

Abstract
Elevated levels of serum unconjugated bilirubin (UCB) in the first weeks of life may lead to long-term neurologic impairment. We previously reported that an early exposure of developing neurons to UCB, in conditions mimicking moderate to severe neonatal jaundice, leads to neuritic atrophy and cell death. Here, we have further analyzed the effect of UCB on nerve cell differentiation and neuronal development, addressing how UCB may affect the viability of undifferentiated neural precursor cells and their fate decisions, as well as the development of hippocampal neurons in terms of dendritic and axonal elongation and branching, the axonal growth cone morphology, and the establishment of dendritic spines and synapses. Our results indicate that UCB reduces the viability of proliferating neural precursors, decreases neurogenesis without affecting astrogliogenesis, and increases cellular dysfunction in differentiating cells. In addition, an early exposure of neurons to UCB decreases the number of dendritic and axonal branches at 3 and 9 days in vitro (DIV), and a higher number of neurons showed a smaller growth cone area. UCB-treated neurons also reveal a decreased density of dendritic spines and synapses at 21 DIV. Such deleterious role of UCB in neuronal differentiation, development, and plasticity may compromise the performance of the brain in later life.
Keywords: astrogliogenesis, hippocampal neurons, neuritic network, neurogenesis, neurospheres, unconjugated bilirubin
INTRODUCTION
The mammalian central nervous system (CNS) is particularly vulnerable to injury during the last half of gestation, when nerve cells proliferation and differentiation give rise to neural structures (Kinney, 2006). If these developmental processes are affected by neurotoxic agents, perturbation of the normal construction of brain circuits can lead to subsequent neurodevelopmental deficits (Mendola et al., 2002).
During development, nerve cell precursors in the ventricular zone give rise to successive waves of neurons and radial glia, followed by astrocytes and oligodendrocytes (Sun et al., 2003; Guillemot, 2007), and perturbations in this developmental sequence can cause neurodevelopmental disorders (Dong and Greenough, 2004; Lasky and Wu, 2005). In developing neurons, axonal elongation and proper formation of neural circuits are dependent on growth cones, motile structures at the tips of growing axons that detect and integrate guidance signals present in the extracellular environment (Yu and Bargmann, 2001; Dontchev and Letourneau, 2003). In the presence of toxic substances or therapeutic agents, growth cones can suffer retraction or collapse (Leong et al., 2001; Radwan et al., 2002), promoting mild to severe alterations of neuronal arborization, which may lead to neurological abnormalities.
The complexity of the dendritic arbor is generally correlated with the number of synaptic inputs (Jan and Jan, 2003). Most CNS excitatory synapses occur on dendritic spines, small protrusions on the dendrites that contain clusters of excitatory neurotransmitter receptors (Yuste et al., 2000). Abnormalities in the number, size, and morphology of dendritic spines are commonly associated with neurodevelopmental disorders including altered learning and memory skills (Leuner and Shors, 2004).
Jaundice, the clinical manifestation of increased levels of unconjugated bilirubin (UCB) in circulation, is correlated with an augmented risk for the emergence of long-term neurodevelopment disabilities (Dalman and Cullberg, 1999; Miyaoka et al., 2000). UCB interacts with cellular membranes (Rodrigues et al., 2002a,b), activates glia with consequent inflammatory response (Fernandes et al., 2004; Falcão et al., 2005; Fernandes et al., 2006; Gordo et al., 2006; Brites et al., 2008), and induces oxidative and nitrosative stress (Brito et al., 2008a,b). In addition, UCB exposure causes the accumulation of extracellular glutamate (Silva et al., 1999; Fernandes et al., 2004; Falcão et al., 2006; Gordo et al., 2006), leading to extensive cellular structural and functional alterations that culminate in cell death through a myriad of pathways (Rodrigues et al., 2002a,b; Silva et al., 2002; Falcão et al., 2006). Recent studies have demonstrated that an early exposure of developing cortical neurons to UCB leads to a reduction of both neurite extension and number of ramifications, effects that were increasingly perpetuated along cell differentiation and contribute to an increased vulnerability to an inflammatory stimulus (Falcão et al., 2007). Interestingly, it was recently shown that Tau microtubule-associated protein, a neuron-specific protein whose function is important in establishing and maintaining neuronal morphology (Behar et al., 1995), is increased in serum of jaundiced newborns and strongly correlated with early-phase UCB encephalopathy (Okumus et al., 2008).
Here, we further explore the action of UCB on neuronal viability, differentiation, and maturation. Exposure of embryonic stem (ES) cell derived neurospheres to UCB decreased cell viability and diminished neurogenesis without affecting astrogliogenesis. Immature neurons treated with UCB showed an irreversible impairment in both axonal and dendritic outgrowth, as well as decreased growth cone area and reduced density of dendritic spines and synapses. Collectively, our results provide an insight into the pathways of neurodevelopmental abnormalities by UCB that may lead to transitory or permanent brain damage, including learning disabilities.
METHODS
Committed Neural Precursor Culture and Treatment
ES cell line 46C expresses enhanced green fluorescent protein (EGFP) under the Sox1 promoter. Proliferation of these cells as floating aggregates (“neurospheres”), cell treatment and in vitro differentiation are outlined in Figure 1(A). Briefly, after 4 days in adherent monoculture in RHB-A™ media (Stem Cell Sciences) on gelatin-coated culture plates, committed neural precursors were induced to form “neurospheres” (Conti et al., 2005) by replating onto uncoated plates (plating density 2 × 105 cells/mL, with around 80% of the cells being Sox1:EGFP positive), in the presence of 10 ng/mL basic fibroblast growth factor (bFGF, Peprotech) and 10 ng/mL epidermal growth factor (EGF, Peprotech). Day 2 suspension cultures were treated with 50 μM UCB or with vehicle (culture medium plus 100 μM human serum albumin to keep UCB in solution) for 48 h at 37°C. Day 4 floating neurospheres were counted and dissociated, and differentiation was induced by plating onto coverslips coated with poly-D-lysine (10 μg/mL) and laminin (2.5 μg/mL) in the absence of bFGF and EGF for 5 days.
Figure 1.
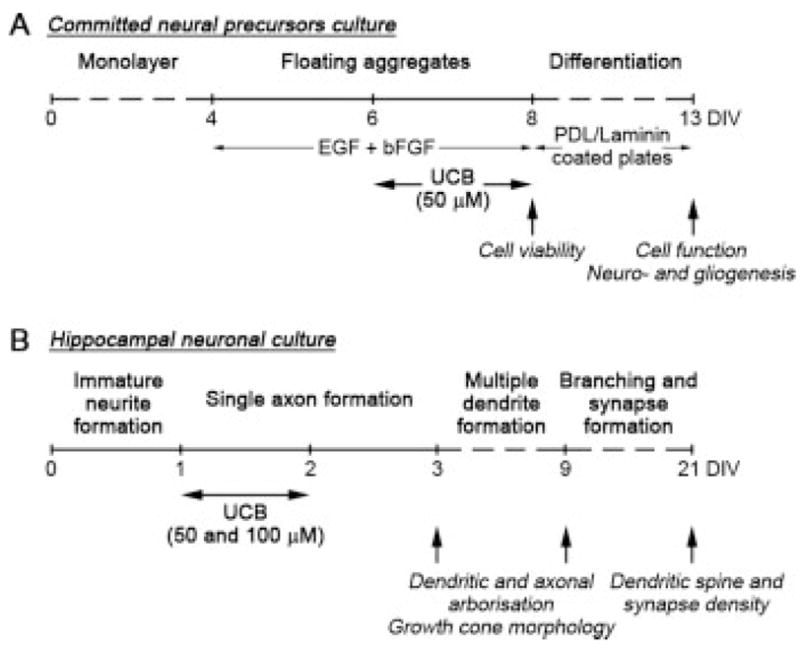
Schematic representation of the timeline for experiments with committed neural precursors and primary hippocampal neuronal cultures. A: Committed neural precursors were obtained from ES monocultures that were adherent for 4 days in vitro (DIV) and then replated onto uncoated plates in the presence of 10 ng/mL bFGF and 10 ng/mL EGF, where they form floating cell aggregates (neurospheres). After 2 days, cells were incubated in the absence (vehicle) or in the presence of 50 μM UCB (plus 100 μM human serum albumin) for 48 h at 37°C. After incubation, cell viability of neurospheres was evaluated and in vitro differentiation was induced by plating neurospheres onto poly-D-lysine and laminin-coated coverslips in the absence of bFGF and EGF. After 5 days of differentiation, mitochondrial function was assessed and neuro- and gliogenesis were also evaluated. B: Primary hippocampal neurons were plated onto poly-D-lysine and laminin-coated coverslips and incubated at 1 DIV in the absence (vehicle) or in the presence of 50 or 100 μM UCB (plus 100 μM human serum albumin) for 24 h at 37°C. Following UCB exposure, the incubation media was replaced with conditioned neuronal growth media without UCB and neurons were analyzed at 3 and 9 DIV for dendritic and axonal arborization and growth cone morphology, or at 21 DIV for dendritic spine and synapse density.
Hippocampal Neuronal Cell Culture and Treatment
Primary hippocampal neurons were prepared from E16 mice as previously described (Lanier et al., 1999). Animals were handled according to the guidelines on care and use of experimental animals set forth by the Institutional Animal Care and Use Committee at the University of Minnesota. Approximately 2 × 104 cells were plated on each 12-mm coverslip coated with poly-D-lysine (100 μg/mL) and laminin (4 μg/mL) in plating medium (MEM with Earle’s salts supplemented with 10 mM HEPES, 10 mM sodium pyruvate, 0.5 mM glutamine, 12.5 μM glutamate, 10% FCS, and 0.6% glucose). After 3 h, the media was replaced with neuronal growth medium (Neurobasal media with B27 supplement and 0.5 mM glutamine). For neurite analysis, neurons were electroporated prior to seeding with a plasmid expressing EGFP under a β-actin promoter (pCAG-EGFP) generated at Lorene M Lanier’s lab (5 μg of plasmid for 1 × 106 cells) using the Mouse Neuron Nucleofector kit (Amaxa). At 1 day in vitro (DIV), cells were incubated with 50 or 100 μM UCB or with vehicle (culture medium plus 100 μM human serum albumin to keep UCB in solution) for 24 h at 37°C. Following UCB exposure, the incubation media was replaced with neuronal growth media conditioned on neuronal cultures at the same state of differentiation and neurons were analyzed at 3, 9, or 21 DIV [Fig. 1(B)].
Cell Viability
Following neurosphere incubation, cell aggregates were counted and dissociated to evaluate cell viability by the trypan blue dye (0.1% w/v) exclusion test and also by propidium iodide staining analysis, using the BD FACSCalibur™ System (BD Biosciences). The assessment of EGFP-positive cells, as a method to quantify Sox1-positive neural precursors, was also performed using the BD FACSCalibur™ System. Standard evaluation of hippocampal neuronal cytotoxicity was performed by measuring the release of lactate dehydrogenase (LDH) by nonviable cells to the incubation medium using the cytotoxicity detection kit as described previously (Silva et al., 2006). All readings were corrected for the possible interference of UCB absorption and the results expressed as percent of LDH release, obtained by treating nonincubated cells with 2% Triton X-100 for 30 min.
MTT Assay
Cellular reduction of MTT [3-(4,5-dimethylthiazol, 2-yl)-2,5-diphenyltetrazolium bromide] (Sigma) was measured in differentiating cells as previously described by us (Silva et al., 2002) as a marker of mitochondrial functionality. Briefly, a stock solution of MTT at 5 mg/mL was freshly prepared and after the incubation period with UCB, supernatants were removed and cells were incubated for 1 h at 37°C with 500 μL of MTT at 0.5 mg/mL, prepared by dilution of the stock solution in RHB-A™ media. After incubation, medium was discarded and MTT formazan crystals were dissolved by addition of 1 mL isopropanol/HCl 0.04 M and gentle shaking for 15 min at room temperature. After centrifugation, absorbance values at 570 nm were determined in a Unicam UV2 spectrophotometer (Unicam Limited, UV2, Cambridge, UK). Results were expressed as percentage of vehicle, which was considered as 100%.
Immunocytochemistry
For committed neural precursors studies, cells were fixed after 5 days in differentiation conditions using 4% paraformaldehyde in PBS (pH 7.4) for 15 min at room temperature and stained for the presence of neurons using antineuronal protein HuC/HuD antibody (1:500, Molecular Probes), and for astrocytes using antiglial fibrillary acidic protein (GFAP) antibody (1:100, Novo Castra) in order to evaluate alterations in neurogenesis and in astrogliogenesis, respectively. Nuclei were stained with Hoechst dye 33258 (Sigma) and pairs of UV and fluorescence images of 10 random microscopic fields (original magnification: 516× that equals 67,500 square microns) were acquired per sample. Immunopositive cells for each cell type and total cells were counted to determine the percentage of positive cells. Results were expressed as percentage over vehicle values.
For hippocampal neurons, cells were fixed at 3, 9, and 21 DIV with PPS (4% paraformaldehyde in PHEM buffer [60 mM PIPES (pH 7.0), 25 mM HEPES (pH 7.0), 10 mM EGTA, 2 mM MgCl2] with 0.12 M sucrose) for 30 min at RT. After rinsing in PBS, coverslips were blocked in 3% fatty acid free bovine serum albumin (BSA) in PBS for 30 min, permeabilized for 10 min in 0.2% triton/PBS, rinsed, and reblocked in 3% BSA/PBS for 30 min. Incubations with primary and secondary antisera were done in the presence of 1% BSA/PBS, and coverslips mounted with 2.5% 1,4-diazabicyclo-[2.2.2]octane/150 mM Tris (pH 8.0)/30% glycerol to reduce photobleaching. Images were captured on an Axiocam HR adapted to an Axiovert Microscope (Zeiss) using Openlab software (Improvision). EGFP-positive neurons were located directly by fluorescence microscopy, F-actin was identified using Alexa 594 or 488 phalloidin (Molecular Probes) and for the other markers the following antisera were used: rabbit anti-MAP2 (Covance); mouse anti-Tau-1 (Chemicon); mouse antitubulin β III (Promega); mouse anti-ERM proteins [a gift from Frank Solomon, generated as previously described (Goslin et al., 1989)]; mouse anti-SV2 (Developmental Studies Hybridoma Bank at the University of Iowa).
Analysis of Neurites, Growth Cone, and Dendritic Spines/Synapses
Axon and dendrite length measurements were performed on stage 3 neurons. For the purpose of this analysis, a neuron was defined as stage 3 if it had a single neurite (the axon) that was at least twice as long as all the other neurites (Strasser et al., 2004). Cells were fixed at 3 and 9 DIV and EGFP-positive neurons were imaged using either a 20× (for 3 DIV) or 10× (for 9 DIV) plan-neofluar objective. Neurites were manually traced using ImageJ v1.40 and the NeuronJ plugin v1.4.0 (Meijering et al., 2004). Dendritic and axonal identities were confirmed by staining neurons with anti-MAP2 for dendrites and with anti-Tau for axon detection.
Growth cone analysis was performed in cells fixed at 3 and 9 DIV, stained with antitubulin β III (Promega), phalloidin to visualize F-actin that identifies the filopodia cytoskeleton, and antibody 13H9 against ERM proteins (Goslin et al., 1989), which link the filopodia to the plasma membrane. Images were taken using a 100× plan-neofluar objective and growth cone area manually traced using ImageJ v1.40.
Dendritic spine and synapse determinations were performed in cells fixed at 21 DIV, stained with anti-MAP2 to identify the dendritic shaft, phalloidin to visualize F-actin at the dendritic spine and anti-SV2 to detect the presynaptic partner. Images were taken using a 100× plan-neofluar objective, and the number of dendritic spines and synapses were counted along the dendritic shafts and expressed as the number of spines per 10 μm of dendrite. Here, all spine like protrusions on dendritic tips were counted as dendritic spines, and a synapse was strictly defined as colocalized presynaptic protein (SV2) and postsynaptic dendritic spines (phalloidin/F-actin). Dendritic spine morphology was also evaluated by the ratio of dendritic spine neck length, measured from the base of the dendrite shaft to the tip of the head in the phalloidin/F-actin channel, versus the spine head width, as an indicator of spine maturation.
For each measurement described earlier, data represent the average of more than 50 different neurons from minimum of three separate experiments.
Statistical Analysis
Results were expressed as mean ± SEM. Differences between groups were determined by one-way ANOVA with Dunnett’s or Bonferroni’s multiple comparisons post tests, using Instat 3.05 (GraphPad Software, San Diego, CA). p < 0.05 was accepted as statistically significant.
RESULTS
UCB reduces the viability of proliferating neurospheres and increases the dysfunction of differentiating cells. Jaundice affects about 80% of premature infants (Truman, 2006). Since neurogenesis continues well into the third trimester in humans (Bhardwaj et al., 2006), this suggests that in jaundice premature babies, UCB may be present during neurogenesis. Therefore, we sought to use an in vitro model of neural precursor generation and differentiation to investigate the effect of UCB on neural stem-cell proliferation and on cell-fate decision choices during development of nerve cells. Pluripotent ES cells can differentiate in vitro into neurons, astrocytes, and oligodendrocytes (Fraichard et al., 1995), following the same order in which they appear in vivo (Okabe et al., 1996). Recent studies have shown that ES-derived neural precursors cells can be grown in free-floating aggregate cultures similar to neurospheres. These cells can be clonally expanded in defined conditions for neural commitment and, in the presence of specific growth factors, can differentiate into neurons or astrocytes (Conti et al., 2005). Therefore, we used ES cell line 46C (Sox1:EGFP) to obtain floating aggregates of neural precursors [Fig. 2(A)]. This ES cell line expresses EGFP under the Sox1 promoter, a gene that is expressed during the neural precursor state, making this cell line ideal for monitoring the acquisition of neural identity by initially undifferentiated cells (Ying and Smith, 2003).
Figure 2.
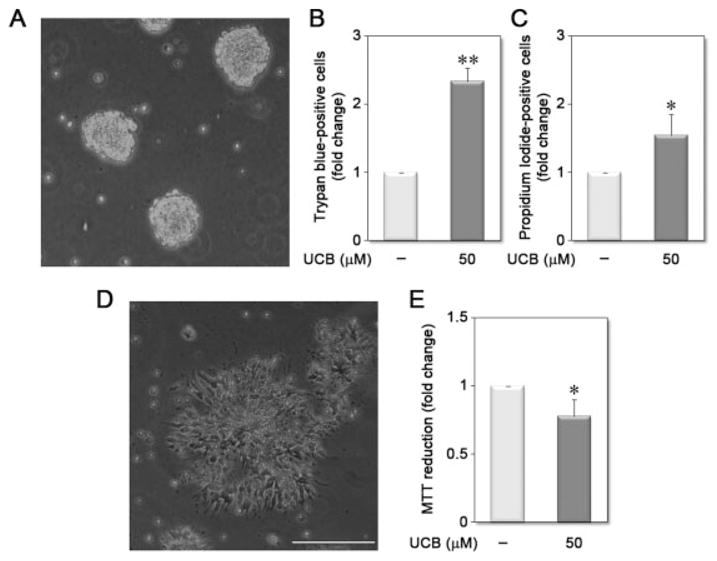
Unconjugated bilirubin (UCB) reduces the viability of proliferating neurospheres and increases the dysfunction of differentiating cells. After 4 days in adherent monoculture, committed neural progenitors derived from mouse embryonic stem cell line 46C were replated onto uncoated plates, in the presence of bFGF and EGF, where they form floating cell aggregates (neurospheres), as observed by phase contrast microscopy (A). Neurospheres were incubated in the absence (vehicle) or in the presence of 50 μM UCB, for 48 h. After incubation, cell aggregates were dissociated to evaluate cell viability by the trypan blue exclusion test (B) and by propidium iodide staining analysis (C). Differentiation of neural precursors is induced by plating the cells into poly-D-lysine and laminin-coated plates, where these cells organize into rosette-shape clusters, as observed by phase contrast microscopy (D). Five days after induction of differentiation, mitochondrial function was measured by MTT reduction (E). Results were expressed as fold change over vehicle. *p < 0.05 and **p < 0.01 vs. respective vehicle. Scale bar equals 1 mm.
The floating 46C neurospheres gradually expanded in size until the end of the incubation period. As expected, we observed that 48-h incubation with 50 μM UCB increased the number of unviable cells in floating aggregates, as observed by either trypan blue exclusion [~2.3-fold, Fig. 2(B)] or propidium iodide staining [~1.5-fold, Fig. 2(C)]. However, UCB did not significantly alter the number of floating aggregates or EGFP-positive cells (results not shown), suggesting that there was no change in the proliferation potential of ES-derived neural precursors.
We next used the MTT reduction assay (a measure of mitochondrial function) to determine whether the cells that survived UCB incubation were functionally active when induced to differentiate into astrocytes and neurons. After dissociating the cell aggregates into single cells, differentiation was induced by plating on poly-D-lysine and laminin-coated plates, where neural precursors formed rosette-shaped cellular structures [Fig. 2(D)]. After 5 DIV, both neurons and astrocytes emerged from these rosettes. When cellular function was tested by MTT assay 5 days after inducing differentiation of the cells, we observed that UCB treatment reduced mitochondrial function of differentiating cells [~30%; Fig. 2(E)], supporting the conclusion that UCB developmental neurotoxicity may have lasting neurological implications.
UCB Decreases Neurogenesis Without Affecting Astrogliogenesis
Having demonstrated that UCB can interfere with neural precursors cell viability, we next sought to determine whether UCB differentially affects neurogenesis and/or astrogliogenesis. Immunocytochemistry with the neuronal marker HuC/D and the astroglial marker GFAP assays were used to distinguish cell types. A 48-h treatment of neural precursor cells with UCB during the proliferation stage (i.e. neurosphere stage) led to a 52.8% decrease in the number of neurons [Fig. 3(A)], even though the cells were differentiated in the absence of UCB. In contrast, UCB did not significantly change the number of astroglial cells [Fig. 3(B)]. Thus, UCB can alter intrinsic cellular mechanisms of stem cell-fate choices contributing to altered neurogenesis during CNS maturation, which may determine defective neuronal functions.
Figure 3.

Unconjugated bilirubin (UCB) decreases neurogenesis without affecting astrogliogenesis. Neurospheres were incubated in the absence (vehicle) or in the presence of 50 μM UCB for 48 h. After incubation, cells were washed with PBS and differentiation was induced by plating aggregates onto poly-D-lysine and laminin-coated coverslips in the absence of bFGF and EGF. After 5 days in vitro, cells were fixed and stained for the presence of (A) neurons using an antibody for neuronal protein HuC/HuD (red in A) and of (B) astrocytes with an antibody for GFAP (red in B), against nuclei staining with Hoechst dye (blue in A and B). Results were expressed as percentage of positive cells over vehicle. **p < 0.01 vs. respective vehicle. Scale bar equals 100 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Treatment of Immature Hippocampal Neurons with UCB Impairs Neuronal Morphogenesis
In vitro and in vivo, pyramidal neurons undergo dramatic morphological changes during maturation. Dotti et al. (1988) divided this differentiation process into five successive stages. Shortly after plating, dissociated neurons attach to the substrate and extend lamellipodia around the soma (Stage 1). The cells then typically form four to five immature neurites (Stage 2). Within 48 h, one neurite begins to elongate rapidly and acquires axonal characteristics (Stage 3). A few days later, the remaining neurites elongate and acquire the characteristics of dendrites (Stage 4). Typically, the axon and dendrites continue to mature, extend collateral branches, and subsequently develop to form synaptic contacts by 9–11 days after plating, finally establishing a neuronal network (Stage 5).
To characterize the effect of UCB on dendritic and axonal elongation and branching, primary cultures of hippocampal neurons were treated at 1 DIV with 50 or 100 μM UCB plus 100 μM human serum albumin [Fig. 1(B)]. After a 24-h treatment, incubation medium was replaced by conditioned growth medium without UCB, and cells were fixed at 3 or 9 DIV and neuronal morphology was analyzed. Since cellular dysfunction and consequent death may be a cause for impaired neuritogenesis, we evaluated cell death at each end point by measuring the release of LDH (Lobner, 2000). Although UCB exposure increased LDH release in about ~1.5-fold when compared with respective control values (~4.1% vs. 2.7% and 5.2% vs. 3.4% for both 3 and 9 DIV, respectively), the amount of cell death did not exceed 10% (data not shown). These findings assure that the majority of neurons were viable throughout the study, thus suggesting that alterations in neuritogenesis are associated with changes in neurite-formation process.
At 3 DIV, vehicle-treated neurons had four to five dendrites with minimal branching and a long axon with more prominent branches [Fig. 4(A)]. By 9 DIV, neuronal maturation was characterized by an increase in dendritic and axonal elongation and numerous branches [Fig. 4(A′)]. Neurons treated with UCB showed a partial degeneration of neurites, often characterized by retraction and fragmentation, with a decreased neurite arborization of the remaining neurons. The effect of UCB was concentration dependent [Fig. 4(B,C)] and was sustained through maturation [Fig. 4(B′–C′)]. Indeed, total neurite output decreased by 10–25% at both 3 DIV [Fig. 4(D)] and 9 DIV [Fig. 4(D′)] after treatment with 50 and 100 μM UCB, respectively.
Figure 4.

Treatment of differentiating hippocampal neurons with unconjugated bilirubin (UCB) reduces neuronal arborization. Examples of embryonic hippocampal neurons treated with vehicle (A and A′), UCB 50 μM (B and B′), or UCB 100 μM (C and C′) for 24 h at 1 DIV and fixed and stained at 3 DIV (A–C) or 9 DIV (A′–C′). Cells were visualized by EGFP fluorescence (shown in black and white and inverted such that EGFP appears black), and neurites were manually traced using ImageJ v1.40 and the NeuronJ plugin v1.4.0. Graphs quantify the effect of UCB on total neurite output at 3 DIV (D) and 9 DIV (D′). *p < 0.05 and **p < 0.01 vs. vehicle. y axis indicates length in microns.
Immunostaining of neurons with MAP2 and Tau-1 was used to identify dendrites and axon, respectively. Although the number and length of dendrites was not significantly changed at 3 DIV, by 9 DIV, treatment with UCB led to a significant decrease in the number (~20%) and length (~15%) of dendrites [Fig. 5(A,B)]. In addition, UCB treatment induced a remarkable reduction in both the number (>20%) and length (>10%) of dendritic branches [Fig. 5(C–D)] at 3 and 9 DIV. UCB exposure also led to a concentration-dependent reduction in axonal length at 9 DIV (>10%) and in the number (>20%) and length of axonal branches (>20%) at 3 and 9 DIV (see Fig. 6). However, UCB appeared to affect dendritic branching more severely than axonal branching, and the effect was most apparent at the highest concentration of UCB at 9 DIV (~60% and ~30% decrease for number and length of dendritic branches, respectively vs. ~25% and ~20% decrease for number and length of axonal branches, respectively). These data support the conclusion that exposure of immature neurons to UCB triggers a reduction in dendritic and axonal arborization, leading to possible aberrations in neuronal connectivity.
Figure 5.

Treatment of differentiating hippocampal neurons with unconjugated bilirubin (UCB) reduces dendritic output. Embryonic hippocampal neurons were treated with vehicle, UCB 50 μM or UCB 100 μM for 24 h at 1 DIV and fixed and stained at 3 DIV or 9 DIV. Cells were visualized by EGFP fluorescence, and neurites were manually traced using ImageJ v1.40 and the NeuronJ plugin v1.4.0. Graphs show the effects of UCB on (A) dendrite number, (B) dendrite length, (C) number of dendrite branches, and (D) length of dendrite branches at 3 and 9 DIV, from at least three independent experiments. *p < 0.05 and **p < 0.01 vs. respective vehicle.
Figure 6.

Treatment of differentiating hippocampal neurons with unconjugated bilirubin (UCB) reduces axonal arborization. Embryonic hippocampal neurons were treated with vehicle, UCB 50 μM or UCB 100 μM for 24 h at 1 DIV and fixed and stained at 3 or 9 DIV. Cells were visualized by EGFP fluorescence and neurites were manually traced using ImageJ v1.40 and the NeuronJ plugin v1.4.0. Graphs show the effects of UCB on (A) axonal length, (B) number of axonal branches, and (C) length of axonal branches at 3 and 9 DIV, from at least three independent experiments. *p < 0.05 and **p < 0.01 vs. respective vehicle.
Treatment of Immature Hippocampal Neurons with UCB Reduces Axonal Growth Cone Area
Axonal growth cones direct axons to their synaptic partner through integration of multiple signaling cues (Yu and Bargmann, 2001). In previous studies, it was shown that the development of cortical axon branches may be correlated with alterations of axonal growth cone behaviors, with large growth cones more likely to pause and form collateral braches and small growth cones more likely to translocate (Szebenyi et al., 2001). Because UCB affected axonal elongation and branching, we decided to investigate if UCB is also able to alter growth cone dynamics. As a first step to address this issue, neurons treated with UCB at 1 DIV were then fixed at 3 or 9 DIV and immunostained with anti-tubulin β III, phalloidin to visualize F-actin, and an antibody to ezrin, moesin, and radaxin (ERMs) that link filopodia to the plasma membrane. At 3 DIV, vehicle-treated growth cones had a large spread morphology with many actin bundles, filopodia, and a characteristic microtubule loop in the central region [Fig. 7(A)]. As expected, by 9 DIV, most growth cones were smaller and had a reduced central region, characteristic of more differentiated neurons (Ramakers et al., 1991).
Figure 7.

Treatment of differentiating hippocampal neurons with unconjugated bilirubin (UCB) reduces growth cone area. A: Representative images of growth cones from neurons treated with vehicle, UCB 50 μM or UCB 100 μM for 24 h at 1 DIV and fixed and labeled at 3 or 9 DIV with antitubulin β III (blue), phalloidin to visualize actin (green), and anti-ERM proteins (red). B: Effect of UCB on growth cone area at 3 and 9 DIV from at least three independent experiments. *p < 0.05 vs. respective vehicle. Scale bar equals 10 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Neurons treated with UCB presented smaller growth cones with a slight, though not significant, decrease in the number of filopodia, without any obvious changes in actin or tubulin organization. However, differences were observed when we measured the spread area of each growth cone. A substantial change in the distribution in growth cone sizes was observed following treatment with UCB [Fig. 7(B)], with cultures showing fewer large growth cones and an increased proportion of growth cones with small spread areas (~10%). Interestingly, this alteration was sustained through 9 DIV and was concentration dependent (~10% and ~20% decrease in area for 50 and 100 μM UCB, respectively). Thus, even though UCB does not have a striking effect on growth cone morphology or cytoskeletal arrangement, the reduction in the number of large growth cones observed in UCB-treated neurons may be related to the decline of branching formation.
Treatment of Immature Hippocampal Neurons with UCB Reduces Spinogenesis
The formation of synapses in vertebrate organisms occurs over a protracted period of development, beginning in the embryo and extending well into the early postnatal period and is tightly coupled to neuronal differentiation and the establishment of neuronal circuitry (Waites et al., 2005). In hippocampal neuronal cultures, initiation of dendritic spines occurs by 9–11 DIV and mature/stable structures can be found by 18–21 DIV. To examine the effect of UCB on dendritic spine formation and synapse localization, 1 DIV neurons were treated with UCB for 24 h, then UCB was replaced with conditioned growth media without UCB, and cells were allowed to develop until 21 DIV. Dendritic spines were visualized using phalloidin to detect F-actin, dendritic shafts stained with MAP2, and synaptic sites were identified by obligatory colocalization of SV2 and actin rich puncta. Because we have determined synapse density by protein colocalization, we cannot report on the functional characteristics of the synapses.
Treatment of immature hippocampal neurons with UCB led to a significant decrease in the density of dendritic spines (~25% for 50 and 100 μM UCB) and of synapses (~25% and ~45% for 50 and 100 μM UCB, respectively) along dendrites [Fig. 8(A–C)]. During maturation, dendritic spine morphology changes from long, thin immature spines to shorter spines tipped by a bulbous head (Lippman and Dunaevsky, 2005). Vehicle-treated neurons exhibited short, mushroom-shaped spines, whereas UCB-treated neurons displayed long, thin spines, resembling a more immature pattern [Fig. 8(A)]. These observations were next analyzed to give a numerical value by calculating the spine neck length to spine head width ratio [Fig. 8(D)]. This ratio was increased in neurons treated with UCB by ~25% and ~35% for 50 and 100 μM UCB, respectively. Together, these data indicate that a short exposure of developing neurons to UCB can adversely affect subsequent dendritic spine formation and synapse establishment. Together, our results suggest that UCB may deleteriously influence morphological processes crucial for proper formation of the neuronal circuits in the brain, including dendrite and axon differentiation, development of dendritic arbor complexity, and dendritic spine formation.
Figure 8.

Treatment of differentiating hippocampal neurons with unconjugated bilirubin (UCB) reduces spinogenesis. A: Representative images of dendrites from neurons treated with vehicle, UCB 50 μM or UCB 100 μM for 24 h at 1 DIV and fixed and labeled at 21 DIV with anti-MAP2 to visualize dendritic shafts (green), phalloidin to visualize actin at the dendritic spines (red), and anti-SV2 to visualize the pre-synaptic terminal (blue). Arrowheads identify a synaptic site by localization of SV2 adjacent to a dendritic spine. Graphics show the effect of UCB on dendritic spine (B) and synapse (C) density, as well as on dendritic spine morphology (D). **p < 0.01 vs. vehicle. Scale bar equals 10 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
Following moderate to severe hyperbilirubinemia, UCB-induced neurological damage may range from reversible alterations to chronic and permanent clinical sequelae or even death, a condition termed kernicterus (Am Academy Pediatrics, 2004). Cohort studies have found an association between milder hyperbilirubinemia and long-term adverse neurodevelopmental effects that are more subtle than the drastic effects of kernicterus (Seidman et al., 1991; Soorani-Lunsing et al., 2001; Zhang et al., 2003). In addition, neonatal hyperbilirubinemia and Gilbert’s syndrome have been linked to the development of mental disorders (Dalman and Cullberg, 1999). Here, we have demonstrated that UCB affects neural precursor viability, impairs neurogenesis, and reduces neurite arborization and synaptic connectivity of developing neurons. Thus, UCB may deleteriously influence several aspects of brain development and function. It is noteworthy that these effects are consistent with the cortical neuropathology observed in schizophrenia, a mental illness that was already suggested to be related with hyperbilirubinemia (Miyaoka et al., 2000). Hence, our study is the first to provide a basis for altered neural development by UCB and its association with long-term changes, leading to uncertain and understudied outcome later in life.
Cellular developmental programs follow different sequences of maturation in the human brain over the last half of gestation and continue after birth (Abraham et al., 2007). Premature infants show increased vulnerability to multiple insults that interfere with basic mechanisms of neuronal and glial maturation during this time frame, leading to delayed synaptogenesis and dendritic arborization compared to the full-term infant brain (Kinney, 2006). Interestingly, hyperbilirubinemia is more prevalent, more severe, and its course more prolonged in preterm infants (Watchko and Oski, 1992; Watchko and Maisels, 2003) and these babies are at increased risk for bilirubin encephalopathy (Cashore and Stern, 1984).
We used neurosphere cultures to investigate the effects of UCB on the viability of undifferentiated precursor neural cells and on the modulation of neurogenesis and astrogliogenesis. Other authors have used neurosphere cultures to study defective astroglial and neuronal functions in fetal alcohol syndrome as a result of altered proliferation and differentiation of CNS progenitor cells (Vemuri and Chetty, 2005). Neurospheres were also used to demonstrate that even low levels of lead can inhibit the proliferation and differentiation of neural precursors, negatively impacting brain structure and function and thus supporting the need for prevention of prenatal lead exposure (Huang and Schneider, 2004). Nevertheless, lead exposure (0.01–10 μM) had no effect on neurosphere viability. In contrast, in the present study, a 48-h exposure of proliferating neural precursors to UCB was shown to decrease cellular viability. This is not surprising because we have previously demonstrated a decrease in the number of viable immature astrocytes and neurons when exposed to UCB (Falcão et al., 2006). However, to the best of our knowledge, no other study has demonstrated the UCB effects on proliferation of neural precursor cells. The harmful effects by UCB were mostly noticed on neuronal differentiation. It is important to note that the neurospheres were exposed to UCB before the cells were dissociated, then induced to differentiate in the absence of UCB. Because dead cells will not adhere under these conditions, all the differentiated cells must arise from precursors that were viable at the time of plating. This supports the conclusion that effects of UCB exposure on neurogenesis are distinct from the effects on precursor viability. Our results suggest that neurogenesis may be more sensitive to UCB than gliogenesis, perhaps because neuronal precursors are more sensitive to UCB injury. Whether UCB affects neurons after differentiation or modulates the expression of cell-cycle regulators required for neural differentiation remains to be determined.
An optimally functioning nervous system depends on growth and elaboration of neuronal processes in order to allow proper communication between neuronal networks (Chen and Ghosh, 2005). Neuronal connectivity requires the growth of axons to their target region and the formation of dendritic trees that extend into specific layers. Within the target region, growth cones at the tips of extending axons are guided to finer target fields including specific subcellular compartments where they form synapses (Redmond, 2008). Our results demonstrate that a transient exposure of developing neurons to UCB, in conditions mimicking a severe condition of neonatal hyperbilirubinemia, produce changes in both dendritic and axonal arborizations, which are sustained throughout neuronal development. The fact that UCB-induced neuronal death is below 10% throughout the time in culture suggests that these effects may be related to changes in the mechanisms of neurite formation, rather than to cellular dysfunction. Interestingly, reduction in dendrite growth following cerebral ischemia was reported to be due to excessive glutamate in the extracellular space (Esquenazi et al., 2002), and we have demonstrated that exposure of neurons to UCB leads to a decreased glutamate uptake (Silva et al., 2002) and a higher secretion (Falcão et al., 2006). Thus, the accumulation of extracellular glutamate may, at least in part, be responsible for the impairment of neurite output upon UCB treatment.
Both in vitro and in vivo, axon elongation and branching generally precedes that of dendrites (Dotti et al., 1988). This fact may account for a more marked action of UCB on total axonal output at 3 DIV, while at 9 DIV it is the dendritic output that appears to be more affected. UCB-induced neurite alterations were mainly observed in branch development and elongation, suggesting a particular action of UCB at this level.
Growth cones are highly sensitive structures that may suffer retraction or collapse and exhibit disintegration of the cytoskeletal structures in the presence of toxic or therapeutic agents (Leong et al., 2001; Radwan et al., 2002). Although neuronal cytoskeletal disassembly was observed in a prior study of our group using much higher UCB to human serum albumin ratio (Silva et al., 2002), in the present conditions, growth cone area was reduced, but no marked abnormalities of growth cone cytoskeleton were noticed. This suggests that, at lower concentrations, UCB affects the cytoskeletal dynamics required for neurite outgrowth and branching without inducing complete growth cone collapse or neurite retraction.
Our data demonstrate that the events triggered by UCB in neurons at 1 DIV not only modify dendrite branching but also reduce dendritic spine and consequently synapse density. Previous reports indicate that UCB exposure leads to an alteration of the electrophysiology of nervous tissue. In vitro studies using hippocampal slices displayed decreased synaptic activity and increased postsynaptic excitability after UCB exposure (Hansen, 1994), whereas UCB induced impairment of specific membrane-bound neurotransmitter uptake was observed in synaptic vesicle membranes (Roseth et al., 1998). In this context, our results suggest that neurons exposed to UCB are not able to form the correct network connectivity, probably as a result of a deficient mechanism of either pre- or postsynaptic assembly.
Immature dendrites are characterized by the presence of filopodia-like spines, whereas more mature dendrites have spines with some type of spine head (Lippman and Dunaevsky, 2005). It is believed that the relative size of the spine head is proportional to its activity and increased strength, probably reflecting the increased number of postsynaptic receptors in the spine. For example, long-term potentiation, the cellular correlate of increased “memory” increases spine head diameter (Hayashi and Majewska, 2005). In accordance, individuals with mental retardation present longer, thinner dendritic spines with a more immature morphology (Ramakers, 2002). UCB-treated neurons exhibited more filopodial-like spines with increased spine neck length to head width ratio; these “immature” dendritic spines may result in a reduction of synaptic strength and account, in part, for cases of mental retardation following severe neonatal hyperbilirrubinemia.
Current knowledge associates mental retardation during childhood and schizophrenia and dementia during early adulthood with altered neurogenesis and loss of dendrites and axons, resulting in changes of neurite patterning and deficient synaptic wiring (Benitez-King et al., 2004; Dierssen and Ramakers, 2006). Many of these long-term effects are developmental and emerge over time (Armstrong, 2006). Common conditions during the perinatal period, including sepsis/inflammation (Marx et al., 2001), hypoxia/ischemia (Park et al., 1996), and iron deficiency (Carlson et al., 2007), have been reported to cause defects in the morphological development of neurites. These effects can be rapid and reversible or permanent, with consequent impairment of neuronal network activity.
A recent study reported that serum Tau levels of jaundiced term newborns are significantly higher in infants that manifested auditory neuropathy, neurologic abnormalities, or electroencephalogram abnormalities than in infants without these abnormalities (Okumus et al., 2008). Tau, a microtubule-associated structural protein, is released into the extracellular space when an axonal injury occurs (Spillantini and Goedert, 1998) and is considered a neurobiochemical marker of neuronal damage (Wunderlich et al., 2006) present in the cerebrospinal fluid of patients with Alzheimer’s disease (Johnson and Hartigan, 1999) or Down’s syndrome (Tapiola et al., 2001). In agreement, cerebellar hypoplasia accompanied by significant reductions in the volume and number of neurons were detected in an in vivo model of kernicterus, the Gunn rat model (Conlee and Shapiro, 1997), whereas neuronal necrosis of pyramidal cell layer of hippocampus is observed at autopsy of kernicteric patients (Perlman et al., 1997), and bilateral lesions of the globus pallidus are observed in surviving children with kernicterus using magnetic resonance imaging (Sugama et al., 2001). However, to our knowledge, no determinant neuroanatomical defects are linked to the manifestation of minor neurological dysfunction throughout the first year of life in cases of moderate hyperbilirubinemia (Soorani-Lunsing et al., 2001). This suggests that in the case of moderate hyperbilirubinemia, neurological dysfuntion may be due to altered neuronal function, rather than cell death. Hence, while it is clear that increased levels of UCB affect neuronal viability, our results indicate that the neurodevelopmental consequences of moderate hyperbilirubinemia may also be due to alterations in neuronal morphology and connectivity. Collectively, our data demonstrate that UCB promotes alterations in neurogenesis, neuritogenesis, and synaptogenesis, giving insights into the mechanisms leading to neurologic impairment that may determine early-phase UCB encephalopathy or even compromise the performance of the brain in later life.
Acknowledgments
Contract grant sponsor: Fundação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal and FEDER; contract grant numbers: POCI/SAU-MMO/55955/2004, PTDC/SAU-NEU/64385/2006, SFRH/BPD/26390/2006, and SFRH/BPD/26381/2006.
Contract grant sponsor: EU FP6 IP FunGenES, LSHG-CT 2003-503-494.
Contract grant sponsor: NINDS, RO1-NS049178.
References
- Abraham H, Veszpremi B, Gomori E, Kovacs K, Kravjak A, Seress L. Unaltered development of the archi- and neocortex in prematurely born infants: Genetic control dominates in proliferation, differentiation and maturation of cortical neurons. Prog Brain Res. 2007;164:3–22. doi: 10.1016/S0079-6123(07)64001-1. [DOI] [PubMed] [Google Scholar]
- Am Academy Pediatrics. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114:297–316. doi: 10.1542/peds.114.1.297. [DOI] [PubMed] [Google Scholar]
- Armstrong FD. Neurodevelopment and chronic illness: Mechanisms of disease and treatment. Mental Retard Dev Disabil Res Rev. 2006;12:168–173. doi: 10.1002/mrdd.20114. [DOI] [PubMed] [Google Scholar]
- Behar L, Marx R, Sadot E, Barg J, Ginzburg I. cis-Acting signals and trans-acting proteins are involved in tau mRNA targeting into neurites of differentiating neuronal cells. Int J Dev Neurosci. 1995;13:113–127. doi: 10.1016/0736-5748(95)00001-w. [DOI] [PubMed] [Google Scholar]
- Benitez-King G, Ramirez-Rodriguez G, Ortiz L, Meza I. The neuronal cytoskeleton as a potential therapeutical target in neurodegenerative diseases and schizophrenia. Curr Drug Targets CNS Neurol Disord. 2004;3:515–533. doi: 10.2174/1568007043336761. [DOI] [PubMed] [Google Scholar]
- Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Bjork-Eriksson T, Nordborg C, et al. Neo-cortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci USA. 2006;103:12564–12568. doi: 10.1073/pnas.0605177103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brites D, Fernandes A, Falcão AS, Brito MA, Silva RFM. Glial and neuronal reactivity to unconjugated bilirubin. In: Binder MD, Hirokawa N, Windhorst U, Hirsch MC, editors. Encyclopedia of Neuroscience, Part 7. Germany: Springer-Verlag GmbH Berlin Heidelberg; 2009. pp. 1726–1730. [Google Scholar]
- Brito MA, Lima S, Fernandes A, Falcão AS, Silva RFM, Butterfield DA, Brites D. Bilirubin injury to neurons: Contribution of oxidative stress and rescue by glycoursodeoxycholic acid. Neurotoxicology. 2008a;29:259–269. doi: 10.1016/j.neuro.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Brito MA, Rosa AI, Falcão AS, Fernandes A, Silva RFM, Butterfield DA, Brites D. Unconjugated bilirubin differentially affects the redox status of neuronal and astroglial cells. Neurobiol Dis. 2008b;29:30–40. doi: 10.1016/j.nbd.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Carlson ES, Stead JD, Neal CR, Petryk A, Georgieff MK. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus. 2007;17:679–691. doi: 10.1002/hipo.20307. [DOI] [PubMed] [Google Scholar]
- Cashore WJ, Stern L. The management of hyperbilirubinemia. Clin Perinatol. 1984;11:339–357. [PubMed] [Google Scholar]
- Chen Y, Ghosh A. Regulation of dendritic development by neuronal activity. J Neurobiol. 2005;64:4–10. doi: 10.1002/neu.20150. [DOI] [PubMed] [Google Scholar]
- Conlee JW, Shapiro SM. Development of cerebellar hypoplasia in jaundiced Gunn rats: A quantitative light microscopic analysis. Acta Neuropathol. 1997;93:450–460. doi: 10.1007/s004010050639. [DOI] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalman C, Cullberg J. Neonatal hyperbilirubinaemia—A vulnerability factor for mental disorder? Acta Psychiatr Scand. 1999;100:469–471. doi: 10.1111/j.1600-0447.1999.tb10899.x. [DOI] [PubMed] [Google Scholar]
- Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006;5 (Suppl 2):48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- Dong WK, Greenough WT. Plasticity of nonneuronal brain tissue: Roles in developmental disorders. Mental Retard Dev Disabil Res Rev. 2004;10:85–90. doi: 10.1002/mrdd.20016. [DOI] [PubMed] [Google Scholar]
- Dontchev VD, Letourneau PC. Growth cones integrate signaling from multiple guidance cues. J Histochem Cytochem. 2003;51:435–444. doi: 10.1177/002215540305100405. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquenazi S, Monnerie H, Kaplan P, Le Roux P. BMP-7 and excess glutamate: Opposing effects on dendrite growth from cerebral cortical neurons in vitro. Exp Neurol. 2002;176:41–54. doi: 10.1006/exnr.2002.7906. [DOI] [PubMed] [Google Scholar]
- Falcão AS, Fernandes A, Brito MA, Silva RFM, Brites D. Bilirubin-induced inflammatory response, glutamate release, and cell death in rat cortical astrocytes are enhanced in younger cells. Neurobiol Dis. 2005;20:199–206. doi: 10.1016/j.nbd.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Falcão AS, Fernandes A, Brito MA, Silva RFM, Brites D. Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol. 2006;112:95–105. doi: 10.1007/s00401-006-0078-4. [DOI] [PubMed] [Google Scholar]
- Falcão AS, Silva RFM, Pancadas S, Fernandes A, Brito MA, Brites D. Apoptosis and impairment of neurite network by short exposure of immature rat cortical neurons to unconjugated bilirubin increase with cell differentiation and are additionally enhanced by an inflammatory stimulus. J Neurosci Res. 2007;85:1229–1239. doi: 10.1002/jnr.21227. [DOI] [PubMed] [Google Scholar]
- Fernandes A, Falcão AS, Silva RFM, Gordo AC, Gama MJ, Brito MA, Brites D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J Neurochem. 2006;96:1667–1679. doi: 10.1111/j.1471-4159.2006.03680.x. [DOI] [PubMed] [Google Scholar]
- Fernandes A, Silva RFM, Falcão AS, Brito MA, Brites D. Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J Neuroimmunol. 2004;153:64–75. doi: 10.1016/j.jneuroim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Fraichard A, Chassande O, Bilbaut G, Dehay C, Savatier P, Samarut J. In vitro differentiation of embryonic stem cells into glial cells and functional neurons. J Cell Sci. 1995;108 (Pt 10):3181–3188. doi: 10.1242/jcs.108.10.3181. [DOI] [PubMed] [Google Scholar]
- Gordo AC, Falcão AS, Fernandes A, Brito MA, Silva RFM, Brites D. Unconjugated bilirubin activates and damages microglia. J Neurosci Res. 2006;84:194–201. doi: 10.1002/jnr.20857. [DOI] [PubMed] [Google Scholar]
- Goslin K, Birgbauer E, Banker G, Solomon F. The role of cytoskeleton in organizing growth cones: A microfilament-associated growth cone component depends upon microtubules for its localization. J Cell Biol. 1989;109:1621–1631. doi: 10.1083/jcb.109.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52. doi: 10.1016/j.pneurobio.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Hansen TWR. Bilirubin in the brain. Distribution and effects on neurophysiological and neurochemical processes. Clin Pediatr (Phila) 1994;33:452–459. doi: 10.1177/000992289403300802. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Majewska AK. Dendritic spine geometry: Functional implication and regulation. Neuron. 2005;46:529–532. doi: 10.1016/j.neuron.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Huang F, Schneider JS. Effects of lead exposure on proliferation and differentiation of neural stem cells derived from different regions of embryonic rat brain. Neurotoxicology. 2004;25:1001–1012. doi: 10.1016/j.neuro.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. The control of dendrite development. Neuron. 2003;40:229–242. doi: 10.1016/s0896-6273(03)00631-7. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer’s disease brain: An update. J Alzheimers Dis. 1999;1:329–351. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- Kinney HC. The near-term (late preterm) human brain and risk for periventricular leukomalacia: A review. Semin Perinatol. 2006;30:81–88. doi: 10.1053/j.semperi.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gates MA, Witke W, Menzies AS, Wehman AM, Macklis JD, Kwiatkowski D, et al. Mena is required for neurulation and commissure formation. Neuron. 1999;22:313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Lasky JL, Wu H. Notch signaling, brain development, and human disease. Pediatr Res. 2005;57:104R–109R. doi: 10.1203/01.PDR.0000159632.70510.3D. [DOI] [PubMed] [Google Scholar]
- Leong CC, Syed NI, Lorscheider FL. Retrograde degeneration of neurite membrane structural integrity of nerve growth cones following in vitro exposure to mercury. Neuroreport. 2001;12:733–737. doi: 10.1097/00001756-200103260-00024. [DOI] [PubMed] [Google Scholar]
- Leuner B, Shors TJ. New spines, new memories. Mol Neurobiol. 2004;29:117–130. doi: 10.1385/MN:29:2:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman J, Dunaevsky A. Dendritic spine morphogenesis and plasticity. J Neurobiol. 2005;64:47–57. doi: 10.1002/neu.20149. [DOI] [PubMed] [Google Scholar]
- Lobner D. Comparison of the LDH and MTT assays for quantifying cell death: Validity for neuronal apoptosis? J Neurosci Methods. 2000;96:147–152. doi: 10.1016/s0165-0270(99)00193-4. [DOI] [PubMed] [Google Scholar]
- Marx CE, Jarskog LF, Lauder JM, Lieberman JA, Gilmore JH. Cytokine effects on cortical neuron MAP-2 immunoreactivity: Implications for schizophrenia. Biol Psychiatry. 2001;50:743–749. doi: 10.1016/s0006-3223(01)01209-4. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Mendola P, Selevan SG, Gutter S, Rice D. Environmental factors associated with a spectrum of neurodevelopmental deficits. Mental Retard Dev Disabil Res Rev. 2002;8:188–197. doi: 10.1002/mrdd.10033. [DOI] [PubMed] [Google Scholar]
- Miyaoka T, Seno H, Itoga M, Iijima M, Inagaki T, Horiguchi J. Schizophrenia-associated idiopathic unconjugated hyperbilirubinemia (Gilbert’s syndrome) J Clin Psychiatry. 2000;61:868–871. doi: 10.4088/jcp.v61n1110. [DOI] [PubMed] [Google Scholar]
- Okabe S, Forsberg-Nilsson K, Spiro AC, Segal M, McKay RD. Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro. Mech Dev. 1996;59:89–102. doi: 10.1016/0925-4773(96)00572-2. [DOI] [PubMed] [Google Scholar]
- Okumus N, Turkyilmaz C, Onal EE, Atalay Y, Serdaroglu A, Elbeg S, Koc E, et al. Tau and S100B proteins as biochemical markers of bilirubin-induced neurotoxicity in term neonates. Pediatr Neurol. 2008;39:245–252. doi: 10.1016/j.pediatrneurol.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3:215–227. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- Perlman JM, Rogers BB, Burns D. Kernicteric findings at autopsy in two sick near term infants. Pediatrics. 1997;99:612–615. doi: 10.1542/peds.99.4.612. [DOI] [PubMed] [Google Scholar]
- Radwan IA, Saito S, Goto F. The neurotoxicity of local anesthetics on growing neurons: A comparative study of lidocaine, bupivacaine, mepivacaine, and ropivacaine. Anesth Analg. 2002;94:319–324. doi: 10.1097/00000539-200202000-00016. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ, De Graan PN, Oestreicher AB, Boer GJ, Corner MA, Gispen WH. Developmental changes in B-50 (GAP-43) in primary cultures of cerebral cortex: Content and phosphorylation of B-50. Int J Dev Neurosci. 1991;9:231–241. doi: 10.1016/0736-5748(91)90043-l. [DOI] [PubMed] [Google Scholar]
- Redmond L. Translating neuronal activity into dendrite elaboration: Signaling to the nucleus. Neurosignals. 2008;16:194–208. doi: 10.1159/000111563. [DOI] [PubMed] [Google Scholar]
- Rodrigues CMP, Solá S, Brites D. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology. 2002a;35:1186–1195. doi: 10.1053/jhep.2002.32967. [DOI] [PubMed] [Google Scholar]
- Rodrigues CMP, Solá S, Castro RE, Laires PA, Brites D, Moura JJG. Perturbation of membrane dynamics in nerve cells as an early event during bilirubin-induced apoptosis. J Lipid Res. 2002b;43:885–894. [PubMed] [Google Scholar]
- Roseth S, Hansen TWR, Fonnum F, Walaas SI. Bilirubin inhibits transport of neurotransmitters in synaptic vesicles. Pediatr Res. 1998;44:312–316. doi: 10.1203/00006450-199809000-00008. [DOI] [PubMed] [Google Scholar]
- Seidman DS, Paz I, Stevenson DK, Laor A, Danon YL, Gale R. Neonatal hyperbilirubinemia and physical and cognitive performance at 17 years of age. Pediatrics. 1991;88:828–833. [PubMed] [Google Scholar]
- Silva R, Mata LR, Gulbenkian S, Brito MA, Tiribelli C, Brites D. Inhibition of glutamate uptake by unconjugated bilirubin in cultured cortical rat astrocytes: Role of concentration and pH. Biochem Biophys Res Commun. 1999;265:67–72. doi: 10.1006/bbrc.1999.1646. [DOI] [PubMed] [Google Scholar]
- Silva RFM, Falcão AS, Fernandes A, Gordo AC, Brito MA, Brites D. Dissociated primary nerve cell cultures as models for assessment of neurotoxicity. Toxicol Lett. 2006;163:1–9. doi: 10.1016/j.toxlet.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Silva RFM, Rodrigues CMP, Brites D. Rat cultured neuronal and glial cells respond differently to toxicity of unconjugated bilirubin. Pediatr Res. 2002;51:535–541. doi: 10.1203/00006450-200204000-00022. [DOI] [PubMed] [Google Scholar]
- Soorani-Lunsing I, Woltil HA, Hadders-Algra M. Are moderate degrees of hyperbilirubinemia in healthy term neonates really safe for the brain? Pediatr Res. 2001;50:701–705. doi: 10.1203/00006450-200112000-00012. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- Strasser GA, Rahim NA, VanderWaal KE, Gertler FB, Lanier LM. Arp2/3 is a negative regulator of growth cone translocation. Neuron. 2004;43:81–94. doi: 10.1016/j.neuron.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Sugama S, Soeda A, Eto Y. Magnetic resonance imaging in three children with kernicterus. Pediatr Neurol. 2001;25:328–331. doi: 10.1016/s0887-8994(01)00306-x. [DOI] [PubMed] [Google Scholar]
- Sun YE, Martinowich K, Ge W. Making and repairing the mammalian brain-signaling toward neurogenesis and gliogenesis. Semin Cell Dev Biol. 2003;14:161–168. doi: 10.1016/s1084-9521(03)00007-7. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Dent EW, Callaway JL, Seys C, Lueth H, Kalil K. Fibroblast growth factor-2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci. 2001;21:3932–3941. doi: 10.1523/JNEUROSCI.21-11-03932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapiola T, Soininen H, Pirttila T. CSF tau and Abeta42 levels in patients with Down’s syndrome. Neurology. 2001;56:979–980. doi: 10.1212/wnl.56.7.979. [DOI] [PubMed] [Google Scholar]
- Truman P. Jaundice in the preterm infant. Paediatr Nurs. 2006;18:20–22. doi: 10.7748/paed.18.5.20.s24. [DOI] [PubMed] [Google Scholar]
- Vemuri MC, Chetty CS. Alcohol impairs astrogliogenesis by stem cells in rodent neurospheres. Neurochem Int. 2005;47:129–135. doi: 10.1016/j.neuint.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- Watchko JF, Maisels MJ. Jaundice in low birthweight infants: Pathobiology and outcome. Arch Dis Child Fetal Neonatal Ed. 2003;88:F455–F458. doi: 10.1136/fn.88.6.F455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watchko JF, Oski FA. Kernicterus in preterm newborns: Past, present, and future. Pediatrics. 1992;90:707–715. [PubMed] [Google Scholar]
- Wunderlich MT, Lins H, Skalej M, Wallesch CW, Goertler M. Neuron-specific enolase and tau protein as neurobiochemical markers of neuronal damage are related to early clinical course and long-term outcome in acute ischemic stroke. Clin Neurol Neurosurg. 2006;108:558–563. doi: 10.1016/j.clineuro.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Ying QL, Smith AG. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003;365:327–341. doi: 10.1016/s0076-6879(03)65023-8. [DOI] [PubMed] [Google Scholar]
- Yu TW, Bargmann CI. Dynamic regulation of axon guidance. Nat Neurosci. 2001;4 (Suppl):1169–1176. doi: 10.1038/nn748. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Holthoff K. From form to function: Calcium compartmentalization in dendritic spines. Nat Neurosci. 2000;3:653–659. doi: 10.1038/76609. [DOI] [PubMed] [Google Scholar]
- Zhang L, Liu W, Tanswell AK, Luo X. The effects of bilirubin on evoked potentials and long-term potentiation in rat hippocampus in vivo. Pediatr Res. 2003;53:939–944. doi: 10.1203/01.PDR.0000061563.63230.86. [DOI] [PubMed] [Google Scholar]