

Leucine aminopeptidase, an exopeptidase that hydrolyzes leucine from the N-terminus of polypeptides, from X. oryzae pv. oryzae was cloned, expressed and crystallized.
Keywords: Xanthomonas oryzae pv. oryzae, leucine aminopeptidase
Abstract
Xanthomonas oryzae pv. oryzae (Xoo) causes the serious disease bacterial blight in rice. The pepA (Xoo0834) gene from Xoo is one of around 100 genes that have been selected for the design of antibacterial drugs. The pepA gene encodes leucine aminopeptidase (LAP), an exopeptidase that catalyzes the hydrolysis of leucine residues from the N-terminus of a protein or peptide. This enzyme was expressed in Escherichia coli, purified and crystallized, and preliminary X-ray structural studies have been carried out. The LAP crystal diffracted to 2.6 Å resolution and belonged to the cubic space group P213. The unit-cell volume of the crystal was compatible with the presence of two monomers in the asymmetric unit.
1. Introduction
Xanthomonas oryzae pv. oryzae (Xoo; Ezuka & Kaku, 2000 ▶) causes the serious disease bacterial blight (BB) in rice and hence is a target for new antibacterial drugs. BB is the most devastating bacterial disease found in rice-cultivating countries and results in serious production losses all over the world, especially in Asia. In 2006, BB caused production losses of rice worth more than $100 million in South Korea alone. No effective drugs are currently available against this disease. Lee et al. (2005 ▶) determined the whole genomic sequence of Xoo, providing information for the rational selection of drug targets. In the first step in this process, 95 genes coding for essential enzymes (Payne et al., 2004 ▶, 2007 ▶) were selected from the 4538 putative genes (Lee et al., 2005 ▶). As three-dimensional structures are important for the rational development of drugs, the pepA (Xoo0834) gene, which encodes leucine aminopeptidase, was cloned and expressed in Escherichia coli.
The aminopeptidases are comprised of various sets of peptidases that play important roles in cell maintenance, growth, development and defence. Aminopeptidases not only catalyze the removal of amino acids from the N-terminus of proteins or peptides, but also function as transcriptional repressors, as viral or toxin receptors, as site-specific recombination factors, in vesicular trafficking etc. (Matsui et al., 2006 ▶). Leucine aminopeptidases (LAPs) are widely distributed cytosolic exopeptidases that catalyze the hydrolysis of amino acids from polypeptide chains (Smith & Hill, 1960 ▶; Hanson & Frohne, 1977 ▶), specifically cleaving leucyl substrates. One of the most intensively studied sets of aminopeptidases are the hexameric leucyl aminopeptidases from animals, plants and microbes (Colloms, 2004 ▶; Sträter & Lipscomb, 2004 ▶; Walling, 2004 ▶). These LAPs belong to the MF clan of metallopeptidases and to the M17 peptidase family (Rawlings & Barrett, 1995 ▶; Rawlings et al., 2006 ▶).
The M17 LAPs have several distinguishing features. These proteins are highly conserved at the amino-acid level, containing two M17 Pfam domains (Rawlings & Barrett, 1995 ▶; Rawlings et al., 2006 ▶). These LAPs have a distinctive feature: the absence of the metallopeptidase motif HEXXH which is present in M1 metallopeptidases. Based on sequence conservation, LAP-enzyme genes have been identified in more than 60 bacterial genomes. Furthermore, while the M17 LAPs are hexameric and bind two metal cations, M1 peptidases with LAP activity are not hexameric and bind a single metal cation. Three-dimensional structures have been determined by X-ray crystallography for the bovine and E. coli M17 LAPs. Diverse expression programs and biological roles have been established for the animal, plant and prokaryotic LAPs and have recently been reviewed (Colloms, 2004 ▶; Sträter & Lipscomb, 2004 ▶; Walling, 2004 ▶).
The target LAP enzyme, which is the product of the pepA (Xoo0834) gene, from Xanthomonas oryzae pv. oryzae discussed in this report is a 51 kDa enzyme consisting of 490 amino acids. The LAP from Xoo shares 47.8% sequence identity and 62.4% sequence similarity with the structurally determined E. coli LAP (Sträter et al., 1999 ▶). It was cloned, expressed, purified and crystallized and preliminary X-ray crystallographic studies have been carried out in order to understand its structural organization and reaction mechanism. It is expected that structural information of LAP may significantly help in the design of a potential antibacterial drug against Xoo.
2. Materials and methods
2.1. Cloning
The Xoo0834 (pepA) gene was amplified via the polymerase chain reaction using the genomic DNA of X. oryzae pv. oryzae cells (Xoo KACC10331 strain). The sequences of the forward and reverse primers designed from the published genome sequence (Lee et al., 2005 ▶) are as follows: 5′-GGG CGG CAT ATG GCC CTG CAA TTC ACC CTG AAC-3′ and 5′-GCC GGG GGA TCC TCA GGC GGC GCG ATC CAG CAA-3′ (the bases in bold designate NdeI and BamHI restriction sites). The PCR-amplified fragment was digested with NdeI and BamHI and was then cloned into a modified pET11a vector previously digested with these enzymes. The modified vector contained six His residues and a tobacco etch virus (TEV) protease cleavage site before the NdeI site in the pET11a vector (Novagen) to facilitate purification of the expressed protein (Doan et al., 2008 ▶). As a result, LAP (Xoo0834) has an extra 18 residues (MGHHHHHHSSENLYFQGH) at its N-terminus; only two residues (GH) remain after TEV cleavage.
2.2. Overexpression and purification
LAP (Xoo0834) was overexpressed in E. coli strain BL21 (DE3) cells. The cells were grown at 295 K to an OD600 of 0.6 in Luria–Bertani medium containing 50 µg ml−1 ampicillin. Protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. Cells were continuously grown for 12 h at 295 K and centrifuged at 6000 rev min−1 (Vision VS24-SMTi V5006A rotor) for 20 min at 277 K. The resulting cell pellet was resuspended in ice-cold lysis buffer (25 mM Tris–HCl pH 7.5, 300 mM NaCl, 15 mM imidazole and 3 mM β-mercaptoethanol) and homogenized with a sonicator (Sonomasher, S&T Science, Korea). The crude cell extract was centrifuged at 15 000 rev min−1 (Vision VS24-SMTi V508A rotor) for 40 min at 277 K. All purification steps were performed at 277 K. The expressed LAP in the clear supernatant fraction was purified in two steps. Firstly, the supernatant was purified using an Ni–NTA His-bind resin column (Qiagen). Unbound proteins were washed from the column using buffer A [25 mM Tris–HCl pH 7.5, 1 M NaCl, 3 mM β-mercaptoethanol, 40 mM imidazole and 20%(v/v) glycerol]. The LAP protein was eluted from the column using buffer B [25 mM Tris–HCl pH 7.5, 300 mM NaCl, 200 mM imidazole, 3 mM β-mercaptoethanol and 20%(v/v) glycerol]. The resulting protein solution was applied onto a Superdex 75S prep-grade column previously equilibrated with 25 mM Tris–HCl pH 7.5, 150 mM NaCl and 3 mM β-mercaptoethanol for further purification by gel filtration. The homogeneity of the purified protein was assessed via SDS–PAGE (Fig. 1 ▶). The eluted protein was extensively dialysed with buffer containing 25 mM Tris–HCl pH 7.5, 3 mM β-mercaptoethanol, 20%(v/v) glycerol. The purified protein was concentrated using a Centriprep (Millipore) to achieve a final concentration of 10 mg ml−1 in buffer A for crystallization screening. 20%(v/v) glycerol was added to the buffers to avoid aggregation and to keep the purified protein soluble and the concentrated protein solution containing 20%(v/v) glycerol was directly used for crystallization.
Figure 1.

SDS–PAGE analysis of LAP (Xoo0834) during purification. Proteins were analyzed on 10% SDS–PAGE and were stained with Coomassie Blue. Lane M, molecular-weight markers (kDa); lane L, purified LAP.
2.3. Crystallization and X-ray data collection
Initial crystallization experiments were carried out by the sitting-drop vapour-diffusion method with various screening kits (Crystal Screens I and II, Index Screen, PEG/Ion Screen and Grid Screen from Hampton Research and Wizard from Emerald BioSystems) and a Hydra II e-drop automated pipetting system (Matrix) using a 1:1 (1 µl:1 µl) ratio of protein to reservoir solution and 200 µl well solution. After 1 d, very small plate-like crystals were observed in four different conditions (Fig. 2 ▶). Of these, only one condition (Hampton Research Index Screen condition E6) gave a crystal of significant size. The best crystal grew after 10 d from a reservoir containing 0.05 M CaCl2, 0.1 M bis-tris pH 6.5, 30%(v/v) PEG monomethyl ether 550 (Fig. 3 ▶). A well grown crystal was flash-cooled in liquid nitrogen in the best crystallization condition supplemented with 20%(v/v) glycerol.
Figure 2.

Crystals of LAP from X. oryzae pv. oryzae (Xoo0834) obtained in sitting-drop setups using a Hydra automated high-throughput automated system with various screening kits. The scale bars represent 0.1 mm. Crystals were obtained using (a) 0.02 M CaCl2, 0.1 M sodium acetate pH 4.6, 15%(v/v) MPD, (b) 0.1 M sodium acetate pH 5, 20%(v/v) MPD and (c) 0.1 M sodium acetate pH 5, 40%(v/v) MPD.
Figure 3.

Crystal of LAP protein obtained by the sitting-drop vapour-diffusion method after 10 d using Index Screen condition E6 [0.05 M CaCl2, 0.1 M bis-tris pH 6.5, 30%(v/v) PEG monomethyl ether 550]. The scale bar represents 0.15 mm.
X-ray diffraction data were collected from the frozen crystal using a Bruker Proteum 300 CCD detector on beamline 6C1 at the Pohang Light Source (PLS), South Korea. The crystal diffracted to 2.6 Å resolution and the data were integrated and scaled using DENZO and SCALEPACK, respectively (Otwinowski & Minor, 1997 ▶). Based on auto-indexing with DENZO, the LAP crystal belonged to the cubic space group P213, with unit-cell parameters a = b = c = 152.1 Å. The final statistics of data collection and processing details are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the last resolution shell.
| Synchrotron | PLS beamline 6C1 |
| Wavelength (Å) | 0.96418 |
| Resolution range (Å) | 50.0–2.6 (2.64–2.60) |
| Space group | P213 |
| Unit-cell parameters (Å) | a = b = c = 152.1 |
| Total No. of reflections | 367823 |
| No. of unique reflections | 36695 |
| Completeness (%) | 100.0 (100.0) |
| Molecules per ASU | 2 |
| VM (Å3 Da−1) | 2.66 |
| Solvent content (%) | 53.8 |
| Average I/σ(I) | 18.3 (3.6) |
| Rmerge† (%) | 15.4 (63.8) |
R
merge = 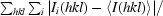
 , where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
, where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
3. Results and discussion
Initial high-throughput crystallization screening of the LAP protein produced a diffraction-quality crystal (Fig. 3 ▶) after 10 d from Index Screen condition E6 [0.05 M CaCl2, 0.1 M bis-tris pH 6.5, 30%(v/v) PEG monomethyl ether 550]. A complete set of diffraction data was collected to 2.6 Å resolution from a single crystal. The systematic absences suggested that the crystal belonged to space group P213. The volume of the asymmetric unit of the LAP crystal was compatible with the presence of two monomers in the unit cell, with a volume per unit molecular weight of the protein of 2.66 Å3 Da−1 and a calculated solvent content of 53.8% (Matthews, 1968 ▶). A preliminary structure solution of the LAP protein was obtained by molecular-replacement (MR) using the MOLREP program (Vagin & Teplyakov, 1997 ▶) with the crystal structure of LAP from E. coli (PDB code 1gyt; full length; Sträter et al., 1999 ▶), which shares 47.8% sequence identity and 62.4% sequence similarity (Fig. 4 ▶), as a search model. The best results were obtained in space group P213, giving a correlation coefficient of 59.3% and an R factor of 42.3%. Examination of the best MR solution structure showed good crystal packing and no clashes were found between symmetry-related molecules. The final model is currently being refined using REFMAC5 (Murshudov et al., 1997 ▶). Our structural data for LAP will provide an insight into its enzymatic reaction mechanisms and may be useful in developing a drug against Xoo.
Figure 4.

Sequence alignment of the LAP (Xoo0834) enzyme and the model LAP from E. coli (PDB code 1gyt). The residues highlighted in red are 100% conserved. The M17 LAP has two Pfam domains, namely the N-terminal domain (blue box) and catalytic domain (green box), which are common to all M17 LAP proteins.
Acknowledgments
We are grateful to Dr K. J. Kim for his assistance at beamline 6C of Pohang Light Source (PLS) in South Korea and to Yun Young Hur from the Cheong Shim International Academy in the Republic of Korea for her technical assistance in much of the laboratory work. This work was supported by a grant (Code No. 20070501034003) from the BioGreen 21 Program, Rural Development Administration, Republic of Korea, by the Ministry of Environment as the ‘GAIA Project’ (173-091-006) and by a Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean government (MEST; No. R01-2008-000-20315-0).
References
- Colloms, S. D. (2004). Handbook of Proteolytic Enzymes, 2nd ed., edited by A. J. Barrett, N. D. Rawlings & J. F. Woessner, pp. 313–315. San Diego: Elsevier/Academic Press.
- Doan, T. T. N., Kim, J.-K., Kim, H., Ahn, Y.-J., Kim, J.-G., Lee, B.-M. & Kang, L.-W. (2008). Acta Cryst. F64, 1115–1117. [DOI] [PMC free article] [PubMed]
- Ezuka, A. & Kaku, H. (2000). Bull. Natl Inst. Agrobiol. Resour.15, 53–54.
- Hanson, H. & Frohne, M. (1977). Methods Enzymol.5, 504–521. [DOI] [PubMed]
- Lee, B. M. et al. (2005). Nucleic Acids Res.33, 577–586. [DOI] [PMC free article] [PubMed]
- Matsui, M., Fowler, J. H. & Walling, L. L. (2006). Biol. Chem.387, 1535–1544. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.277, 307–326. [DOI] [PubMed]
- Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. (2007). Nature Rev. Drug Discov.6, 29–40. [DOI] [PubMed]
- Payne, D. J., Gwynn, M. N., Holmes, D. J. & Rosenberg, M. (2004). Methods Mol. Biol.266, 231–259. [DOI] [PubMed]
- Rawlings, N. D. & Barrett, A. J. (1995). Methods Enzymol.248, 183–228. [DOI] [PubMed]
- Rawlings, N. D., Morton, F. R. & Barrett, A. J. (2006). Nucleic Acids Res.34, D270–D272. [DOI] [PMC free article] [PubMed]
- Smith, E. L. & Hill, R. L. (1960). The Enzymes, 2nd ed., edited by P. D. Boyer, H. Lardy & K. Myrback, Vol. 4, pp. 37–62. New York: Academic Press.
- Sträter, N. & Lipscomb, W. N. (2004). Handbook of Proteolytic Enzymes, 2nd ed., edited by A. J. Barrett, N. D. Rawlings & J. F. Woessner, pp. 896–899. San Diego: Elsevier/Academic Press.
- Sträter, N., Sherratt, D. J. & Colloms, S. D. (1999). EMBO J.18, 4513–4522. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Walling, L. L. (2004). Handbook of Proteolytic Enzymes, 2nd ed., edited by A. J. Barrett, N. D. Rawlings & J. F. Woessner, pp. 901–904. San Diego: Elsevier/Academic Press.