

Abstract
Context: Autoimmune polyendocrine syndrome type 1 (APS1) is an autosomal recessive disorder caused by mutations in the autoimmune regulator (AIRE) gene. Hypoparathyroidism occurs in 80% of patients with APS1 and has been suggested to result from an autoimmune reaction against the calcium-sensing receptor (CaSR) in parathyroid cells. Anti-CaSR binding antibodies have previously been detected in patients with APS1.
Objective: The aim of this study was to determine whether anti-CaSR antibodies present in APS1 patients could modulate the response of the CaSR to stimulation by Ca2+.
Results: The results indicated that two of the 14 APS1 patients included in the study had anti-CaSR antibodies that stimulated the receptor. These antibodies were detected by their ability to increase both Ca2+-dependent extracellular signal-regulated kinase phosphorylation and inositol phosphate accumulation in human embryonic kidney 293 cells expressing the CaSR.
Conclusion: An important implication of the present results is that although the majority of APS1 patients do not have CaSR-stimulating antibodies, there may be a small but substantial minority of patients in whom the hypoparathyroid state is the result of functional suppression of the parathyroid glands rather than their irreversible destruction.
The presence of calcium-sensing receptor stimulating antibodies in patients with autoimmune polyendocrine syndrome type 1 is discussed.
Autoimmune polyendocrine syndrome type 1 (APS1) is a rare autosomal recessive disorder (1) caused by mutations in the autoimmune regulator (AIRE) gene (2,3), which encodes a putative transcription factor (4,5). The disease, also known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), is characterized by multiple organ-specific autoimmunity and ectodermal manifestations (1,6). In the majority of cases, disease components include mucocutaneous candidiasis, hypoparathyroidism, and Addison’s disease, with type 1 diabetes mellitus, alopecia, vitiligo, autoimmune hepatitis, and pernicious anemia occurring less frequently (1,6). APS1 patients typically display a wide variety of antibodies against enzymes found in the affected organs, including 17-α-hydroxylase (7) 21-hydroxylase (8,9) and side-chain cleavage enzyme (9), all of which are present in the adrenal cortex. Antibodies against pancreatic glutamic acid decarboxylase 65 and tyrosine phosphatase-like protein IA-2, which are prevalent in autoimmune type 1 diabetes mellitus, can also be detected in individuals with APS1 (10,11). Other identified autoantigens include tryptophan hydroxylase (12), tyrosine hydroxylase (13), and aromatic l-amino acid decarboxylase (14), which are present in APS1 patients with intestinal dysfunction, alopecia, and autoimmune hepatitis, respectively. In addition, the transcription factors SOX9 and SOX10 are vitiligo-associated autoantigens in APS1 (15). More recently, interferon antibodies have been identified in almost all patients with this condition, making them a diagnostic marker for APECED (16,17).
Hypoparathyroidism occurs in 80% of patients with APS1 and is associated with hypocalcemia, hyperphosphatemia, and low serum levels of PTH. Early reports suggested that the clinical symptoms and biochemical manifestations of hypoparathyroidism could result from humoral immune responses to parathyroid cells (18,19,20). Subsequently, parathyroid autoantigens targeted by autoantibodies in APS1 patients have been identified as the NACHT leucine-rich-repeat protein 5 (NALP5) (21) and the G protein-coupled calcium-sensing receptor (CaSR) (22,23,24,25), which plays a pivotal role in maintaining calcium homeostasis by sensing circulating calcium levels and regulating PTH synthesis and release as well as renal calcium excretion (26).
Recently, we confirmed the presence of anti-CaSR antibodies in a cohort of patients with APS1 using a novel immunoprecipitation assay (24). The aim of the current study was to identify anti-CaSR antibodies in APECED patients that affect the activity of the CaSR, because antibody-induced activation of the receptor could result in low levels of PTH and thus contribute to the clinical picture of hypoparathyroidism in APS1. The effects of patient IgG on the function of the CaSR were assessed by measuring their effects on both Ca2+-dependent extracellular signal-regulated kinase (ERK1/2) phosphorylation and inositol phosphate (IP) accumulation in human embryonic kidney 293 (HEK293) cells expressing the CaSR.
Patients and Methods
Patients and controls
Fourteen patients (seven males, seven females; mean age, 18 yr; range, 10–47 yr) were studied. All patients had Addison’s disease and mucocutaneous candidiasis. Thirteen patients had hypoparathyroidism. Other autoimmune diseases present were: premature ovarian failure, five; alopecia, three; vitiligo, five; type 1 diabetes mellitus, three; autoimmune hypothyroidism, one; and pernicious anemia, two. All patients carried mutations in both alleles of the AIRE gene (3). In our previous study, anti-CaSR antibodies were detected in 12 of these patients using immunoprecipitation assays, whereas the remaining two were negative (24).
Twenty healthy individuals (nine males, 11 females; mean age, 32 yr; range, 24–48 yr) who had no present or past history of autoimmune disorders were the control subjects. No individual had anti-CaSR antibodies when tested in immunoprecipitation assays (24).
The study was approved by the Ethics Committee of Tampere University Hospital, Tampere, Finland, and the APS1 patients participated in the study after obtaining informed consent, given either by the patient or by their parents. Sera were stored at −20 C.
IgG preparation
IgG was isolated from sera by affinity column chromatography using protein G-Sepharose 4 Fast Flow (Amersham Biosciences AB, Uppsala, Sweden) according to the manufacturer’s instructions. Eluted IgG fractions were extensively dialyzed against PBS (pH 7.4; Sigma, Poole, UK) and concentrated using an Amicon Concentrator (Amicon Inc., Beverly, MA). IgG samples were sterilized with a Millex Filter Unit (Millipore Corp., Bedford, MA), and the final concentrations were measured by photometry at 280 nm. All IgG samples were stored at 10 mg/ml at −20 C.
Cell culture
HEK293 cells and HEK293 cells stably expressing the CaSR (HEK293-CaSR) (27) were cultured in Dulbecco’s DMEM medium (Invitrogen, Paisley, UK) containing 4.5 g/liter pyruvate, 10% (vol/vol) fetal calf serum, 100 U/ml penicillin G, 100 μg/ml streptomycin, 2 mm l-glutamine (all from Invitrogen) at 37 C in a 95% humidified atmosphere of 5% CO2. Transfections of HEK293 cells using a calcium phosphate transfection system (Invitrogen) were carried out exactly as previously detailed (24). Plasmid pDEST26/CaSR, containing human CaSR cDNA, was obtained from RZPD (German Resource Centre for Genome Research, Berlin, Germany). Plasmid pcMCHR1 carrying human melanin-concentrating hormone receptor (MCHR1) cDNA is described elsewhere (28).
Cell extracts were prepared from untransfected and transfected HEK293 cells as detailed elsewhere (24). Total protein content of cell extracts was determined using a Bio-Rad Protein Assay Kit (Bio-Rad Laboratories Ltd., Hemel Hempstead, UK). Extracts were diluted where appropriate to contain equivalent amounts of total protein and stored at −80 C until required.
Measurement of CaSR-stimulated inositol-1-phosphate (IP1) accumulation
The response of HEK293-CaSR cells to Ca2+ was assessed by measuring intracellular IP1 accumulation (27). HEK293-CaSR cells were plated in 24-well plates at 4 × 105 cells per well in 500 μl of culture medium and incubated overnight at 37 C. Cell monolayers were washed first with serum-free medium and then with buffer containing 146 mm NaCl, 2.5 mm KCl, 10 mm LiCl2, 0.2 mm MgCl2, 2.8 mm glucose, and 10 mm HEPES (pH 7.4). Cells were subsequently incubated for 60 min at 37 C in 200 μl of buffer alone or containing varying concentrations of Ca2+ (as CaCl2). After treatment, cells were lysed for 30 min at 37 C with 50 μl of 2.5% IP-One ELISA Kit Lysis Reagent (CIS Bio International, Gif-sur-Yvette, France). The accumulation of intracellular IP1 was measured according to an IP-One ELISA Kit (CIS Bio International), an immunoassay based on competition between free IP1 and IP1-horseradish peroxidase (HRP) conjugate for binding to anti-IP1 monoclonal antibody. The results for IP1 accumulation were expressed as: percentage inhibition of IP1-HRP binding = [1 − IP1-HRP binding in stimulated cells/IP1-HRP binding in unstimulated cells] × 100. An increase in IP1 accumulation in the HEK293-CaSR cells is reflected by an increase in the percentage inhibition of IP1-HRP binding.
For measuring the effects of APS1 patient and control IgG on CaSR-stimulated IP1 accumulation, HEK293-CaSR cell monolayers were incubated with IgG at a 1:20 dilution in 100 μl of buffer for 10 min at 37 C. Cells were then incubated for 60 min at 37 C with a further 100 μl of buffer alone or buffer containing Ca2+ before measuring IP1 accumulation. Each experiment included HEK293-CaSR cells stimulated with Ca2+ alone and HEK293-CaSR cells left untreated with IgG and Ca2+. For dilution experiments, IgG samples were tested at dilutions of 1:50, 1:100, 1:200, 1:500, and 1:1000 with Ca2+. To determine the effects of heat treatment, IgG samples were treated at 100 C for 10 min before testing at a dilution of 1:20.
For absorption experiments, IgG samples were incubated with either untransfected or transfected (with pDEST26/CaSR or pcMCHR1) HEK293 cell extract (300 μg of total protein) at 4 C for 16 h in buffer containing 150 mm NaCl, 10 mm Tris-HCl (pH 7.4), 1% (vol/vol) Triton X-100 and Protease Inhibitor Cocktail (Sigma). IgG samples without cell extract were incubated under the same conditions. After absorption, samples were centrifuged at 45,000 × g for 1 h at 4 C, and the supernatants were recovered. For effects upon CaSR-stimulated IP1 accumulation, IgG samples were tested at a 1:20 dilution.
Measurement of CaSR-stimulated ERK1/2 phosphorylation
The response of HEK293-CaSR cells to Ca2+ was assessed by measuring the phosphorylation of ERK1/2 (27). HEK293-CaSR cells were plated in 96-well plates at 1.5 × 104 cells per well in 100 μl of culture medium and incubated overnight at 37 C. Cells were then incubated with serum-free medium overnight. Cell monolayers were washed first with serum-free medium and then with buffer. Cells were subsequently incubated for 60 min at 37 C in 100 μl of buffer alone or buffer containing varying concentrations of Ca2+. After treatment, cells were fixed for 20 min at room temperature with 100 μl of 4% Cell Fixing Buffer according to a Cellular Activation of Signaling ELISA (CASE) Kit (SuperArray Bioscience Corporation, Frederick, MD). The phosphorylation of ERK1/2 was then measured according to a CASE Kit (SuperArray Bioscience Corporation), and the results were expressed as: ratio of phosphorylated ERK1/2:total ERK1/2.
For measuring the effects of APS1 patient and control IgG on CaSR-stimulated ERK1/2 phosphorylation, HEK293-CaSR cell monolayers were incubated with IgG at a 1:20 dilution in 100 μl of buffer for 10 min at 37 C. Cells were then incubated for 60 min at 37 C with a further 100 μl of buffer alone or buffer containing Ca2+ before measuring ERK1/2 phosphorylation. Each experiment included HEK293-CaSR cells stimulated with Ca2+ alone and HEK293-CaSR cells left untreated with IgG and Ca2+.
Statistical analyses
Statistical analyses were performed using unpaired Student’s t tests. The prevalence of antibody reactivity to the CaSR was compared between APS1 patients and controls using Fisher’s exact test for 2 × 2 contingency tables. In all tests, P values <0.05 (two-tailed) were regarded as significant.
Results
Effects of APS1 patient IgG on CaSR-stimulated IP1 accumulation
The response to Ca2+ of the CaSR expressed in HEK293 cells was assessed by measuring intracellular IP1 accumulation (27) as described in Patients and Methods. To determine the effects of IgG from APS1 patients (n = 14) and healthy controls (n = 20) on CaSR function, HEK293-CaSR cells were incubated with IgG samples at a 1:20 dilution before no stimulation or stimulation with various concentrations of Ca2+ (0.5, 1.5, 3.0, and 5.0 mm). Each experiment included HEK293-CaSR cells stimulated with Ca2+ alone and HEK293-CaSR cells left untreated with IgG and Ca2+. IP accumulation was measured using the IP-One ELISA Kit as detailed in Patients and Methods, and the percentage inhibition of IP1-HRP binding was calculated.
Of the 20 control IgG samples analyzed, none had any effect upon the levels of IP1 accumulation when compared with stimulation by Ca2+ alone (Fig. 1). Similar results were obtained for 12 of the 14 APS1 patient IgG samples tested (Fig. 1). In contrast, IgG samples from two APS1 patients (APS1-2 and APS1-15) significantly increased the levels of IP1 accumulation at Ca2+ concentrations of 0.5, 1.5, and 3.0 mm when compared with Ca2+ stimulation alone (P values <0.0001) (Fig. 1). The IgG from these two patients had no effect on IP1 accumulation, however, when no Ca2+ was added to the buffer, indicating that some degree of receptor activation was needed to see the functional effect of the IgG. The results indicated the presence of CaSR-stimulating activity in these two APS1 patient IgG samples. Both of these patients were positive for anti-CaSR binding antibodies (24) and had hypoparathyroidism as part of their APS1.
Figure 1.

Effect of APS1 patient and control IgG on the response of the CaSR to Ca2+ stimulation by measuring IP1 accumulation. Changes in IP1 accumulation were measured in response to no Ca2+ and Ca2+ at 0.5, 1.5, 3.0, and 5.0 mm in HEK293-CaSR cells preincubated with either APS1 patient (n = 14) or control (n = 20) IgG samples at a 1:20 dilution. IP accumulation was measured in the IP-One ELISA Kit as detailed in Patients and Methods. The results show the mean percentage inhibition of IP1-HRP binding (±sd) in three experiments in which HEK293-CaSR cells were preincubated with IgG from APS1 patients 2, 3, 4, 5, controls 1, 2, 3, 4, 5, and no IgG (A); IgG from APS1 patients 2, 6, 7, 8, 9, controls 6, 7, 8, 9, 10, and no IgG (B); IgG from APS1 patients 2, 10, 11,12, 13, controls 11, 12, 13, 14, 15, and no IgG (C); and IgG from APS1 patients 2, 14, 15, controls 16, 17, 18, 19, 20, and no IgG (D).
To determine the effects of diluting IgG on CaSR-stimulated IP1 accumulation, HEK293-CaSR cells were incubated with IgG samples from APS1 patients APS1-2 and APS1-15 (both with CaSR-stimulating activity) and from two controls at dilutions of 1:20, 1:50, 1:100, 1:200, 1:500, and 1:1000 before stimulation with Ca2+ at 1.5 mm. The limiting dilutions for the detection of CaSR-stimulating activity in IgG samples from APS1 patients APS1-2 and APS1-15 was 1:200 (Fig. 2): only IgG dilutions of up to 1/200 were noted to significantly increase the levels of IP1 accumulation when compared with Ca2+ stimulation alone (P values <0.05) (Fig. 2).
Figure 2.

Effect of diluting APS1 patient and control IgG on the response of the CaSR to Ca2+ stimulation. Changes in IP1 accumulation were measured in response to Ca2+ at 1.5 mm in HEK293-CaSR cells preincubated with IgG samples at dilutions of 1:20, 1:50, 1:100, 1:200; 1:500, and 1:1000. IP accumulation was measured in the IP-One ELISA Kit as detailed in Patients and Methods. The results show the mean percentage inhibition of IP1-HRP binding (±sd) of three experiments for IgG from APS1 patients APS1-2 and APS1-15 and from two controls.
To determine the effects of heat treatment of the IgG from APS1 patients APS1-2 and APS1-15 and from two controls on CaSR-stimulated IP1 accumulation, HEK293-CaSR cells were incubated with IgG samples (heat treated and untreated) at a dilution of 1:20 before stimulation with Ca2+ at 1.5 mm. Heat treatment of IgG samples from APS1 patients APS1-2 and APS1-15 before testing significantly reduced the CaSR-stimulating effects of the samples on HEK293-CaSR cells compared with untreated IgG (P < 0.05) (Fig. 3). This result indicates that the effects of the IgG samples on HEK293-CaSR cells were not simply due to contaminating Ca2+ or some other nonspecific, heat-stable factor.
Figure 3.
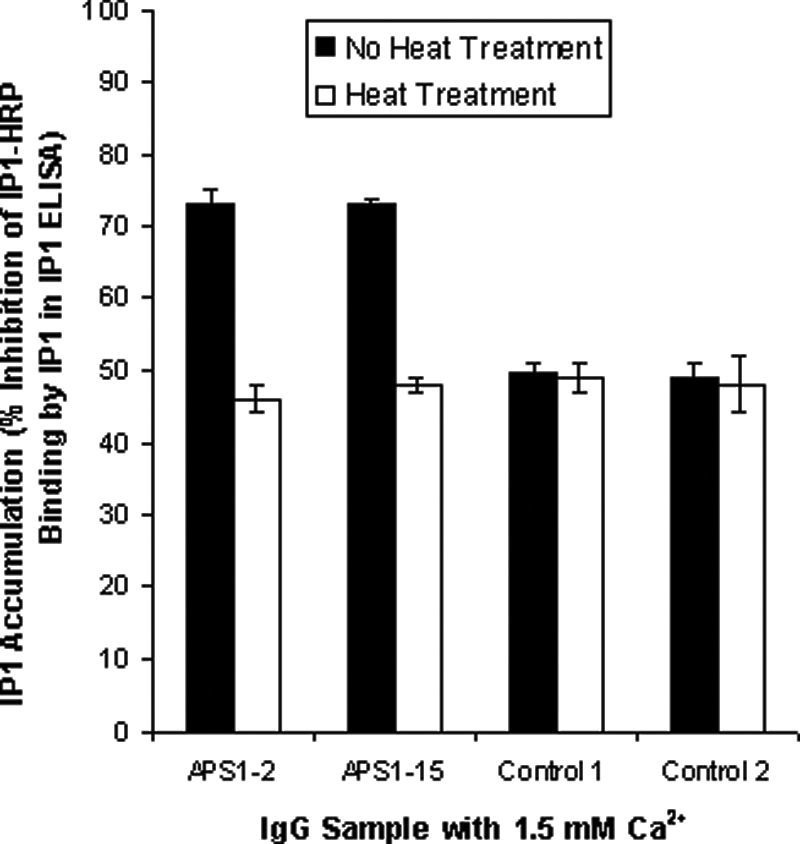
Effects of heat treating APS1 patient and control IgG on the response of the CaSR to Ca2+ stimulation. Changes in IP1 accumulation were measured in response to Ca2+ at 1.5 mm in HEK293-CaSR cells preincubated with IgG samples at a dilution of 1:20. Both heat-treated and untreated IgG samples were tested. IP accumulation was measured in the IP-One ELISA Kit as detailed in Patients and Methods. The results show the mean percentage inhibition of IP1-HRP binding (±sd) of three experiments for heat-treated and untreated IgG from APS1 patients APS1-2 and APS1-15 and from two controls.
To confirm the specificity of the CaSR-stimulating activity in the two IgG samples, absorption experiments were undertaken. Samples of IgG from APS1 patients APS1-2 and APS1-15 and from two controls were preabsorbed with extract from either HEK293 cells or HEK293 cells expressing the CaSR or the MCHR1 resulting from transfection with either pDEST26/CaSR or pcMCHR1, respectively. Preabsorbed and unabsorbed IgG samples were then incubated at a 1:20 dilution with HEK293-CaSR cells before stimulation with 1.5 mm Ca2+. Preabsorption of IgG from APS1 patients APS1-2 and APS1-15 with extract from either HEK293 cells or HEK293 cells expressing MCHR1 did not significantly decrease the stimulatory effect of the samples upon HEK293-CaSR cells compared with unabsorbed IgG samples (P > 0.05) (Fig. 4). In contrast, the stimulatory effect upon HEK293-CaSR cells was significantly reduced in both APS1 patient IgG samples preabsorbed with HEK293 cells expressing the CaSR compared with unabsorbed IgG samples (P < 0.05) (Fig. 4). The absorption experiments indicated that the stimulatory activity present in the IgG samples from both APS1 patients reflected the presence of antibodies that specifically interacted with the CaSR.
Figure 4.

Effect of preabsorbing APS1 patient and control IgG on the response of the CaSR to Ca2+ stimulation. Changes in IP1 accumulation were measured in response to Ca2+ at 1.5 mm in HEK293-CaSR cells preincubated with IgG samples at a dilution of 1:20. IgG samples had been preabsorbed by cell extracts of either HEK293 cells or HEK293 cells transfected with either pDEST26/CaSR or pcMCHR1. Unabsorbed IgG samples were also tested. IP accumulation was measured in the IP-One ELISA Kit as detailed in Patients and Methods. The results show the mean percentage inhibition of IP1-HRP binding (±sd) of three experiments for IgG from APS1 patients APS1-2 and APS1-15 and from two controls.
Effect of APS1 patient IgG on CaSR-stimulated ERK1/2 phosphorylation
The response to Ca2+ of the CaSR expressed in HEK293 cells was assessed by measuring ERK1/2 phosphorylation (27) as detailed in Patients and Methods. To determine the effects of IgG from APS1 patients APS1-2 and APS1-15 on CaSR function with respect to ERK1/2 phosphorylation, HEK293-CaSR cells were incubated with IgG samples at a 1:20 dilution before no stimulation or stimulation with various concentrations of Ca2+ (0.5, 1.5, 3.0, and 5.0 mm). Each experiment included HEK293-CaSR cells stimulated with Ca2+ alone and HEK293-CaSR cells left untreated with either IgG or Ca2+. The phosphorylation of ERK1/2 was measured according to a CASE Kit as in Patients and Methods, and the ratio of phosphorylated ERK1/2:total ERK1/2 was determined. None of the two controls was observed to have an effect upon the levels of ERK1/2 phosphorylation when compared with Ca2+ stimulation alone (Fig. 5). In contrast, IgG samples from the two APS1 patients (APS1-2 and APS1-15) were noted to significantly increase ERK1/2 phosphorylation at Ca2+ concentrations of 0.5 and 1.5 mm when compared with Ca2+ stimulation alone (P values <0.05) (Fig. 5). As with the stimulatory effect of IgG from these two patients on IP1 accumulation, no effect of the IgG was observed in the presence of nominal zero Ca2+. The results indicated the presence of CaSR-stimulating activity in IgG samples from the two APS1 patients.
Figure 5.
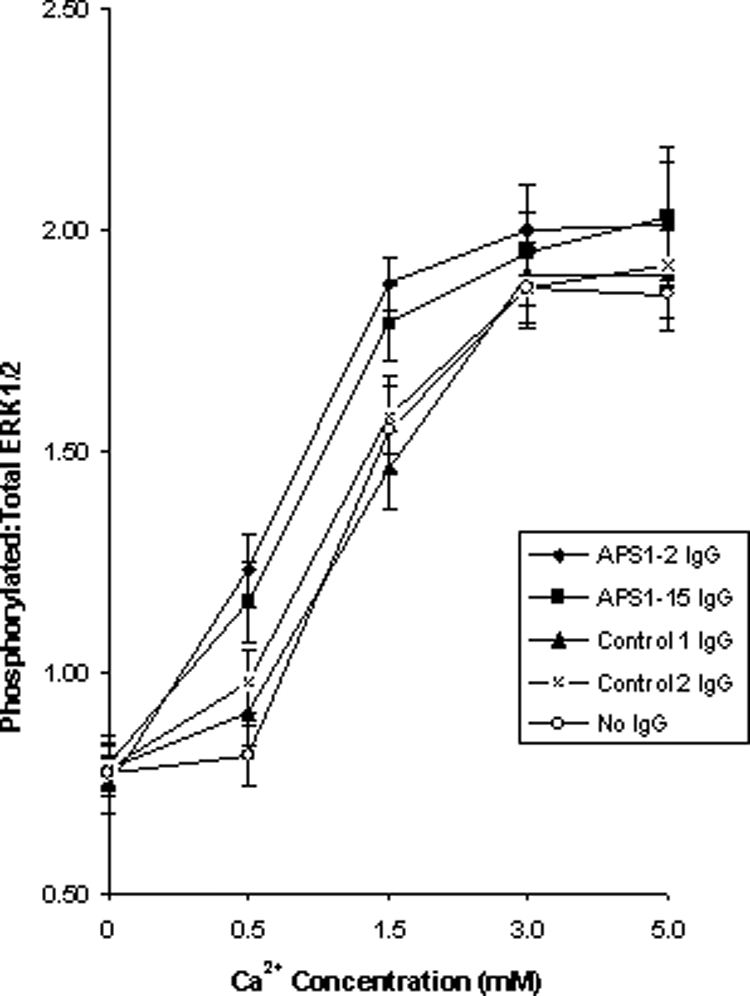
Effect of APS1 patient and control IgG on the response of the CaSR to Ca2+ stimulation by measuring ERK1/2 phosphorylation. Changes in ERK1/2 phosphorylation were measured in response to no Ca2+ and Ca2+ at 0.5, 1.5, 3.0, and 5.0 mm in HEK293-CaSR cells preincubated with either APS1 patient (n = 2) or control (n = 2) IgG samples at a 1:20 dilution. The phosphorylation of ERK1/2 was measured according to a CASE Kit as in Patients and Methods. The results show the mean ratio of ERK1/2 phosphorylation (±sd) of three experiments for preincubation of HEK293-CaSR cells with IgG from APS1 patients APS1-2 and APS1-15, IgG from two controls and no IgG.
Discussion
Previously, we identified anti-CaSR antibodies in patients with APS1 using a novel immunoprecipitation assay (24) supporting the view that the CaSR can be a target for autoantibodies in APECED patients (22,23,25). The aim of the present study was to determine whether anti-CaSR antibodies in APS1 patients were able to modulate the function of the CaSR because such antibodies are more likely to have a direct role in disease pathogenesis than antibodies that simply bind the receptor. Indeed, in several autoimmune disorders, antibodies have been demonstrated to affect the functioning of cell receptors resulting in disease manifestations. For example, thyroid-stimulating antibodies activate the TSH receptor and elicit hyperthyroidism in Graves’ disease by mimicking the binding of TSH, and antibodies directed against the acetylcholine receptor block the acetylcholine-binding site and provoke accelerated receptor degradation causing myasthenia gravis (29,30). In the case of the CaSR, antibody-induced activation could result in low levels of PTH and the resultant hypoparathyroidism that is associated with APS1.
Overall, our results indicated that two of 13 (14%) of the hypoparathyroid APS1 patient IgG samples tested exhibited readily detectable and statistically significant activation of the CaSR. Although such antibodies were not found at a significantly increased prevalence in the APS1 patient group when compared with healthy controls in our comparatively small sample size (P = 0.16), nonetheless these results are compatible with the appearance of stimulating anti-CaSR antibodies in a subset of patients. The anti-CaSR binding antibodies previously demonstrated in the remaining 10 APS1 patients (24) may bind at a site on the receptor that has no effect upon functionality. An analogy might be with the detection of stimulatory antibodies in only a subset of the entire patient population with TSH receptor autoantibodies (29). Alternatively, CaSR-modulating activities in these samples may be too low to detect in the assays used in this study.
Of interest, the two APS1 patient IgG samples that stimulated IP1 accumulation and ERK1/2 activity did so only in the presence of Ca2+ in the experimental buffer. This observation is similar to that seen with the so-called type 2, calcimimetic CaSR activators (31). The latter require the presence of physiological levels of extracellular Ca2+ to activate the CaSR. In the cases of both the activating IgG studied here and the calcimimetics, binding of antibody or calcimimetic may act in an allosteric manner to favor the active conformation of the receptor, but Ca2+ must be present to initiate productive signal transduction.
In addition to APS1, anti-CaSR antibodies have been demonstrated in individuals affected by isolated autoimmune hypoparathyroidism (AH) (22,23,27,32) and autoimmune hypocalciuric hypercalcemia (AHH) (33,34,35). With respect to CaSR function, anti-CaSR activating and blocking antibodies have only been detected previously in two patients with AH and five patients with AHH, respectively (27,33,34). In addition, a unique anti-CaSR antibody from an individual with AHH has been reported to both stimulate the Ca2+-dependent accumulation of inositol phosphates and inhibit the Ca2+-dependent phosphorylation of ERK1/2 (35). The anti-CaSR antibodies detected in APS1 patients in this study were similar in their effects upon Ca2+-dependent receptor activity to those identified earlier in individuals with AH (27): IgG samples increased both Ca2+-dependent IP accumulation and ERK1/2 phosphorylation in HEK293 cells expressing the receptor.
An important implication of the present results is that although the majority of APS1 patients do not have CaSR-stimulating antibodies, there may be a small but substantial minority of patients in whom the hypoparathyroid state is the result of functional suppression of the parathyroid glands rather than their irreversible destruction. The documentation of the inactivating anti-CaSR antibodies in AHH proves that the presence of anti-CaSR antibodies need not produce destruction of the parathyroid glands (33). In addition, our earlier demonstration of the presence of activating antibodies to the CaSR in one patient with transient hypoparathyroidism as well as in a second hypoparathyroid patient with a morphologically intact parathyroid gland despite many years of hypoparathyroidism proves that activating antibodies also need not irreversibly destroy the parathyroids (27). In such cases, it is possible that a calcilytic CaSR inhibitor might be of diagnostic and/or therapeutic utility. Because such agents acutely stimulate PTH secretion by blocking the inhibitory effect of calcium on the CaSR, demonstrating a PTH response in a hypoparathyroid subject administered a calcilytic could document the presence of residual functioning parathyroid tissue. Moreover, if such functioning tissue were present, the calcilytic might be of therapeutic utility if it were able to block the suppressive effect of the antibody on parathyroid function.
To summarize, the present study is the first to define specific anti-CaSR antibodies in patients with APS1 that simulate the receptor, albeit in only two of 13 such patients with hypoparathyroidism, and also provides further evidence for the CaSR as a humoral immune target in APECED.
Acknowledgments
We thank Professor J. Perheentupa (The Hospital for Children and Adolescents, Helsinki University Hospital, FIN-00029 Helsinki, Finland) for kindly supplying the APS1 patient clinical details.
Footnotes
This work was supported by a European Union Framework Program 6 Grant (EURAPS; to A.P.W.). E.M.B. was supported by National Institutes of Health Grants DK67111 and DK078331.
Disclosure Summary: E.H.K., N.G.G., K.J.E.K., and P.F.W. have nothing to declare. A.P.W. has received lecture fees from Merck. E.M.B. has a financial interest in the calcimimetic, Sensipar, and has given lectures for Athena Diagnostics, Inc.
First Published Online October 16, 2009
For editorial see page 4655
Abbreviations: AH, Autoimmune hypoparathyroidism; AHH, hypocalciuric hypercalcemia; AIRE, autoimmune regulator; APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; APS1, autoimmune polyendocrine syndrome type 1; CaSR, calcium-sensing receptor; ERK1/2, extracellular signal-regulated kinase; HEK293, human embryonic kidney 293; HRP, horse-radish peroxidase; IP, inositol phosphate; IP1, inositol-1-phosphate; MCHR1, melanin-concentrating hormone receptor 1.
References
- Perheentupa J 2006 Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab 91:2843–2850 [DOI] [PubMed] [Google Scholar]
- Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N 1997 Positional cloning of the APECED gene. Nat Genet 17:393–398 [DOI] [PubMed] [Google Scholar]
- The Finnish-German APECED Consortium 1997 An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 17:399–403 [DOI] [PubMed] [Google Scholar]
- Gibson TJ, Ramu C, Gemünd C, Aasland R 1998 The APECED polyglandular autoimmune syndrome protein, AIRE-1, contains the SAND domain and is probably a transcription factor. Trends Biochem Sci 23:242–244 [DOI] [PubMed] [Google Scholar]
- Aasland R, Gibson TJ, Stewart AF 1995 The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci 20:56–59 [DOI] [PubMed] [Google Scholar]
- Neufeld M, Maclaren N, Blizzard R 1980 Autoimmune polyglandular syndromes. Pediatr Ann 9:154–162 [PubMed] [Google Scholar]
- Krohn K, Uibo R, Aavik E, Peterson P, Savilahti K 1992 Identification by molecular cloning of an autoantigen associated with Addison’s disease as steroid 17 α-hydroxylase. Lancet 339:770–773 [DOI] [PubMed] [Google Scholar]
- Winqvist O, Karlsson FA, Kämpe O 1992 21-Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet 339:1559–1562 [DOI] [PubMed] [Google Scholar]
- Uibo R, Aavik E, Peterson P, Perheentupa J, Aranko S, Pelkonen R, Krohn KJ 1994 Autoantibodies to cytochrome P450 enzymes P450scc, P450c17, and P450c21 in autoimmune polyglandular disease types I and II and in isolated Addison’s disease. J Clin Endocrinol Metab 78:323–328 [DOI] [PubMed] [Google Scholar]
- Tuomi T, Björses P, Falorni A, Partanen J, Perheentupa J, Lernmark A, Miettinen A 1996 Antibodies to glutamic acid decarboxylase and insulin-dependent diabetes in patients with autoimmune polyendocrine syndrome type I. J Clin Endocrinol Metab 81:1488–1494 [DOI] [PubMed] [Google Scholar]
- Gylling M, Tuomi T, Björses P, Kontiainen S, Partanen J, Christie MR, Knip M, Perheentupa J, Miettinen A 2000 β-cell autoantibodies, human leukocyte antigen II alleles, and type 1 diabetes in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab 85:4434–4440 [DOI] [PubMed] [Google Scholar]
- Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J, Husebye E, Kämpe O, Rorsman F 1998 Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet 352:279–283 [DOI] [PubMed] [Google Scholar]
- Hedstrand H, Ekwall O, Haavik J, Landgren E, Betterle C, Perheentupa J, Gustafsson J, Husebye E, Rorsman F, Kämpe O 2000 Identification of tyrosine hydroxylase as an autoantigen in autoimmune polyendocrine syndrome type I. Biochem Biophys Res Commun 267:456–461 [DOI] [PubMed] [Google Scholar]
- Gebre-Medhin G, Husebye ES, Gustafsson J, Winqvist O, Goksøyr A, Rorsman F, Kämpe O 1997 Cytochrome P450IA2 and aromatic L-amino acid decarboxylase are hepatic autoantigens in autoimmune polyendocrine syndrome type I. FEBS Lett 412:439–445 [DOI] [PubMed] [Google Scholar]
- Hedstrand H, Ekwall O, Olsson MJ, Landgren E, Kemp EH, Weetman AP, Perheentupa J, Husebye E, Gustafsson J, Betterle C, Kämpe O, Rorsman F 2001 The transcription factors SOX9 and SOX10 are vitiligo autoantigens in autoimmune polyendocrine syndrome type 1. J Biol Chem 276:35390–35395 [DOI] [PubMed] [Google Scholar]
- Oftedal BE, Wolff AS, Bratland E, Kämpe O, Perheentupa J, Myhre AG, Meager A, Purushothaman R, Ten S, Husebye ES 2008 Radioimmunoassay for autoantibodies against interferon omega; its use in the diagnosis of autoimmune polyendocrine syndrome type I. Clin Immunol 129:163–169 [DOI] [PubMed] [Google Scholar]
- Kisand K, Link M, Wolff AS, Meager A, Tserel L, Org T, Murumägi A, Uibo R, Willcox N, Trebusak Podkrajsek K, Battelino T, Lobell A, Kämpe O, Lima K, Meloni A, Ergun-Longmire B, Maclaren NK, Perheentupa J, Krohn KJ, Scott HS, Husebye ES, Peterson P 2008 Interferon autoantibodies associated with AIRE deficiency decrease the expression of IFN-stimulated genes. Blood 112:2657–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posillico JT, Wortsman J, Srikanta S, Eisenbarth GS, Mallette LE, Brown EM 1986 Parathyroid cell surface autoantibodies that inhibit parathyroid hormone secretion from dispersed human parathyroid cells. J Bone Miner Res 1:475–483 [DOI] [PubMed] [Google Scholar]
- Brandi ML, Aurbach GD, Fattorossi A, Quarto R, Marx SJ, Fitzpatrick LA 1986 Antibodies to bovine parathyroid cells in autoimmune hypoparathyroidism. Proc Natl Acad Sci USA 83:8366–8369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattorossi A, Aurbach GD, Sakaguchi K, Cama A, Marx SJ, Streeten EA, Fitzpatrick LA, Brandi ML 1988 Anti-endothelial cell antibodies: detection and characterisation in sera from patients with autoimmune hypoparathyroidism. Proc Natl Acad Sci USA 85:4015–4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimohammadi M, Björklund P, Hallgren A, Pöntynen N, Szinnai G, Shikama N, Keller MP, Ekwall O, Kinkel SA, Husebye ES, Gustafsson J, Rorsman F, Peltonen L, Betterle C, Perheentupa J, Akerström G, Westin G, Scott HS, Holländer GA, Kämpe O 2008 Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med 358:1018–1028 [DOI] [PubMed] [Google Scholar]
- Li Y, Song YH, Rais N, Connor E, Schatz D, Muir A, Maclaren N 1996 Autoantibodies to the extracellular domain of the calcium sensing receptor in patients with acquired hypoparathyroidism. J Clin Invest 97:910–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Ploix C, Orgiazzi J, Desbos A, Moreira A, Vidal H, Monier JC, Bienvenu J, Fabien N 2004 Calcium-sensing receptor autoantibodies are relevant markers of acquired hypoparathyroidism. J Clin Endocrinol Metab 89:4484–4488 [DOI] [PubMed] [Google Scholar]
- Gavalas NG, Kemp EH, Krohn KJ, Brown EM, Watson PF, Weetman AP 2007 The calcium-sensing receptor is a target of autoantibodies in patients with autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab 92:2107–2114 [DOI] [PubMed] [Google Scholar]
- Pelletier-Morel L, Fabien N, Mouhoub Y, Boitard C, Larger E 2008 Hyperparathyroidism in a patient with autoimmune polyglandular syndrome. Intern Med 47:1911–1915 [DOI] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC 1993 Cloning and characterisation of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366:575–580 [DOI] [PubMed] [Google Scholar]
- Kifor O, McElduff A, LeBoff MS, Moore Jr FD, Butters R, Gao P, Cantor TL, Kifor I, Brown EM 2004 Activating antibodies to the calcium-sensing receptor in two patients with autoimmune hypoparathyroidism. J Clin Endocrinol Metab 89:548–556 [DOI] [PubMed] [Google Scholar]
- Kemp EH, Waterman EA, Hawes BE, O'Neill K, Gottumukkala RV, Gawkrodger DJ, Weetman AP, Watson PF 2002 The melanin-concentrating hormone receptor 1, a novel target of autoantibody responses in vitiligo. J Clin Invest 109:923–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weetman AP, McGregor AM 1994 Autoimmune thyroid disease: further developments in our understanding. Endocr Rev 15:788–830 [DOI] [PubMed] [Google Scholar]
- Hoedemaekers AC, van Breda Vriesman PJ, De Baets MH 1997 Myasthenia gravis as a prototype autoimmune receptor disease. Immunol Res 16:341–354 [DOI] [PubMed] [Google Scholar]
- Nemeth EF 2002 Pharmacological regulation of parathyroid hormone secretion. Curr Pharm Des 8:2077–2087 [DOI] [PubMed] [Google Scholar]
- Goswami R, Brown EM, Kochupillai N, Gupta N, Rani R, Kifor O, Chattopadhyay N 2004 Prevalence of calcium sensing receptor autoantibodies in patients with sporadic idiopathic hypoparathyroidism. Eur J Endocrinol 150:9–18 [DOI] [PubMed] [Google Scholar]
- Kifor O, Moore Jr FD, Delaney M, Garber J, Hendy GN, Butters R, Gao P, Cantor TL, Kifor I, Brown EM, Wysolmerski J 2003 A syndrome of hypocalciuric hypercalcemia caused by autoantibodies directed at the calcium-sensing receptor. J Clin Endocrinol Metab 88:60–72 [DOI] [PubMed] [Google Scholar]
- Pallais JC, Kifor O, Chen YB, Slovik D, Brown EM 2004 Acquired hypocalciuric hypercalcemia due to autoantibodies against the calcium-sensing receptor. N Engl J Med 351:362–369 [DOI] [PubMed] [Google Scholar]
- Makita N, Sato J, Manaka K, Shoji Y, Oishi A, Hashimoto M, Fujita T, Iiri T 2007 An acquired hypocalciuric hypercalcemia autoantibody induces allosteric transition among active human Ca-sensing receptor conformations. Proc Natl Acad Sci USA 104:5443–5448 [DOI] [PMC free article] [PubMed] [Google Scholar]