

Abstract
By activating the Toll-like receptor 4-nuclear factor-κB signal transduction pathway, the bacterial endotoxin lipopolysaccharide (LPS) induces anorexia, weight loss, fever, and other components of the sickness response. By comparison, the hormones leptin and insulin cause anorexia without sickness via a central mechanism involving the phosphatidylinositol-3 kinase signaling pathway. In the current study, we investigated whether a common Toll-like receptor 4 and phosphatidylinositol-3 kinase signaling intermediate, atypical protein kinase Cζ/λ (aPKC), contributes to changes of energy balance induced by these stimuli. Immunohistochemistry analysis revealed that aPKC is expressed in the arcuate and paraventricular nuclei of the hypothalamus, key sites of leptin, insulin, and LPS action. Although administration of LPS, insulin, and leptin each acutely increased hypothalamic aPKC activity at doses that also reduce food intake, LPS treatment caused over 10-fold greater activation of hypothalamic a PKC signaling than that induced by leptin or insulin. Intracerebroventricular pretreatment with an aPKC inhibitor blocked anorexia induced by LPS but not insulin or leptin. Similarly, LPS-induced hypothalamic inflammation (as judged by induction of proinflammatory cytokine gene expression) and neuronal activation in the paraventricular nucleus (as judged by c-fos induction) were reduced by central aPKC inhibition. Although intracerebroventricular aPKC inhibitor administration also abolished LPS-induced fever, it had no effect on sickness-related hypoactivity or weight loss. We conclude that although hypothalamic aPKC signaling is not required for food intake inhibition by insulin or leptin, it plays a key role in inflammatory anorexia and fever induced by LPS.
Inhibition of atypical protein kinase C activity in the brain prevents fever and anorexia induced by lipopolysaccharide.
Immune activation by systemic illness elicits a state of profound negative energy balance that may be adaptive in the acute response to infection (1) but can contribute to excess morbidity and mortality if prolonged (2,3,4). Sickness responses include behavioral and autonomic features such as fever, anorexia, weight loss, and hypoactivity. The mechanisms underlying these coordinated responses involve the conversion of immune-generated signals to neural outputs and their subsequent integration with adiposity-related inputs that govern energy homeostasis such as leptin and insulin (5,6,7). This convergence of metabolic and inflammatory signals occurs in various hypothalamic regions including the arcuate and paraventricular nuclei (PVN).
Progress toward clarifying mechanisms underlying sickness responses has been facilitated by animal studies using the bacterial endotoxin lipopolysaccharide (LPS). Systemic LPS administration in rodents recapitulates the metabolic alterations of sickness by binding to Toll-like receptor 4 (TLR4), which in turn activates the transcription factor nuclear factor-κB (NF-κB) and consequent production of numerous inflammatory mediators including cytokines IL-1β, IL-6, and TNF-α (7,8,9). Current evidence suggests that central nervous system (CNS) immune responses are required for LPS-induced anorexia and hypothalamic inflammation (10,11). For example, mice deficient in TLR4 or the TLR4-associated signaling molecule MyD88 display near-complete insensitivity to LPS, even when transplanted with wild-type donor bone marrow that restores the peripheral immune response. Although CNS administration of cytokine antagonists can curtail features of sickness (8,12), no single cytokine has proven critical for the full sickness response (8), suggesting a variety of inflammatory mediators may contribute.
Protein kinase Cζ and -λ (aPKC) are the two highly homologous members of the atypical subfamily of PKC isoforms that differ from classical PKCs in their calcium and diacylglycerol-independent regulation (13,14). The insulin and leptin signaling intermediate phosphatidylinositol-3 kinase (PI3K) is a major regulator of aPKC activity (13). Insulin stimulation of PI3K activates aPKC (along with Akt) to facilitate Glut4 translocation to the plasma membrane in muscle and fat (13). In immune signaling, aPKC can modulate the NF-κB pathway through phosphorylation of inhibitory-κBα (IκBα) and the p65 subunit of NF-κB itself, actions that enhance NF-κB signaling (15,16). Through binding to the scaffolding protein p62, aPKC can also interact with receptor-interacting protein and TNF receptor-associated factor 6, signaling modules immediately downstream of both TLR4 and the TNF-α receptor (17,18). Lastly, in macrophages, LPS induction of signaling downstream of TLR4 is dependent on aPKC activation (19,20). Thus, aPKC is uniquely positioned to mediate convergent intracellular immune and metabolic responses.
The evidence for aPKC involvement in inflammatory signaling has been generated primarily in cultured cells with reduction of aPKC signaling by either dominant-negative aPKC overexpression or administration of a specific peptide inhibitor that mimics the autoinhibitory pseudosubstrate domain of aPKC (21). Comparatively, in vivo studies have yielded less consistent results. Whereas PKCλ knockout mice die in early embryogenesis from cell polarity defects (22), cell lines from PKCλ knockout chimeras show no deficits in response to either LPS or TNF-α (22). In contrast, B-lymphocyte NF-κB signaling depends on intact aPKC activity (23,24). Although both PKCζ and -λ are expressed in the brain (25,26), little is known about either their regulation by PI3K or TLR4 or their role in CNS inflammation and its effect on energy homeostasis.
In this study, we sought to clarify mechanisms of LPS-induced sickness in the rat by focusing on the role of aPKC in CNS inflammatory signaling. We demonstrate expression of aPKC protein in the hypothalamus that is strongly up-regulated by LPS. Furthermore, using a specific inhibitor of aPKC, we demonstrate that LPS-mediated neuronal activation and hypothalamic cytokine generation require aPKC activity. In cultured hypothalamic neurons, we find that the action of LPS to induce NF-κB target genes similarly requires aPKC. Finally, we show in rats that most, but not all, sickness responses to LPS are dependent on intact aPKC enzymatic activity in the CNS.
Materials and Methods
Animals
Male Wistar rats weighing 250–300 g (Harlan, Indianapolis, IN) were individually housed in ventilated Plexiglass cages in a temperature-controlled room with 12-h light, 12-h dark cycles. Water and standard rat chow were available ad libitum except where noted. All experiments involved groups of weight-matched animals. Where indicated, rats were stereotaxically implanted with 26-gauge cannulae (Plastics One, Roanoke, VA) in the third cerebral ventricle (3V) by established methods (27). Intracerebroventricular (icv) injections used 2-min infusions of 2 μl. All study protocols were approved by the Animal Care Committees at the University of Washington and University of Cincinnati and conformed to standards in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996).
Reagents
These studies used leptin (Dr. A. F. Parlow, National Hormone and Peptide Program, Torrance, CA), insulin (HumulinR; Eli Lilly, Indianapolis, IN), LPS (Escherichia coli serotype 055:B5; Sigma, St. Louis, MO), myristoylated aPKC pseudosubstrate inhibitor (INH) (Invitrogen, Carlsbad, CA), and the PI3K inhibitor, LY294002 (Calbiochem, La Jolla, CA).
Animal protocols
Protocol 1: effect of LPS, leptin, and insulin on hypothalamic aPKC activity
Four-hour-fasted rats (n = 9/group) received either ip saline (volume 0.75 ml) or LPS (50 μg/kg) (the minimum dose that reliably induces both hypothalamic inflammation and anorexia) and were euthanized 2 h later by decapitation after exposure to inhaled CO2. An approximately 200-mg rectangular block of mediobasal hypothalamus defined caudally by the mammillary bodies, rostrally by the optic chiasm, laterally by the optic tracts, and superiorly by the apex of the hypothalamic third ventricle (28) was excised for aPKC activity measurements.
Separately, overnight-fasted, 3V-cannulated rats (n = 8–12/group) received icv injection of leptin (10 μg), insulin (10 mU), or saline followed 2 h later by the killing of the animals, extraction of hypothalamic tissue, and processing of lysates for both Western blot and aPKC activity assay.
Western analyses were conducted as described (29) using the following antibodies: 1) rabbit anti-PKCζ/λ (Santa Cruz Biotechnologies, Santa Cruz, CA); 2) rabbit anti-Akt (Millipore, Billerica, MA); and 3) rabbit anti-phospho-serine-473-Akt (New England BioLabs, Ipswich, MA). An in vitro assay for aPKC kinase activity was performed as described (29). Briefly, aPKC protein immunoprecipitated with the rabbit anti-PKCζ/λ antibody (Santa Cruz Biotechnologies) was collected on Sepharose-AG beads (Santa Cruz Biotechnologies) and incubated for 8 min at 30 C in buffer containing 50 mm Tris/HCl (pH 7.5), 100 μm Na3VO4, 100 μm Na4P2O7, 1 mm NaF, 100 μm phenylmethylsulfonyl fluoride, 4 μg phosphatidylserine (Sigma), 50 μm [γ-32P]ATP (PerkinElmer, Waltham, MA), 5 mm MgCl2 and, as substrate, 40 μm serine analog of the PKC-ε pseudosubstrate (Invitrogen). After incubation, 32P-labeled substrate was trapped on P-81 filter papers and counted. Measurements were normalized to saline controls.
Protocol 2: effect of aPKC inhibition on LPS-induced neuronal activation in the PVN
We first identified a dose of aPKC pseudosubstrate INH that inhibits hypothalamic aPKC activity. Overnight-fasted, 3V-cannulated rats (n = 6/group) received icv INH (2 nmol) or saline pretreatment 1 h before icv insulin (10 mU) or saline, and hypothalamic tissue was processed for aPKC activity as described above. Whereas hypothalamic aPKC activity did not differ between INH/saline and saline/saline groups, INH pretreatment abolished insulin-induced enhancement of aPKC activity (data not shown). Therefore, the 2 nmol dose of INH was used in all icv studies.
3V-cannulated rats (n = 4–6/group) fasted 4 h were pretreated with icv INH (2 nmol) or saline 1 h before ip LPS (50 μg/kg) or saline. Two hours after ip injection, animals were perfused via cardiac puncture with 4% paraformaldehyde/PBS. Brains were removed, postfixed overnight, equilibrated in 25% sucrose, and snap frozen on dry ice-cooled isopentane for c-fos immunohistochemistry as described below.
Protocol 3: effect of INH on LPS-induced cytokine gene expression
3V-cannulated rats (n = 6/group) fasted 4 h received icv INH (2 nmol) or saline 1 h before ip LPS (100 μg/kg) or saline. Two hours later, medial hypothalamic blocks and spleens were processed as above for proinflammatory cytokine mRNA levels.
Protocol 4: effect of INH on LPS-induced sickness responses
Temperature and ambulatory activity.
One week after ip temperature transponder implantation (MiniMitter, Sun River, OR), 3V-cannulated rats (n = 6/group) received icv INH (2 nmol) or saline 1 h before ip LPS (100 μg/kg) or saline given at dark cycle onset. The rats were placed in individual metabolic cages (Columbus Instruments, Columbus, OH) equipped for continuous measurements of core body temperature and ambulatory activity (determined from the number of adjacent infrared beam breaks).
Food intake and body weight.
Four-hour-fasted, 3V-cannulated rats (n = 8/group) received icv INH (2 nmol) or saline 1 h before ip LPS (50 μg/kg) or saline at dark cycle onset. Body weight and food intake were measured after 4 and 24 h.
Protocol 5: effect of PI3K inhibition on LPS-induced anorexia
3V-cannulated rats (n = 8/group) fasted 4 h received icv LY294002 (1 nmol) or vehicle (5% dimethylsulfoxide) 1 h before ip LPS (50 μg/kg) or saline at dark cycle onset. Body weight and cumulative food intake were measured after 4 and 24 h.
Protocol 6: effect of INH on leptin-induced anorexia
Protocol 4 was followed with icv leptin (4 μg) or vehicle (10 mm NaHCO3) substituted for ip injections.
Protocol 7: effect of INH on insulin-induced anorexia
Protocol 4 was followed using male Long-Evans rats (Harlan) with icv insulin (10 mU) or saline given 5 h before dark cycle onset. The pharmacological dose of insulin was chosen because it reliably generates anorexia that can be blocked by LY294002 (28).
Immunohistochemistry
Coronal sections (14 μm) through the hypothalamus were processed for aPKC and c-fos immunoreactivity using standard immunohistochemical procedures. Sections blocked in 5% normal goat or donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA) were incubated overnight at 4 C with rabbit anti-PKCζ/λ (1:1000), goat anti-PKCλ (1:500; Santa Cruz Biotechnologies), or rabbit anti-c-fos (Ab5, 1:5000; Oncogene, San Diego, CA). Biotinylated goat antirabbit or donkey antigoat IgG secondary antibody, ABC reagent, and diaminobenzidine substrate (Vector Laboratories, Burlingame, CA) were used per the manufacturer’s instructions. Images were captured on an Eclipse E600 upright microscope (Nikon, Tokyo, Japan) equipped with a Spot RT color digital camera (Diagnostic Instruments, Sterling Heights, MI). c-fos-positive cells were quantified on eight anatomically matched sections at 100-μm intervals through the full rostrocaudal extent of the PVN.
Cell culture
N-43/5 cells, a hypothalamic cell line derived from melanocortin neurons (30), were cultured in monolayer in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (HyClone Laboratories, Logan, UT), 20 mm glucose, and penicillin/streptomycin and maintained at 37 C in an atmosphere of 5% CO2. Cells were pretreated for 1 h with medium containing INH (10 or 30 μm) or medium alone followed by incubation for 1 h with or without LPS (1 μg/ml). Real-time PCR was performed on mRNAs extracted using Trizol (Sigma). Cell viability was assessed using Trypan blue exclusion.
Real-time PCR
Splenic, hypothalamic, and cell culture RNA extraction, quantification, and reverse transcription were performed as described (10). Real-time PCR for IκBα, IL-6, IL-1β, TNF-α, peroxisome proliferator and activator-γ (PPAR-γ), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was conducted on an ABI prism 7900HT using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA) using the following primers: IκBα forward, 5′-TGCCTGGCCAGTGTAGCAGTCTT-3′, reverse, 5′-CAAAGTCACCAAGTGCTCCACGAT-3′; IL-6 forward, 5′-CAGAGGATACCACCCACAACAGA-3′, reverse, 5′-CAGTGCATCATCGCTGTTCATACA-3′; IL-1β forward, 5′-TACAAGGAGAGACAAGCAACGACA-3′, reverse, 5′-GATCCACACTCTCCAGCTGCA-3′; TNF-α forward, 5′-GCTCCCTCTCATCAGTTCCA-3′, reverse, 5′-CTCCTCTGCTTGGTGGTTTG-3′; PPAR-γ forward, 5′-AGGCCGAGAAGGAGAAGCTGTTG-3′, reverse, 5′-TGGCCACCTCTTTGCTCTGCTC-3′; GAPDH forward, 5′-AACGACCCCTTCATTGAC-3′, reverse, 5′-TCCACGACATACTCAGCAC-3′. Expression levels analyzed with the Sequence Detection System software (Applied Biosystems) were normalized to GAPDH mRNA content then expressed as fold relative to saline or media alone controls.
Statistical analysis
Data are presented as mean ± sem. For between-subjects comparisons, statistical significance was determined by unpaired two-tailed t tests or two-way ANOVA using least significant differences post hoc comparisons. Temperature data were analyzed by both repeated measures ANOVA and two-way ANOVA on 3-h binned data. P < 0.05 was considered statistically significant.
Results
Expression of aPKC in the hypothalamus
To characterize the hypothalamic distribution of aPKC in male Wistar rats, we performed an immunohistochemical analysis of aPKC protein expression using a polyclonal antibody that recognizes both aPKC isoforms. Immunoreactive aPKC protein was abundant in neurons of the arcuate nucleus (Fig. 1A) as well as the PVN, in which it was restricted primarily to the medial parvocellular region (Fig. 1B) (26). Consistent with previous studies (25), aPKC protein was localized predominantly in the cytoplasmic compartment of immunopositive neurons (Fig. 1, A and B).
Figure 1.
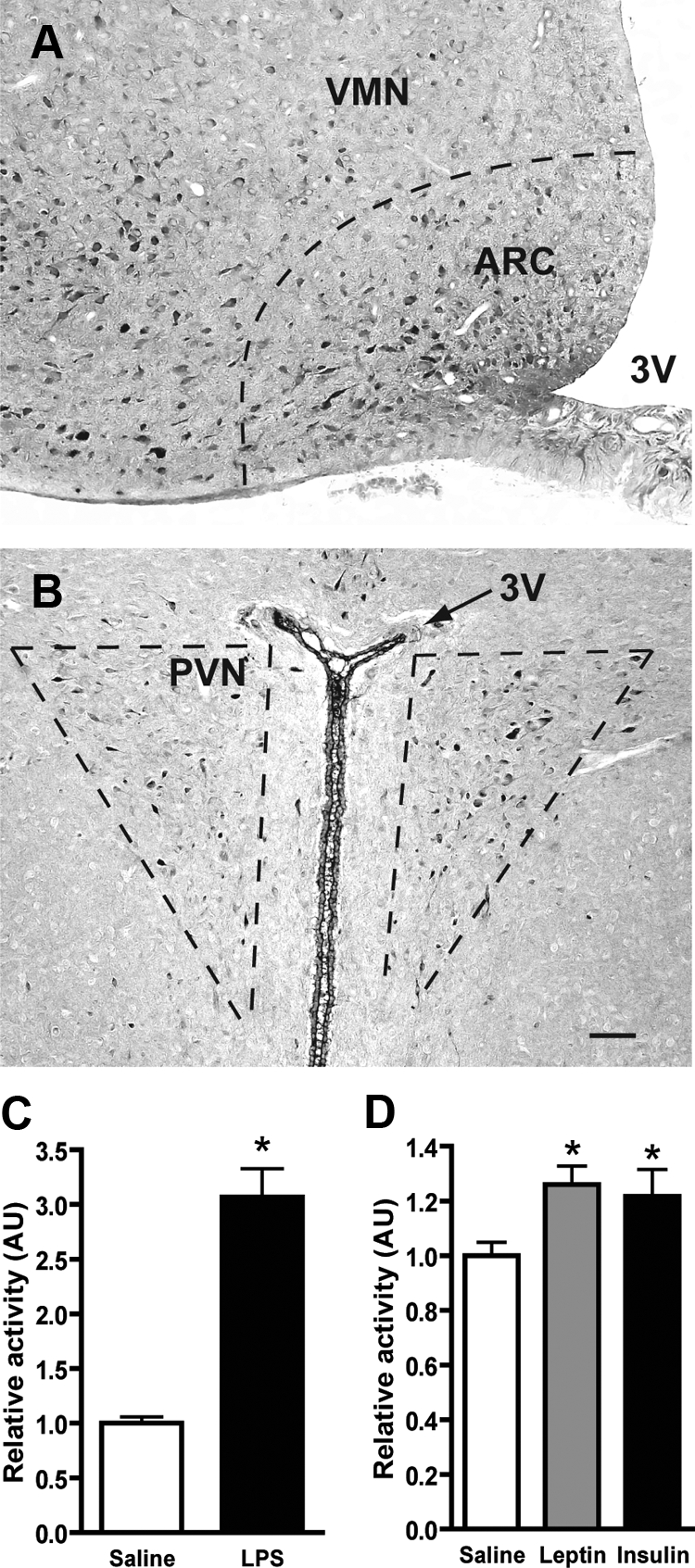
Localization of aPKC in hypothalamus and response to LPS, leptin, and insulin. Representative images from frozen sections of perfused adult male rat brain stained with a polyclonal aPKC antibody. A, In the midcaudal region of the ventral hypothalamus, aPKC expression is largely restricted to neurons of the arcuate nucleus (ARC). Staining is also observed in the ventrolateral portion of the neighboring ventromedial nucleus (VMN). B, More restricted expression is seen in the PVN within the medial parvocellular region. Scale bar, 100 μm for both images. C, After a 4-h fast, rats (n = 9/group) were injected ip with saline or LPS (50 μg/kg) followed 2 h later by the animals being killed. In vitro aPKC kinase activity was determined on mediobasal hypothalamus extracts immunoprecipitated with a polyclonal aPKC antibody. Data (mean + sem) was normalized to saline-injected controls. *, P < 0.001. D, After a 4-h fast, 3V-cannulated rats (n = 8–12/group) were injected icv with saline, leptin (10 μg), or insulin (10 mU) followed by the animals being euthanized at 30, 45, and 20 min, respectively. aPKC kinase activity was determined as in C. *, P < 0.05 relative to saline control. AU, Arbitrary units.
A series of control experiments verified the specificity of the staining including preimmune serum-treated slides showing no staining, a highly similar pattern of immunoreactivity with a PKCλ-specific polyclonal antibody and Western blot analysis of adenovirally expressed PKC isoforms showing specific binding to the expected aPKC bands (data not shown).
Induction of hypothalamic aPKC activity by LPS, leptin, and insulin
To determine whether LPS activated hypothalamic aPKC at doses that cause anorexia and other sickness responses, we measured aPKC enzymatic activity in the hypothalamus of rats 2 h after ip injection of LPS (50 μg/kg). Relative to rats receiving ip saline, LPS-treated animals responded with a 3-fold increase in hypothalamic aPKC activity (Fig. 1C; P < 0.001) with no change in the total amount of aPKC protein (data not shown). By comparison, although both insulin and leptin stimulated hypothalamic aPKC activity after icv injection (∼23% increase for both; Fig. 1D; P < 0.05), this effect was much smaller than was seen after ip LPS. In the same samples, icv insulin doubled the level of phosphorylated Akt, another target of PI3K (data not shown).
Role of aPKC in LPS-induced neuronal activation in the PVN requires aPKC activity
The effector arm of the LPS sickness response includes PVN neurons whose activation can be measured by induction of the early response gene, c-fos (31). To assess the role of aPKC in LPS-mediated PVN neuron activation, we performed pretreatment icv injections of either saline vehicle or a cell-permeable pseudosubstrate inhibitor (INH) specific for aPKC (21) followed by ip administration of either LPS or saline and measured the c-fos response in the PVN. As expected, PVN c-fos expression was minimal in ip saline-injected controls whether they received icv saline (12 ± 3; Fig. 2, A and E) or icv INH pretreatment (22 ± 4; Fig. 2, B and E). In contrast, ip LPS (100 μg/kg) after icv saline pretreatment resulted in a marked increase of c-fos positive neurons in the PVN (148 ± 25; Fig. 2, C and E). This effect was reduced by 62% in animals that received icv INH pretreatment (54 ± 11; Fig. 2, D and E), primarily in medial parvocellular neurons. In contrast, neurons of the thalamic paraventricular nucleus were also activated by LPS but showed no difference in the number of LPS-stimulated c-fos-positive cells between icv saline- and icv INH-treated animals (data not shown).
Figure 2.

Role of aPKC activity in LPS induction of c-fos in the PVN. A–D, Representative c-fos immunohistochemistry of PVN-containing sections from rats pretreated with icv saline (Sal) or INH (2 nmol) followed by ip saline or LPS (100 μg/kg). A and B, Both Sal/Sal and INH/Sal show minimal fos immunoreactivity. C, Sal/LPS animals show the expected increase in intensity and number of stained cells throughout the PVN. D, INH/LPS animals have less fos-positive neurons in the PVN, mostly due to reduction in the medial parvocellular group. E, Quantification of c-fos immunohistochemistry. Bars, mean + se of cell counts averaged from eight sections through the PVN in at least four animals/group. *, P < 0.05 vs. Sal/Sal and LPS/INH; **, P < 0.05 vs. Sal/Sal and Sal/LPS.
Role of aPKC activity in hypothalamic inflammation
To determine whether aPKC activity was a mediator of hypothalamic inflammation stimulated by LPS, we assessed the effect of icv aPKC inhibition on hypothalamic expression of IκBα and IL-6 in rats treated with systemic LPS or saline (Fig. 3, A and B). The peripheral inflammatory response was monitored using splenic IκBα and IL-6 expression (Fig. 3, C and D). In animals pretreated with icv saline, ip LPS (100 μg/kg) induced a modest increase in IκBα expression (∼27%; P < 0.05; Fig. 3A) and a doubling of IL-6 mRNA in mediobasal hypothalamus at 2 h (Fig. 3B). In spleen, no significant differences were observed for IκBα mRNA content, but IL-6 mRNA levels increased by nearly 5-fold (Fig. 3, C and D) as did expression of IL-1β and TNF-α (data not shown). By comparison, icv INH prevented the LPS-induced increase of both IκBα and IL-6 expression in the hypothalamus but not the spleen (Fig. 3, A–D), revealing that the central effect of aPKC inhibition was not due to reduced peripheral inflammation. The level of hypothalamic PPARγ expression remained unchanged by INH treatment (data not shown), showing that INH did not cause a general repression of gene transcription. Compared with icv saline pretreatment, however, icv INH diminished hypothalamic IκBα gene expression by 40% in rats treated with ip saline (Fig. 3A), and a statistically insignificant decrease was observed with IL-6 as well (Fig. 3B). These results revealed a mild degree of inflammation in icv saline-treated animals, a conclusion corroborated by evidence that cannulated but uninjected rats exhibit much lower hypothalamic levels of proinflammatory cytokines (data not shown). Thus, aPKC inhibition in the CNS attenuated both the modest inflammatory response caused by icv saline injection and the much greater activation of the hypothalamic NF-κB pathway triggered by systemic LPS administration.
Figure 3.

Effect of central aPKC inhibition on LPS induction of hypothalamic inflammation. 3V-cannulated rats (n = 6/group) were treated with icv saline (Sal) or INH (2nmol) followed by ip saline or LPS (100 μg/kg) and euthanized 2 h later for collection of mediobasal hypothalamus (A and B) and spleen (C and D). Extracts were processed for quantification using real-time PCR of IκBα (A and C) and IL-6 (B and D) gene expression relative to GAPDH mRNA and normalization to Sal/Sal levels. At the 2-h time point, splenic IκBα mRNA is not significantly changed with any condition (P = ns). *, P < 0.05 compared with Sal/Sal and INH/LPS in A and B and compared with Sal/Sal in D.
Involvement of aPKC in LPS-triggered cytokine gene expression in cultured hypothalamic neurons
To determine whether aPKC inhibition could suppress LPS-induced inflammation specifically in neurons, we used a hypothalamic cell line, N-43/5, originally derived from melanocortin neurons of the arcuate nucleus (30). Cells were treated with LPS (1 μg/ml) for 1 h followed by harvesting of mRNA for real-time PCR analysis. The NF-κB target genes IκBα and IL-6 were induced 5.5- and 8-fold, respectively, by LPS compared with untreated cells (Fig. 4, A and B). Pretreatment with INH reduced IκBα and IL-6 gene expression significantly (Fig. 4, A and B) without evidence of cellular toxicity or changes in global gene expression (data not shown; see Materials and Methods). These results suggest that in hypothalamic neurons, aPKC activity is required for intact LPS-induced inflammation.
Figure 4.

Role of aPKC activity in NF-κB pathway activation by LPS in cultured N-43/5 hypothalamic neurons. A and B, N-43/5 cells were cultured overnight in serum-free medium followed by treatment with INH (0, 10, or 30 μm), LPS (1 μg/ml), or the combination for 1 h. Quantification of IκBα (A) and IL-6 (B) gene expression by real-time PCR with relative to GAPDH mRNA and normalizated to INH 0 μm control cells. Bars, Mean + se from three experiments with duplicate wells. *, P < 0.05 compared with all other treatments; **, P < 0.05 compared with control treatments and LPS alone.
CNS aPKC contribution to the LPS fever response
The febrile response to LPS is a well-studied manifestation of sickness (1,7,32) deriving from the cytokine-driven generation of prostaglandins in the anterior hypothalamus (1,32). Given the requirement for aPKC activation in the induction of hypothalamic IL-6 production by LPS, we predicted that icv INH pretreatment would blunt LPS-induced fever. To test this hypothesis, we used implanted temperature transponders to generate continuous measures of core body temperature for 12 h in 3V-cannulated rats (Fig. 5A). Animals were injected icv with saline or INH followed by ip injection of saline or LPS (100 μg/kg) 1 h before the onset of the dark cycle. Although icv saline caused a modest, transient temperature elevation above untreated animals (average 38.22 vs. 37.88) (Fig. 5, A and B; data not shown), consistent with the onset of low-grade hypothalamic inflammation (Fig. 3), this effect was small compared with the effect of systemic LPS in the icv saline/ip LPS group. During h 3–6 and h 6–9 after ip injection, these animals exhibited a temperature that was increased by 0.6 and 0.4 C, respectively, compared with the icv saline/ip saline group (P < 0.05) (Fig. 5, A and B). The key finding of this study was that whereas icv INH had no significant effect compared with icv saline in the absence of LPS, it fully blocked LPS-induced fever (Fig. 5, A and B).
Figure 5.

Role of aPKC activity in body temperature and locomotor responses to systemic LPS. 3V-cannulated rats bearing temperature transponders (n = 6/group) were treated with icv saline (Sal) or INH (2 nmol) followed by ip saline or LPS (100 μg/kg) 1 h before the onset of the dark cycle (time = 0 h). A, Temperature traces were obtained via continuous monitoring until the end of the dark cycle (time = 12 h). Data points have been averaged to 15-min intervals for clarity. *, P < 0.05 compared with all other treatments for duration of time points covered by bar above Sal/LPS condition. B, Integration of the continuous temperature information in A using 3-h bins. *, P < 0.05 compared with other conditions. C, Animals treated as above were simultaneously monitored for ambulatory activity by measuring laser beam breaks within the cage. Bars represent summation of beam breaks over the entire 12-h dark cycle. *, P < 0.05 relative to Sal/Sal.
Along with fever, LPS rapidly reduces locomotor activity as part of the sickness response. To determine whether aPKC activation contributed to this aspect of sickness, we studied animals in a metabolic cage apparatus that used infrared beam break monitoring to measure ambulatory activity. Unlike the fever response, central inhibition of aPKC with icv INH did not alter ambulatory activity reduction by LPS (Fig. 5C).
aPKC involvement in food intake regulation by LPS, leptin, and insulin
To determine whether LPS-induced anorexia required CNS signaling by aPKC, we infused icv INH (or saline) before ip LPS (or saline) and monitored food intake over a 24-h period. In rats receiving icv saline, ip LPS (50 μg/kg) caused an approximately 40% decrease of food intake at 4 h relative to ip saline (5.8 vs. 9.8 g, P < 0.01, Fig. 6A). Consistent with a key role for aPKC in LPS-induced anorexia, icv INH pretreatment restored food intake to control levels in LPS-injected animals [9.5 vs. 9.8 g, P = ns (not significant), Fig. 6A] but had no effect in the absence of LPS (10.4 vs. 9.8 g, P = ns). The 24-h data from icv INH/ip LPS rats showed a similar pattern with food intake being restored to control levels by icv INH pretreatment (Fig. 6B). Despite this effect, icv INH did not reduce LPS-induced weight loss at 24 h (−5.5 vs. −6.2 g; icv saline/ip LPS vs. icv INH/ip LPS; P = ns), a process known to involve catabolism of both lean and fat mass compartments via a mechanism that is mediated, at least in part, independently of reduced food intake (10).
Figure 6.

Role of PI3K/aPKC signaling in feeding effects of LPS, leptin, and insulin. 3V-cannulated rats (n = 8/group) were pretreated with icv saline (Sal) or INH (2 nmol) followed by ip saline or LPS (50 μg/kg) just before the dark cycle, and food intake was monitored over 24 h. Effects of INH pretreatment on LPS-induced anorexia at 4 h (A) and 24 h (B) are shown. *, P < 0.05 compared with all other conditions. C, 3V-cannulated rats (n = 8/group) were treated with icv vehicle (5% dimethylsulfoxide; Veh) or LY294002 (LY; 1 nmol) followed by ip saline or LPS (50 μg/kg) just before the dark cycle, and food intake was monitored over 24 h. P = 0.058 at 4 h. *, P < 0.05 vs. Veh/Sal and INH/LPS; **, P < 0.05 vs. Veh/Sal and Veh/LPS. D, 3V-cannulated rats (n = 8/group) pretreated with icv saline (Sal) or INH (2 nmol) followed by icv vehicle (0.1 m Na citrate) (Veh) or leptin (4 μg) (Lep) just before the dark cycle were monitored for food consumption over 24 h. P = ns for Sal/Lep vs. INH/Lep. *, P < 0.05 vs. Sal/Veh E, 3V-cannulated rats (n = 8/group) were pretreated with icv saline (Sal) or INH (2 nmol) followed by icv saline or insulin (4 mU) (Ins) 4 h before dark cycle onset. *, P < 0.05 vs. Sal/Sal.
As described above, PI3K is a major upstream regulator of aPKC activity, and both leptin and insulin increase hypothalamic PI3K activity (see Fig. 1). To determine whether LPS-induced anorexia was similarly dependent on CNS PI3K signaling, we repeated the above study in a separate group of rats using icv injection of the relatively specific PI3K inhibitor LY294002 in place of icv INH. Similar to the results obtained with icv INH, central administration of LY294002 prevented LPS-induced anorexia at 4 h and partially restored food intake to control levels at 24 h (Fig. 6C). Like aPKC inhibition, blockade of PI3K did not curtail 24-h body weight loss due to LPS (−6.4 vs. −6.3 g; icv saline/ip LPS vs. icv LY/ip LPS; P = ns).
We and others previously reported that pharmacological doses of leptin and insulin reduce food intake through a mechanism involving hypothalamic PI3K signaling (6). Because both hormones increase hypothalamic aPKC as well as PI3K activity (see Fig. 1 and data not shown), we predicted that aPKC was required for anorexia induced by either leptin or insulin. Intracerebroventricular leptin (4 μg) reduced food intake by about 30 and 45% at 4 and 24 h, respectively (Fig. 6D). Unlike the effect of central aPKC inhibition on ip LPS treatment (see Fig. 6A), however, icv INH had no effect on leptin-induced anorexia at either time point (Fig. 6D). Likewise, icv INH did not alter the approximately 30% food intake reduction by icv insulin (10 mU) (Fig. 6E). Thus, despite both the ability of insulin and leptin to activate hypothalamic aPKC activity (albeit modestly) and clear evidence of a role for aPKC in peripheral insulin action (13), anorexia induced by insulin and leptin appear to involve mechanisms that are dependent on PI3K but are independent of aPKC.
Discussion
To elucidate mechanisms underlying sickness behaviors, we investigated the role of aPKC in the CNS response to the sickness-inducing inflammatory stimulus, LPS. Here we report that aPKC is expressed in key medial hypothalamic nuclei for energy balance and autonomic regulation and that hypothalamic aPKC activation by LPS is critical for elaboration of some, but not all, components of the sickness response. Specifically, LPS-induced anorexia, fever, and activation of c-fos neurons in the PVN, a key output nucleus for sickness responses, are strongly attenuated by icv pretreatment with a specific aPKC inhibitor. In contrast, LPS-induced suppression of locomotor activity remains unaltered by central aPKC inhibition. These observations implicate aPKC signal transduction as a divergence point among neurocircuits through which distinct components of the sickness response are mediated. Furthermore, with no requirement for aPKC activity in leptin- or insulin-mediated anorexia, aPKC action in the CNS also appears to distinguish signal transduction pathways mediating inflammatory anorexia from those triggered by adiposity-related inputs.
Our findings also shed light on mechanisms that link central inflammatory mechanisms to sickness responses such as fever and anorexia. Specifically, animals treated centrally with an aPKC inhibitor display both reduced hypothalamic inflammation and diminished fever and anorexic responses to a systemic LPS challenge. This effect, which occurs despite unaltered peripheral inflammatory responses (measured by splenic cytokine mRNA), suggests that hypothalamic inflammation is a driving force for metabolic adaptations to sickness. This result parallels findings from bone marrow transplant studies showing that intact TLR4/MyD88 signaling in the brain, but not the periphery, is required for sickness responses to LPS (10,11). Our current findings combined with recent reports (10,11) suggest that with LPS administration, circulating cytokines enter the CNS due to disruption of the blood-brain barrier (33,34) but fail to induce the full sickness response in the setting of either aPKC inhibition or TLR4 signaling deficiency in the CNS. This outcome may reflect failure to amplify the cytokine signal within the CNS or, in the case of aPKC inhibition, an inability to propagate the signal beyond a common downstream effector.
Although the mechanisms whereby central inhibition of aPKC diminishes the sickness response initiated by LPS remain to be determined, our findings have translational implications for illness-related wasting syndromes. Although anorexia alone cannot fully explain inflammatory cachexia (10,34), a sustained reduction of intake negatively impacts the course of many chronic illnesses. Indeed, anorexia and cachexia are significant sources of morbidity and mortality accounting for up to 20% of cancer deaths and correlating with poor prognosis in sepsis, heart failure, kidney disease, and emphysema (2,3,4). Unfortunately, current therapies are largely ineffective, reflecting our limited understanding of underlying mechanisms. Our findings raise the possibility that a strategy using central aPKC inhibition may provide an approach to reducing the burden of inflammatory anorexia, although additional interventions will likely be needed to alleviate cachexia-related weight loss.
The sickness response encompasses a variety of behaviors and metabolic adaptations with integration in the hypothalamus. Our examination of the physiologic response to LPS suggests that some components of this coordinated response are regulated independently. Specifically, the finding that aPKC inhibition prevented LPS-induced fever and anorexia without affecting reduced ambulatory activity or weight loss resembles the effect of LPS treatment in MyD88 knockout mice (35). These animals are protected from reduced food and water intake, fever, and hypoactivity but show modest weight loss with LPS administration due to loss of lean body mass (cachexia). These observations, along with the finding that IL-1 is required for melanocortin-induced ambulatory activity (36), suggest a bifurcation in the pathway downstream of TLR4 with MyD88/aPKC-dependent and independent branches controlling distinct elements of the sickness response. Consistent with this possibility, LPS activation of CRH neurons in the PVN (part of the hypothalamic-pituitary-adrenal stress axis) specifically requires MAPK activity (37). Alternatively, inflammatory signals may trigger anorexia through direct effects on arcuate nucleus or PVN neurons using TLR4-MyD88-aPKC signaling and inducing cachexia-related weight loss through cells in other brain regions or at peripheral sites. In this study, we provide initial evidence that inhibition of aPKC activity in cultured hypothalamic neurons can directly limit LPS-induced cytokine generation. However, whether neurons or other brain cell types (e.g. microglia) are the primary sources of hypothalamic cytokines during inflammatory illness remains unknown.
An alternative explanation for the differences in components of the sickness response centers on the role of microglia, the primary immune cells of the CNS. Microglia respond robustly to LPS and have a well-documented ability to regulate neuronal inflammatory responses (38,39). It is conceivable that aPKC or MyD88 action in microglia causes generation and release of small molecules that potently alter neuronal function, such as prostaglandin E2, implicated in both fever and anorexia (34,40). Indeed, cytokine-mediated up-regulation of prostaglandin synthesis enzymes is dependent on aPKC activity in a number of cell types (41,42). In this model, direct LPS action on neurons in circumventricular regions of the CNS lacking a blood-brain barrier could trigger weight loss through alterations in energy use without anorexia, whereas other components of the sickness response might depend on aPKC-mediated effects in microglia. Future studies using mice with cell-specific, targeted deletions of aPKC or other TLR4 pathway genes should prove helpful in testing these competing hypotheses.
Despite pronounced differences in other biological effects, leptin, insulin, and LPS share the capacity to reduce food intake and promote weight loss through convergent signaling mechanisms that involve intermediates such as PI3K (6,38). In addition, LPS administration raises circulating leptin levels (43), and functional leptin signaling is required for some components of the sickness response (44,45). Conversely, leptin requires CNS IL-1β signaling to reduce food intake (46). Although icv studies are inherently pharmacological rather than physiological in nature, it is still noteworthy that aPKC is not a shared mediator of anorexia between leptin/insulin and LPS. This finding implies the existence of divergent mechanisms downstream of PI3K that can affect hypothalamic control of energy balance. Indeed, both Forkhead box O1 (FOXO1) and mammalian target of rapamycin (mTOR) can mediate leptin and insulin action in the hypothalamic arcuate nucleus (47,48). Whether LPS/TLR4 signaling involves these PI3K targets is an important unanswered question.
This study provides support for aPKC involvement in the metabolic consequences of inflammatory signaling, joining a growing literature that has established aPKC both as a regulator of NF-κB pathway activity and a downstream mediator of TLR4 signaling in LPS-dependent processes (14,15,16,17,18,19,20,23,24,49). Although prior investigations have largely used cell culture systems, several studies have used in vivo approaches. In a series of manuscripts, Peng et al. (49,50) describe activation of Kupffer cell aPKC in a TLR4-dependent manner in rat models of pancreatitis and experimental sepsis, although the authors do not report on disease outcomes with altered aPKC signaling interventions. Recently Farese and colleagues (51) demonstrated activation of hepatic NF-κB through aPKC in high-fat-fed mice. Overexpression of dominant-negative aPKC in the liver of these animals reduced hepatic NF-κB activity and consequently ameliorated systemic insulin resistance and hyperlipidemia. Future work by our group will assess the therapeutic utility of central aPKC inhibition in experimental sepsis and preclinical cancer models.
In summary, our study extends existing literature connecting aPKC with inflammatory regulation and demonstrates a specific requirement for aPKC activity in the CNS for generation of certain sickness behaviors in response to systemic inflammation. This finding provides a molecular target to enable cell-specific dissection of sickness response neurocircuitry and to generate CNS-targeted therapies for amelioration of illness-related anorexia.
Acknowledgments
We acknowledge the expert technical assistance of Alex Cubelo, Loan Nguyen, J. D. Fischer, and Iaela David in the conduct of this investigation. We thank members of the Schwartz laboratory for helpful discussions regarding these data and this manuscript.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants DK068384, DK052989, and DK083042 (to M.W.S.), DK074758 (to B.E.W.), and DK065969; Veterans Affairs Merit Review Program (to R.V.F.); and fellowships from the American Diabetes Association and NIH (DK007247) (to J.P.T.). Metabolic cage measurements were supported by the Clinical Nutrition Research Unit (DK035816), the Diabetes Endocrine Research Center (P30 DK17047), and the Mouse Metabolic Phenotyping Center (U24 DK076126).
Disclosure Summary: The authors have nothing to disclose.
First Published Online October 9, 2009
Abbreviations: aPKC, Atypical protein kinase C; CNS, central nervous system; IκBα, inhibitory-κBα; icv, intracerebroventricular; INH, inhibitor; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; ns, not significant. PI3K, phosphatidylinositol-3 kinase; PPAR, peroxisome proliferator and activator; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PVN, paraventricular nuclei; TLR4, Toll-like receptor 4; 3V, third cerebral ventricle.
References
- Kluger MJ 1991 Fever: role of pyrogens and cryogens. Physiol Rev 71:93–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosaeus I, Daneryd P, Lundholm K 2002 Dietary intake, resting energy expenditure, weight loss and survival in cancer patients. J Nutr 132:3465S–3466S [DOI] [PubMed] [Google Scholar]
- Morley JE, Thomas DR, Wilson MM 2006 Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr 83:735–743 [DOI] [PubMed] [Google Scholar]
- Tan BH, Fearon KC 2008 Cachexia: prevalence and impact in medicine. Curr Opin Clin Nutr Metab Care 11:400–407 [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG 2000 Central nervous system control of food intake. Nature 404:661–671 [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW 2006 Central nervous system control of food intake and body weight. Nature 443:289–295 [DOI] [PubMed] [Google Scholar]
- Dantzer R, Kelley KW 2007 Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 21:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R 2002 Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25:154–159 [DOI] [PubMed] [Google Scholar]
- O'Neill LA 2008 The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Ogimoto K, Tang J, Harris Jr MK, Raines EW, Schwartz MW 2007 Evidence that lipopolysaccharide-induced anorexia depends upon central, rather than peripheral, inflammatory signals. Endocrinology 148:5230–5237 [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M 2005 Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci 25:1788–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layé S, Gheusi G, Cremona S, Combe C, Kelley K, Dantzer R, Parnet P 2000 Endogenous brain IL-1 mediates LPS-induced anorexia and hypothalamic cytokine expression. Am J Physiol Regul Integr Comp Physiol 279:R93–R98 [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Standaert ML 2005 Atypical protein kinase C in insulin action and insulin resistance. Biochem Soc Trans 33:350–353 [DOI] [PubMed] [Google Scholar]
- Moscat J, Rennert P, Diaz-Meco MT 2006 PKCζ at the crossroad of NF-κB and Jak1/Stat6 signaling pathways. Cell Death Differ 13:702–711 [DOI] [PubMed] [Google Scholar]
- Diaz-Meco MT, Dominguez I, Sanz L, Dent P, Lozano J, Municio MM, Berra E, Hay RT, Sturgill TW, Moscat J 1994 ζPKC induces phosphorylation and inactivation of IκB-α in vitro. EMBO J 13:2842–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Diaz-Meco MT, Moscat J 2003 Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J 22:3910–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J 2000 The atypical PKC-interacting protein p62 channels NF-κB activation by the IL-1-TRAF6 pathway. EMBO J 19:1576–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J 1999 The interaction of p62 with RIP links the atypical PKCs to NF-κB activation. EMBO J 18:3044–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuschieri J, Umanskiy K, Solomkin J 2004 PKC-ζ is essential for endotoxin-induced macrophage activation. J Surg Res 121:76–83 [DOI] [PubMed] [Google Scholar]
- Procyk KJ, Rippo MR, Testi R, Hofmann F, Parker PJ, Baccarini M 2000 Lipopolysaccharide induces jun N-terminal kinase activation in macrophages by a novel Cdc42/Rac-independent pathway involving sequential activation of protein kinase Cζ and phosphatidylcholine-dependent phospholipase C. Blood 96:2592–2598 [PubMed] [Google Scholar]
- Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL 1993 A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J Biol Chem 268:1982–1986 [PubMed] [Google Scholar]
- Soloff RS, Katayama C, Lin MY, Feramisco JR, Hedrick SM 2004 Targeted deletion of protein kinase Cλ reveals a distribution of functions between the two atypical protein kinase C isoforms. J Immunol 173:3250–3260 [DOI] [PubMed] [Google Scholar]
- Leitges M, Sanz L, Martin P, Duran A, Braun U, García JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J 2001 Targeted disruption of the ζPKC gene results in the impairment of the NF-κB pathway. Mol Cell 8:771–780 [DOI] [PubMed] [Google Scholar]
- Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, Adachi T, Patke A, Santana A, Tarakhovsky A 2003 Essential role of Src-family protein tyrosine kinases in NF-κB activation during B cell development. Nat Immunol 4:274–279 [DOI] [PubMed] [Google Scholar]
- Naik MU, Benedikz E, Hernandez I, Libien J, Hrabe J, Valsamis M, Dow-Edwards D, Osman M, Sacktor TC 2000 Distribution of protein kinase Mζ and the complete protein kinase C isoform family in rat brain. J Comp Neurol 426:243–258 [DOI] [PubMed] [Google Scholar]
- Oster H, Eichele G, Leitges M 2004 Differential expression of atypical PKCs in the adult mouse brain. Brain Res Mol Brain Res 127:79–88 [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Sipols AJ, Marks JL, Sanacora G, White JD, Scheurink A, Kahn SE, Baskin DG, Woods SC, Figlewicz DP 1992 Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology 130:3608–3616 [DOI] [PubMed] [Google Scholar]
- Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers Jr MG, Seeley RJ, Schwartz MW 2003 Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 52:227–231 [DOI] [PubMed] [Google Scholar]
- Kanoh Y, Sajan MP, Bandyopadhyay G, Miura A, Standaert ML, Farese RV 2003 Defective activation of atypical protein kinase Cζ and λ by insulin and phosphatidylinositol-3,4,5-(PO4)(3) in skeletal muscle of rats following high-fat feeding and streptozotocin- induced diabetes. Endocrinology 144:947–954 [DOI] [PubMed] [Google Scholar]
- Mayer CM, Fick LJ, Gingerich S, Belsham DD2009 Hypothalamic cell lines to investigate neuroendocrine control mechanisms. Front Neuroendocrinol 30:405–423 [DOI] [PubMed] [Google Scholar]
- Rivest S, Laflamme N 1995 Neuronal activity and neuropeptide gene transcription in the brains of immune-challenged rats. J Neuroendocrinol 7:501–525 [DOI] [PubMed] [Google Scholar]
- Hopkins SJ 2007 Central nervous system recognition of peripheral inflammation: a neural, hormonal collaboration. Acta Biomed 78(Suppl 1):231–247 [PubMed] [Google Scholar]
- Banks WA 2001 Anorectic effects of circulating cytokines: role of the vascular blood-brain barrier. Nutrition 17:434–437 [DOI] [PubMed] [Google Scholar]
- Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A 2001 Inflammatory response: pathway across the blood-brain barrier. Nature 410:430–431 [DOI] [PubMed] [Google Scholar]
- Ogimoto K, Harris Jr MK, Wisse BE 2006 MyD88 is a key mediator of anorexia, but not weight loss, induced by lipopolysaccharide and interleukin-1β. Endocrinology 147:4445–4453 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Ogimoto K, Schwartz MW 2006 Role of hypothalamic interleukin-1β (IL-1β) in regulation of energy homeostasis by melanocortins. Peptides 27:265–273 [DOI] [PubMed] [Google Scholar]
- Singru PS, Sánchez E, Acharya R, Fekete C, Lechan RM 2008 Mitogen-activated protein kinase contributes to lipopolysaccharide-induced activation of corticotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus. Endocrinology 149:2283–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhans W 2007 Signals generating anorexia during acute illness. Proc Nutr Soc 66:321–330 [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH 2009 Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145 [DOI] [PubMed] [Google Scholar]
- Johnson PM, Vogt SK, Burney MW, Muglia LJ 2002 COX-2 inhibition attenuates anorexia during systemic inflammation without impairing cytokine production. Am J Physiol Endocrinol Metab 282:E650–E656 [DOI] [PubMed] [Google Scholar]
- Di Mari JF, Mifflin RC, Adegboyega PA, Saada JI, Powell DW 2003 IL-1α-induced COX-2 expression in human intestinal myofibroblasts is dependent on a PKCζ-ROS pathway. Gastroenterology 124:1855–1865 [DOI] [PubMed] [Google Scholar]
- Miller BW, Baier LD, Morrison AR 1997 Overexpression of protein kinase C-ζ isoform increases cyclooxygenase-2 and inducible nitric oxide synthase. Am J Physiol 273:C130–C136 [DOI] [PubMed] [Google Scholar]
- Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet 3rd DJ, Flier JS, Lowell BB, Fraker DL, Alexander HR 1997 Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 185:171–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden LM, du Plessis I, Poole S, Laburn HP 2006 Interleukin-6 and leptin mediate lipopolysaccharide-induced fever and sickness behavior. Physiol Behav 89:146–155 [DOI] [PubMed] [Google Scholar]
- Rummel C, Inoue W, Sachot C, Poole S, Hübschle T, Luheshi GN 2008 Selective contribution of interleukin-6 and leptin to brain inflammatory signals induced by systemic LPS injection in mice. J Comp Neurol 511:373–395 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Ogimoto K, Morton GJ, Williams DL, Schwartz MW 2007 Central interleukin-1 (IL1) signaling is required for pharmacological, but not physiological, effects of leptin on energy balance. Brain Res 1144:101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ 2006 Hypothalamic mTOR signaling regulates food intake. Science 312:927–930 [DOI] [PubMed] [Google Scholar]
- Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, Kim YB, Lee KU 2006 Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci 9:901–906 [DOI] [PubMed] [Google Scholar]
- Peng Y, Sigua CA, Rideout D, Murr MM 2008 Deletion of toll-like receptor-4 down-regulates protein kinase C-ζ and attenuates liver injury in experimental pancreatitis. Surgery 143:679–685 [DOI] [PubMed] [Google Scholar]
- Peng Y, Sigua CA, Gallagher SF, Murr MM 2007 Protein kinase C-ζ is critical in pancreatitis-induced apoptosis of Kupffer cells. J Gastrointest Surg 11:1253–1261 [DOI] [PubMed] [Google Scholar]
- Sajan MP, Standaert ML, Nimal S, Varanasi U, Pastoor T, Mastorides S, Braun U, Leitges M, Farese R2009 The critical role of atypical protein kinase C in activating hepatic SREBP-1c and NFκB in obesity. J Lipid Res 50:1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]