Freiser HE, editor. Plenum; New York: 1980. p. 58. [Google Scholar]
- 20.Buck RP. J Electroanal Chem. 1987;219:23. [Google Scholar]
- 21.Sun L, Weber SG. Polym Mater Sci Eng. 1997;76:614. [Google Scholar]
- 22.Cadogan A, Gao Z, Lewenstam A, Ivaska A. Anal Chem. 1992;64:2496. [Google Scholar]
- 23.Momma T, Yamamoto M, Komaba S, Osaka T. J Electroanal Chem. 1996;407:91. [Google Scholar]
- 24.Gyurcsanyi RE, Nyback A-S, Toth K, Nagy G, Ivaska A. Analyst. 1998;123:1339. [Google Scholar]
- 25.Gyurcsanyi RE, Rangisetty N, Clifton S, Pendley BD, Linder E. Talanta. 2004;63:89. doi: 10.1016/j.talanta.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Sutter J, Lindner E, Gyurcsanyi RE, Pretsch E. Anal Bioanal Chem. 2004;380:7. doi: 10.1007/s00216-004-2737-4. [DOI] [PubMed] [Google Scholar]
- 27.Bobacka J, Lindfors T, McCarrick M, Ivaska A, Lewenstam A. Anal Chem. 1995;67:3819. [Google Scholar]
- 28.Lindfors T, Ivaska A. Anal Chem. 2004;76:4387. doi: 10.1021/ac049439q. [DOI] [PubMed] [Google Scholar]
- 29.Bobacka J, McCarrick M, Lewenstam A, Ivaska A. Analyst. 1994;119:1985. [Google Scholar]
- 30.Chumbimuni-Torres KY, Rubinova N, Radu A, Kubota LT, Bakker E. Anal Chem. 2006;78:1318. doi: 10.1021/ac050749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai C-Z, Fierke MA, Stein A, Bühlmann P. Anal Chem. 2007;79:4621. doi: 10.1021/ac070132b. [DOI] [PubMed] [Google Scholar]
- 32.Stein A, Li F, Denny NR. Chem Mater. 2008;20:649. [Google Scholar]
- 33.Lee KT, Lytle JC, Ergang NS, Oh SM, Stein A. Adv Funct Mater. 2005;15:547. [Google Scholar]
- 34.Crespo GA, Macho S, Rius FX. Anal Chem. 2008;80:1316. doi: 10.1021/ac071156l. [DOI] [PubMed] [Google Scholar]
- 35.Michalska A, Konopka A, Maj-Zurawska M. Anal chem. 2003;75:141. doi: 10.1021/ac025916y. [DOI] [PubMed] [Google Scholar]
- 36.Konopka A, Sokalski T, Michalska A, Lewenstam A, Maj-Zurawska M. Anal Chem. 2004;76:6410. doi: 10.1021/ac0492158. [DOI] [PubMed] [Google Scholar]
- 37.Michalska A, Dumanska J, Maksymiuk K. Anal Chem. 2003;75:4964. [Google Scholar]
- 38.Michalska AJ, Appaih-Kusi C, Heng LY, Walkiewicz S, Hall EAH. Anal Chem. 2004;76:2031. doi: 10.1021/ac0353132. [DOI] [PubMed] [Google Scholar]
- 39.Sutter J, Radu A, Peper S, Bakker E, Pretsch E. Anal Chim Acta. 2004;523:53. [Google Scholar]
- 40.Heng LY, Toth K, Hall EAH. Talanta. 2004;63:73. doi: 10.1016/j.talanta.2003.12.051. [DOI] [PubMed] [Google Scholar]
- 41.Chumbimuni-Torres KY, Rubinova N, Radu A, Kubota LT, Bakker E. Anal Chem. 2006;78:1318. doi: 10.1021/ac050749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konopka A, Sokalski T, Lewenstam A, Maj-Zurawska M. Electroanalysis. 2006;18:2232. [Google Scholar]
- 43.Ergang NS, Lytle JC, Lee KT, Oh SM, Smyrl WH, Stein A. Adv Mater. 2006;18:1750. [Google Scholar]
- 44.Wang Z, Ergang NS, Al-Daous M, Stein A. Chem Mater. 2005;17:6805. [Google Scholar]
- 45.Wang Z, Li F, Ergang NS, Stein A. Chem Mater. 2006;18:5543. [Google Scholar]
- 46.Morf WE. The Principles of Ion-Selective Electrodes and of Membrane Transport. Elsevier; New York: 1981. [Google Scholar]
- 47.Meier PC. Anal Chim Acta. 1982;136:363. [Google Scholar]
- 48.Environmental Protection Agency. EPA Drinking Water Criteria Document for Silver. EPA; Washington DC, EPA CASRN: 1989. pp. 7440pp. 7422–7444. [Google Scholar]
- 49.Bobacka J, Lahtinen T, Nordman J, Haggstrom S, Rissanen K, Lewenstam A, Ivaska A. Electroanalysis. 2001;13:723. [Google Scholar]
- 50.Bobacka J, Vaananen V, Lewenstam A, Ivaska A. Talanta. 2004;63:135. doi: 10.1016/j.talanta.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Oesch U, Simon W. Anal Chem. 1980;52:692. [Google Scholar]
- 52.Fu B, Bakker E, Yun JH, Yang VC, Meyerhoff ME. Anal Chem. 1994;66:2250. doi: 10.1021/ac00086a009. [DOI] [PubMed] [Google Scholar]
- 53.Ceresa A, Sokalski T, Pretsch E. J Electroanal Chem. 2001;501:70. [Google Scholar]
- 54.Meier PC, Morf WE, Laubli M, Simon W. Anal Chim Acta. 1984;1984:1. [Google Scholar]
- 55.Amemiya S, Bühlmann P, Pretsch E, Rusterholz B, Umezawa Y. Anal Chem. 2000;72:1618. doi: 10.1021/ac991167h. [DOI] [PubMed] [Google Scholar]
- 56.Szigeti Z, Malon A, Vigassy T, Csokai V, Grun A, Wygladacz K, Ye N, Xu C, Chebny VJ, Bitter I, Rathore R, Bakker E, Pretsch E. Anal Chim Acta. 2006;572:1. doi: 10.1016/j.aca.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]