

Abstract
The Lats2 tumor suppressor protein has previously been implicated in promoting p53 activation in response to mitotic apparatus stress, by preventing Mdm2-driven p53 degradation. We now report that Lats2 also plays a role in an ATR-Chk1-mediated stress checkpoint in response to oncogenic H-Ras. Activated mutant H-Ras triggers the translocation of Lats2 from centrosomes into the nucleus, coupled with an increase in Lats2 protein levels. This leads to induction of p53 activity, upregulation of proapoptotic genes, downregulation of antiapoptotic genes and eventually apoptotic cell death. Many of the cells that survive apoptosis undergo senescence. However, a fraction of the cells escape this checkpoint mechanism, despite maintaining high mutant H-Ras expression. These escapers display increased genome instability, as evidenced by a substantial fraction of cells with micronuclei and cells with polyploid genomes. Interestingly, such cells exhibit markedly reduced levels of Lats2, in conjunction with enhanced hypermethylation of the Lats2 gene promoter. Our findings suggest that Lats2 might play an important role in quenching H-Ras-induced transformation, while silencing of Lats2 expression might serve as a mechanism to enable tumor progression.
Keywords: Ras, p53, Lats2, apoptosis, polyploidy
Introduction
Cancer arises through the accumulation of genetic changes that enhance the growth and survival of developing tumor cells. Activating mutations in the Ras pathway occur in approximately 30% of all human cancers (Ding et al. 2008). However, mutation of Ras alone is insufficient to induce tumor development. To a large measure, this is due to the fact that aberrant activation of oncogenic Ras triggers a series of protective cellular responses aimed at curtailing its potential cancer-promoting effects. This protective response is dependent on, and often orchestrated by, a number of pivotal tumor suppressor proteins. For instance, deregulated Ras activity can induce cellular senescence, a process shown to depend at least in part on p53 and on the products of the INK4a tumor suppressor locus (Serrano 1997). More recent evidence implicates Ras-induced p53-dependent senescence as a pivotal cancer-inhibitory mechanism also in animal models and in humans (Bartek et al. 2007; Ventura et al. 2007; Xue et al. 2007; Halazonetis et al. 2008). Consistent with this view, the tumor suppressor p53 is mutated at a high frequency in many tumor types. Even in those cancers that retain wild-type p53, its activity is likely to be compromised by genetic or epigenetic deficiencies in upstream pathways (Vazquez et al. 2008; Wang and El-Deiry 2008).
Recently, we described a novel tumor suppressor axis, Lats2 (Large Tumor Suppressor 2)-Mdm2-p53, which is critical for the maintenance of proper chromosome number in the face of mitotic apparatus stress (Aylon et al. 2006). In response to such stress, Lats2 protein departs the centrosomes, where it normally resides, and translocates to the nucleus. In the nucleus, Lats2 binds the negative regulator of p53, Mdm2, leading to inhibition of p53 degradation and induction of a p53-driven transcriptional response. Within this process is also embedded a positive feedback loop, wherein the Lats2 gene itself is directly transcriptionally activated by p53, leading to a gradual and continuous increase in Lats2 protein levels. This axis underpins a checkpoint mechanism that acts to prevent the proliferation of cells with polyploid genomes.
A number of studies suggest a specific involvement of Lats2 in protecting cells from Ras driven transformation and tumorigenesis. Using a system of V-Ras-transformed NIH3T3, Li et al (2003) found that overexpression of Lats2 could suppress tumorigenesis in nude mice. Subsequently, Voorhoeve et al (2006) reported that downregulation of Lats2 via overexpression of miR-372/3 could bypass H-Ras-induced senescence in primary human fibroblasts.
We now provide evidence that H-Ras activation affects the Lats2 tumor suppressor in a three-pronged manner. Initially, acute signaling propagated from oncogenic Ras leads to a pronounced upregulation of Lats2, through a combination of transcriptional and posttranscriptional mechanisms. This underpins an ATR-Lats2-p53-dependent replicative stress checkpoint response that promotes apoptosis. Following this wave of apoptosis, Lats2-dependent senescence acts as a second line of defense against H-Ras activation. Finally, cells surviving sustained oncogenic H-Ras activity are found to have neutralized the Lats2-p53 tumor suppressor pathway by hypermethylation of the Lats2 gene promoter. These cells emerge with features characteristic of transformation, such as polyploidy, enhanced cell migration and anchorage-independent growth. Remarkably, reconstitution of Lats2 expression leads to a p53-dependent reversal of these transformed features and leads to induction of apoptosis. These findings illustrate the importance of Lats2 in quenching H-Ras-induced transformation and provide experimental evidence that silencing of Lats2 expression may serve as a mechanism to enable tumor progression.
Materials and Methods
Plasmids
The plasmids used are summarized in Supplemental Table 1.
RNA analysis
Total RNA was isolated using NucleoSpin RNA II kit (Macherey-Nagel) or mirVana miRNA isolation kit (Ambion). qRT-PCR was performed as described (Aylon et al. 2006). For small RNAs (5S and miR-373), 2.5 μg total RNA was converted to cDNA using specific primers for reverse transcription (Ambion) and MMLV reverse transcriptase. Expression levels were determined using miR-373 specific primers relative to 5S RNA (Ambion). Primer sequences are listed in Supplemental Table 2.
Protein analysis
Western blot analysis was performed using standard procedures, employing the following antibodies: Lats2 (LA-2; Yabuta et al. 2007), GAPDH (Chemicon), H-Ras (sc-520, Santa Cruz); p53 (mix of DO1+PAb1801) and p21 (sc-397, Santa Cruz). Immunofluorescence was performed as described (Aylon et al. 2006). Lats2 was detected with anti-KPM/Lats2 (Santa Cruz) or LA-2 (Yabuta et al. 2007). Protein signals were quantified using ImageJ software and normalized to the corresponding GAPDH signals.
Senescence was quantified by staining with a β-gal activity kit (Cell Signaling) following 4 or 10 days of hygromycin and blasticidin selection.
Cell culture, transfections, viral infections, Cell cycle analysis, Promoter methylation, Cell migration and Soft agar colony assays are described in Supplementary Materials and Methods.
Results
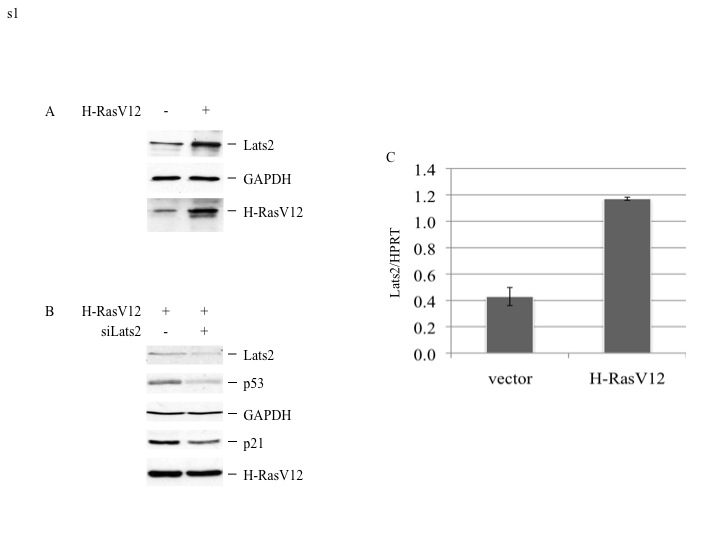
To explore the functional links between oncogenic H-Ras activation and Lats2, we made use of WI-38 human lung embryonic fibroblasts that had been immortalized with telomerase and subsequently subjected to long term passaging in culture (WI-38/hTERTfast; Milyavsky et al. 2003). Four days after infection of these cells with a recombinant retrovirus encoding constitutively activated mutant H-RasV12, a marked increase in endogenous Lats2 protein was evident (Fig 1A). Lats2 mRNA increased nearly twofold (Fig 1B, black bars), partially accounting for the observed elevation in Lats2 protein. We also quantified unspliced, intron-containing Lats2 precursor mRNA, expected to more reliably capture differences in transcription rates (Phelps et al. 2006; Ponzio et al. 2007). This analysis revealed a nearly 4.5 fold increase in Lats2 precursor mRNA in response to H-RasV12 expression (Fig 1B, gray bars). A similar trend was observed also in non-immortalized WI-38 cells (Supplementary Fig S1). Hence, oncogenic H-Ras stimulates Lats2 gene transcription and accumulation of Lats2 protein in both primary and immortalized WI-38 cells.
Figure 1. H-Ras overexpression causes an increase in endogenous Lats2.
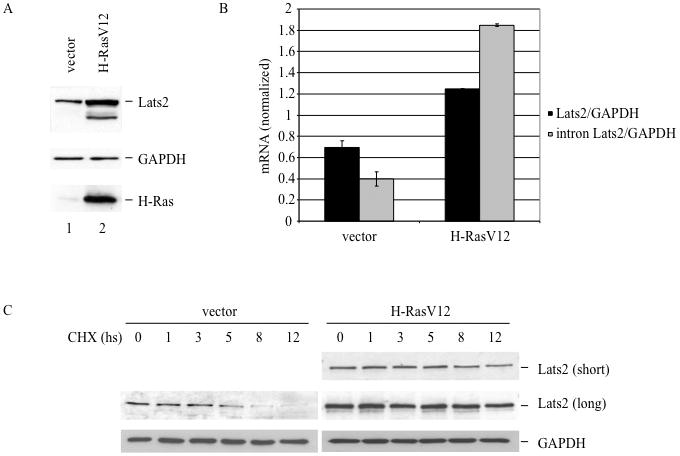
(A) WI-38 cells were infected with H-RasV12 or vector only. Lysates of hygromycin-resistant cells were analyzed four days after infection by Western blot to visualize Lats2 protein, GAPDH and H-Ras. (B) Cells were infected as in (A). RNA was prepared from each culture four days after infection and analyzed by qRT-PCR. Values were normalized to GAPDH mRNA. “Lats2” refers to the product of primers amplifying the exon3-exon4 junction whereas “intron Lats2” amplifies a region within intron 3 of Lats2. (C) Cells were infected as in (A), treated four days after infection with 80μg/ml cycloheximide (CHX) for the indicated time periods, and then harvested for Western blot analysis. For easier comparison, both a short and long exposure of the Lats2 blot are presented.
The microRNA miR-373 can target directly Lats2 mRNA (Voorhoeve et al. 2006). Indeed, H-RasV12 overexpression downregulated miR-373 levels (Supplementary Fig S2), suggesting that repression of miR-373 expression by activated H-Ras also might, in principle, promote Lats2 induction. However, inhibition of miR-373 by a specific locked nucleic acid (LNA) oligonucleotide or overexpression of a pre-miR-373 precursor had very little effect on Lats2 protein levels in these cells (data not shown). It remains possible that, in other cell types with higher levels of endogenous miR-373, repression of miR-373 by activated H-Ras does play a role in modulating Lats2.
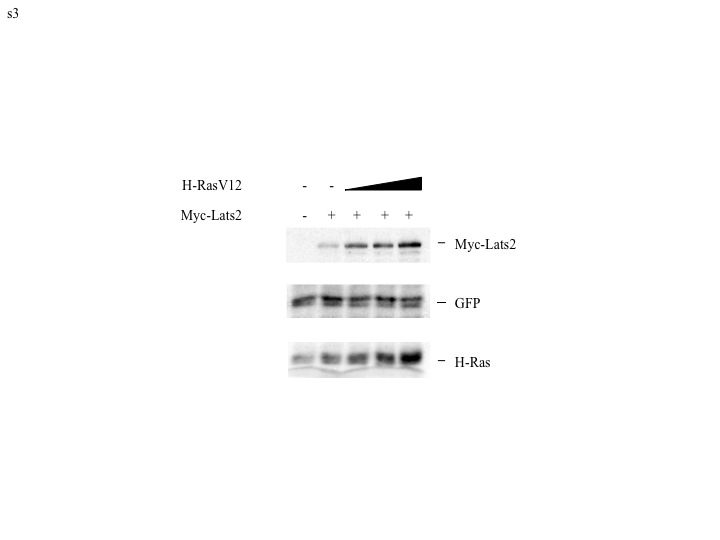
The fold increase of Lats2 protein (Fig. 1A) exceeded that of Lats2 mRNA (Fig. 1B). Therefore, to determine whether H-RasV12 also had an effect on Lats2 protein stability, a cycloheximide chase experiment was performed. As seen in Fig. 1C, endogenous Lats2 protein stability was indeed elevated by H-RasV12. Whereas the half-life of Lats2 was approximately 5 hours in control cells, it was extended to about 8 hours in cells infected with H-RasV12. Moreover, in human breast cancer-derived MCF7 cells, transient transfection of a Lats2 expression plasmid together with increasing amounts of an H-RasV12 plasmid led to a dose-dependent increase in the levels of the exogenous Lats2 protein (Supplementary Fig. S3). Together, these data indicate that multiple mechanisms, transcriptional as well as post-transcriptional, contribute to Lats2 protein upregulation by oncogenic H-Ras.
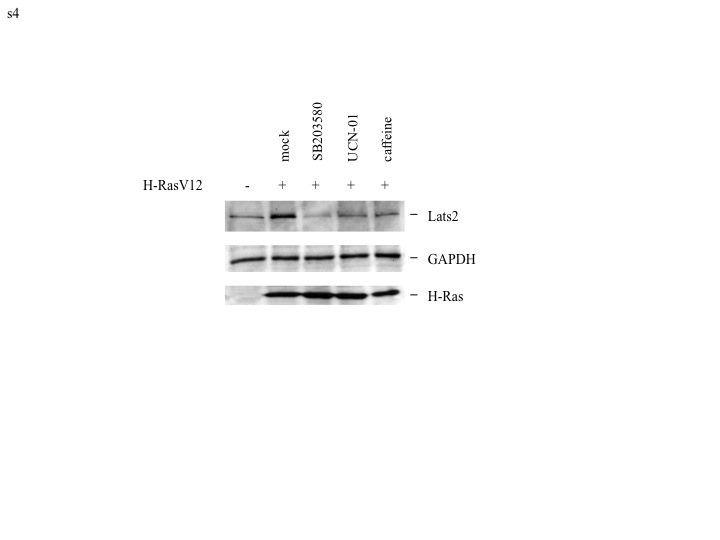
The protein kinases ATM and Chk2 recently have been proposed to mediate cellular senescence in response to DNA damage occurring downstream to aberrant oncogene activation, thereby setting a barrier against oncogene-driven transformation (reviewed in Bartek et al. 2007). However, Lats2 is not activated in response to overt DNA damage (Aylon et al. 2006). On the other hand the ATM-related kinase, ATR, known to play an important role in the response to DNA replication stress, has also been implicated in H-Ras signaling (Abulaiti et al. 2006; Fikaris et al. 2006; Mallette et al. 2007). We therefore investigated whether ATR might be involved in relaying the H-Ras-driven signal for Lats2 induction. Indeed, expression of H-RasV12 caused a marked increase of phosphorylated ATR (Fig 2A, lanes 1,5), confirming that H-Ras-induced oncogenic stress leads to ATR activation in these cells. Strengthening the possibility that ATR mediates the pertinent signal, treatment of H-RasV12 expressing cells with caffeine, a compound that inhibits ATR (as well as ATM, although less specifically), decreased basal levels of Lats2 and impeded Lats2 accumulation (Fig 2A, compare lanes 5,7). ATR-mediated phosphorylation of the Chk1 kinase on serine 345 is key to the propagation of replication stress signals (Jiang et al. 2003). Indeed, oncogenic H-Ras led to a substantial induction of Chk1 Ser345 phosphorylation, suggested by the appearance of a slower migrating form of Chk1 and formally proven with phosphospecific antibodies (Fig 2A, compare lanes 1 and 5). These phosphorylation events were abrogated by treatment with caffeine (compare lanes 5 and 7). Importantly, treatment with the Chk1 inhibitor UCN-01 (lane 8) significantly diminished basal as well as H-RasV12-induced Lats2 protein. Similar effects were observed in HCT116 colorectal cancer-derived cells (Supplementary Fig S4), supporting the generality of this signaling pathway. In contrast, the involvement of the stress kinase p38 varied among different cell lines; whereas treatment with the p38 inhibitor SB203580 abrogated H-RasV12-induced Lats2 accumulation in HCT116 cells (Supplementary Fig. S4), it failed to do so in the non-transformed WI-38 cells (Figure 2A). It is also noteworthy that both these cell types lack functional p14ARF tumor suppressor protein (Burri et al. 2001; Milyavsky et al. 2003), indicating that, at least in WI-38 and HCT116 cells, ARF is dispensable for the induction of Lats2 by oncogenic Ras. Together, these findings imply that oncogenic H-Ras generates ATR-Chk1-dependent stress, which induces Lats2 accumulation.
Figure 2. Chk1 and ATR are implicated in Lats2 accumulation in response to activated H-Ras.
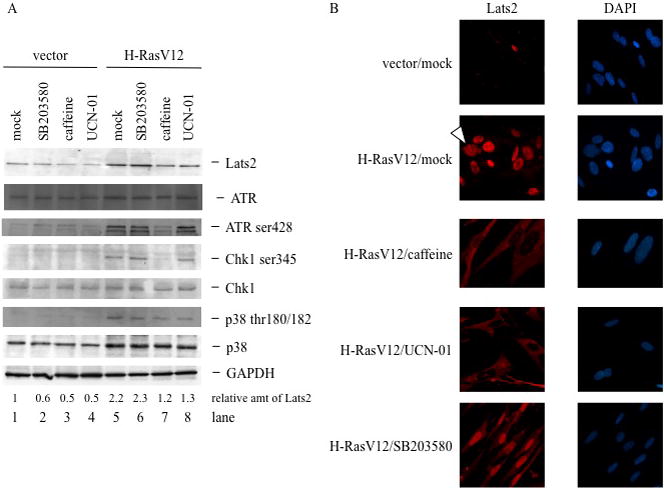
(A) Cells were infected as in Fig. 1A. Four days after infection, hygromycin-resistant cultures were treated overnight with either 10μM SB 203580, 0.5μM UCN-01, or 100μM caffeine, or left non-treated (mock). Cell lysates were subjected to Western blot analysis to visualize ATR, phospho-ATR, total Chk1, phospho-Chk1, p38 and phopho-p38. (B) Cells were processed and treated as in (A), fixed and immunostained to visualize Lats2. Nuclear DNA was visualized by DAPI staining. Arrowhead denotes cell with micronuclei. Protein signals were quantified using ImageJ software and normalized to the corresponding GAPDH signals.
Lats2 is predominantly a centrosomal protein (Toji et al. 2004). Upon damage to the mitotic apparatus Lats2 departs from the centrosome and accumulates in the nucleus, where it binds Mdm2 and stabilizes p53 (Aylon et al. 2006). Remarkably, nuclear accumulation of Lats2, which occurs only rarely and sporadically in unperturbed WI-38/hTERTfast cells (Fig 2B, vector/mock), was dramatically elicited by transient overexpression of H-RasV12 (Fig 2B, H-RasV12/mock). Furthermore, as previously observed (Denko et al. 1995; Abulaiti et al. 2006), cells expressing oncogenic H-RasV12 tended to form micronuclei; these micronuclei contained Lats2 (arrowhead). Interestingly, inhibition of ATR by caffeine or of Chk1 by UCN-01 completely abolished Lats2 nuclear accumulation (Fig 2B). Instead, with both inhibitors, Lats2 was predominantly distributed throughout the cytoplasm. These observations suggest that H-RasV12-induced nuclear translocation of Lats2 is a multistep process; the first step involves release of Lats2 from the centrosomes into the cytoplasm, while in the second step cytoplasmic Lats2 is transported into the nucleus. While both steps are induced by oncogenic H-Ras, only the second one appears to be dependent on ATR and Chk1. Our data also suggests that centrosome-confined Lats2 may not be readily accessible to antibodies, accounting for the very weak staining in control cells (Fig. 2B, vector/mock) as compared to the more robust signal on Western blots (Fig. 2A).
Treatment of H-RasV12-overexpressing WI-38 cells with the p38 kinase inhibitor, SB203580, elicited a mixed phenotype. While the majority of Lats2 protein became nuclear, a portion remained cytoplasmic (Fig 2B). Interestingly, in HCT116 cells, where SB203580 abrogated Lats2 upregulation in response to H-RasV12 expression (Supplementary Fig S4), it also prevented its nuclear accumulation (Supplementary Fig S5). In sum, we find that constitutively active H-Ras generates a signal that dislodges Lats2 from the centrosomes and drives it into the nucleus in an ATR-Chk1 dependent manner. Depending on cell context, p38 may also participate in this pathway.
In response to mitotic apparatus damage, Lats2 stabilizes p53 by alleviating Mdm2-dependent p53 ubiquitylation and degradation (Aylon et al. 2006). Likewise, oncogenic activation also caused Lats2-dependent p53 accumulation. Whereas knockdown of Lats2 did not affect basal p53 levels (Fig. 3A, lanes 1,2), it strongly attenuated p53 induction by H-RasV12 (lanes 3,4). p21 is a canonical p53-response gene that is markedly upregulated in response to H-Ras (Fig. 3B, lanes 1,3). In Lats2-depleted cells, p21 induction by H-RasV12 was completely abolished at both protein (Fig 3B, lanes 3,4) and RNA (Fig 3C) levels. Modulation of Lats2 also influenced the transcriptional program of other p53 target genes. Thus, transient overexpression of H-RasV12 resulted in increased transcription of the proapoptotic gene Bax and decreased expression of the antiapoptotic gene Birc3/cIAP2 (Fig 3C). Remarkably, Lats2 knock-down (Fig. 3D) reversed this trend, restoring both Bax and Birc3 mRNA to nearly basal levels (Fig. 3C). A similar picture was observed in HCT116 cells (Supplementary Fig S6). Moreover, in an isogenic HCT116 cell line lacking functional p53, knockdown of Lats2 had no effect on p21 or Birc3 mRNA levels (HCT-/-, Supplementary Fig S6), indicating that the transcriptional input of Lats2 downstream to H-Ras activation was strictly p53-dependent. Interestingly, similar to what has been observed in cells under mitotic apparatus stress (Aylon et al. 2006), induction of Lats2 mRNA by oncogenic H-Ras was entirely dependent on p53 (Fig 3D). In all above aspects, Lats2 fits the bill of a classic tumor suppressor protein in that it responds to H-Ras activation by enhancing the expression of cell cycle-inhibitory and proapoptotic genes and diminishing that of antiapoptotic genes.
Figure 3. p53 response to H-RasV12 overexpression is dependent on Lats2.
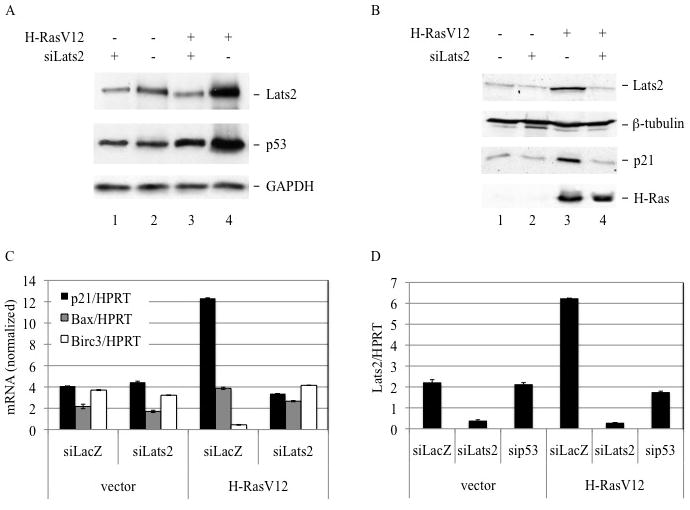
(A) WI-38 cells were infected as in Fig. 1A. Four days after infection, hygromycin-resistant cells were transiently transfected with Lats2 siRNA or control siRNA. Cell lysates were prepared 24 hours after transfection and analyzed by Western blot to visualize Lats2, p53 and GAPDH. (B) Cells were processed as in (A) and subjected to Western blot analysis with antibodies against Lats2, β-tubulin, p21 and H-Ras. (C) Cells were processed as in (A). RNA was prepared 24 hours after transfection and subjected to qRT-PCR analysis with primers specific for the indicated genes. Values were normalized to HPRT mRNA. (D) Cells were processed as in (A), except that transfection was with either LacZ siRNA, Lats2 siRNA or p53 siRNA. RNA was prepared 24 hours after transfection and subjected to qRT-PCR analysis with primers specific for the indicated genes. Values were normalized to HPRT mRNA.
We next explored the biological consequences of the Ras-Lats2-p53 pathway. Within 4 days, expression of H-RasV12 led to a nearly six-fold increase in apoptosis in WI-38 cells (2.3% to 13.4%), as assessed by the presence of cells with a sub-G1 (<2N) DNA content (Fig 4A, top panels). Remarkably, Lats2 knock-down reduced apoptosis to nearly basal levels, with a concurrent elevation in the number of polyploid (>4N) cells (from 10.8% to 16.6%, Fig. 4A, top panels). Treatment with caffeine, which inhibited the nuclear accumulation of Lats2 in response to H-RasV12, had a similar effect as Lats2 knock-down; specifically, caffeine diminished H-RasV12-driven apoptosis and increased the percentage of polyploid cells (Fig 4B). Hence, ATR-mediated nuclear accumulation of Lats2 is essential to efficiently induce apoptosis and prevent genomic instability in response to oncogenic H-Ras.
Figure 4. Lats2 affects the biological outcome of H-RasV12 overexpression.
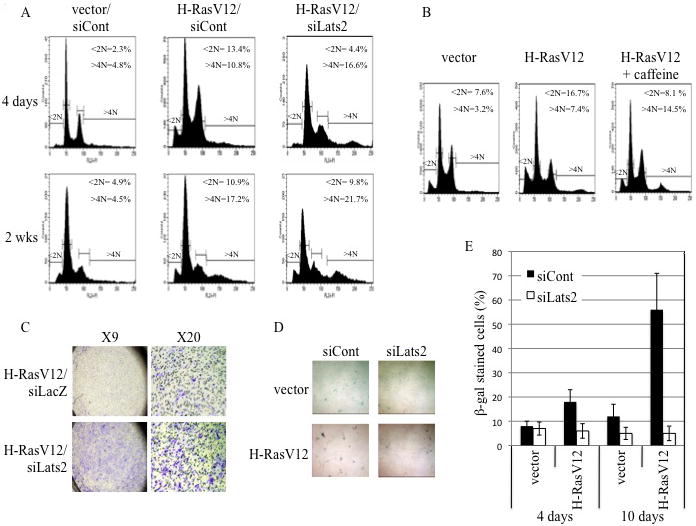
(A) WI-38 cells were infected with various combinations of recombinant retroviruses encoding H-RasV12, Lats2 shRNA (siLats2) or mouse-specific PUMA shRNA (siCont) or empty retrovirus (vector). Cells were fixed, stained with PI and taken for FACS analysis either 4 days after infection and hygromycin and blasticidin selection or after an additional two weeks (2 wks) of growth in culture with antibiotic selection. Positions and percentages of sub-G1 cells (<2N) and polyploid cells (>4N) are indicated. Data is representative of at least three independent experiments. (B) Cells were infected with empty retrovirus (vector) or H-RasV12 and selected with hygromycin. Four days after infection, cultures were treated for 24 hours with 100μM caffeine or left untreated. Cells were fixed, stained with PI and subjected to FACS analysis. (C) Cells were infected and selected as in (B). Four days after infection, cells were transiently transfected with Lats2 siRNA or lacZ siRNA. 24 hours after transfection equal amounts of cells were plated in transwells and subjected in duplicate to migration assays. Wells were photographed at two magnifications as indicated. Data is representative of at least three independent experiments. (D) Cells were infected and processed as in (A). Cultures were stained for β-gal activity after 10 days of combined hygromycin and blasticidin selection. (E) Cells were infected and selected as in (D). Data represents percentage of β-gal positive cells after 4 and 10 days of H-RasV12 expression, based on counting of blue and white cells from three randomly chosen fields from each condition. Error bars represent standard deviation.
Similar to its effect on H-RasV12-driven induction of polyploidy, Lats2 knock-down also enhanced H-RasV12-dependent cell migration (Fig 4C), indicating that Lats2 acts to counteract also this malignancy-related effect of oncogenic H-Ras. Together, these data indicate that Lats2 obstructs the survival and propagation of genomically unstable cells and restricts their malignant potential.
Like apoptosis, senescence serves as a barrier to prevent oncogene-driven cellular transformation (Lowe 1999; Bartkova et al. 2006; Kim et al. 2007). Previously, Lats2 was found as a target of miR-372/3 in a screen for bypassing H-RasV12-induced, p53-dependent senescence in primary BJ cells (Voorhoeve et al. 2006), suggesting that Lats2 may mediate the H-RasV12 signal that triggers p53 also in senescence. To address this possibility, we employed senescence-associated β-galactosidase (SA β-gal) staining to monitor cell senescence at different times after infection with H-RasV12. Four days after infection, when H-RasV12 primarily elicited apoptosis, a mild induction of H-RasV12-induced senescence could be observed (Fig. 4E). Much more dramatic senescence was seen at ten days after infection with H-RasV12 (Fig. 4D,E; the siCont/H-RasV12 panel in Fig. 4D contains a much smaller total number of cells than the other 3 panels). Remarkably, the oncogene-induced senescence was completely abrogated by knockdown of Lats2. Together, these data suggest two waves of Lats2-dependent barriers to combat H-RasV12 oncogenesis. First, the bulk of cells undergo Lats2-dependent apoptosis; then, many of the survivors undergo Lats2-dependent senescence.
Our findings imply that acute signaling, propagated from oncogenic H-Ras, evokes a Lats2-p53-dependent checkpoint response that triggers apoptosis and senescence. It is conceivable that continued expression of activated H-Ras might exert a selective pressure favoring cells that have inactivated this checkpoint response. Such “escapers” are more likely to emerge with features of transformation, e.g. genomic instability, increased proliferation and augmented migration and invasiveness. Indeed, when H-RasV12-infected WI-38 cells were maintained in culture for more extended periods, they exhibited increased genome instability. As illustrated in Fig. 4A, cultures surviving 2 weeks of H-RasV12 expression bear a larger portion of polyploid cells (from 4.5% to 17.2% >4N DNA content without or with H-RasV12, respectively; bottom panels). In these cells, apoptosis levels were slightly lower and, notably, were unaffected by Lats2 knock-down. It is conceivable that such “escapers” might already constitute the majority of cells that have failed to undergo senescence by day 10 (see Fig. 4D). Together, these observations suggest that “escaper” cells might have successfully downregulated the Lats2 response, resulting in enhanced survival and increased genome instability.
To explore more directly the notion that Lats2 dysfunction might underlie the “escaper” phenotype, we examined microscopically the pattern of Lats2 protein distribution in such cells. Indeed, whereas H-Ras activation led to extensive nuclear accumulation of Lats2 in the days immediately following infection (see Fig. 2B), this effect was lost upon more prolonged cultivation (Fig. 5A, third row). Furthermore, such cells exhibited persistent centrosomal Lats2, which failed to translocate to the nucleus even after nocodazole treatment (Fig. 5A, fourth row), unlike the typical response (Aylon et al. 2006; see also Fig. 5A, second row). Hence, prolonged expression of oncogenic H-Ras selects for cells in which the Lats2 response has been numbed. As previously reported (Abulaiti et al. 2006), long-term H-RasV12 expressors displayed an elevated incidence of micronuclei (Fig 5A, DAPI), which became even more pronounced following nocodazole treatment. This further argues that numbing of the Lats2 response enables the survival and propagation of genomically unstable cells.
Figure 5. Chronic expression of H-RasV12 selects for Lats2 silencing.
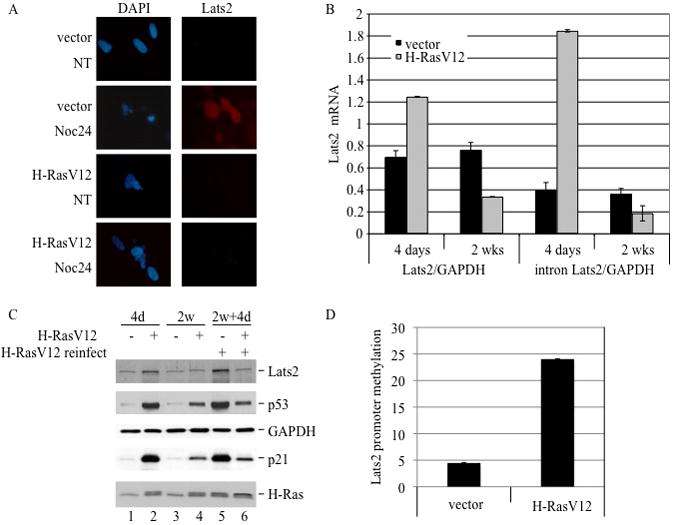
(A) WI-38 cells were infected with either vector only or H-RasV12. After three weeks of selection, cells were treated overnight with 200ng/ml nocodazole (NOC) or DMSO only (NT). Fixed cells were immunostained to visualize Lats2. Nuclear DNA was visualized by DAPI staining. (B) Cells were infected as in (A). RNA was analyzed by qRT-PCR following 4 days or 2 weeks of selection, using exon-junction Lats2 primers (left) or intronic Lats2 primers (right). In both instances, values were normalized to GAPDH mRNA. (C) Cells were infected as in (A). After two weeks of growth under hygromycin selection, cells were infected with a recombinant retrovirus expressing H-RasV12 carrying puromycin resistance and grown for an additional 4 days (2w+4d). Cell lysates were subjected to Western blot analysis to visualize Lats2, p53, GAPDH, p21 and H-Ras. (D) Cells were infected as in (A). After two weeks of selection, high molecular weight DNA was extracted, treated with bisulfate and analyzed by qRT-PCR using methylation-specific primers. Values were normalized to non-methylated ALU sequence and a total-methylated SssI sample.
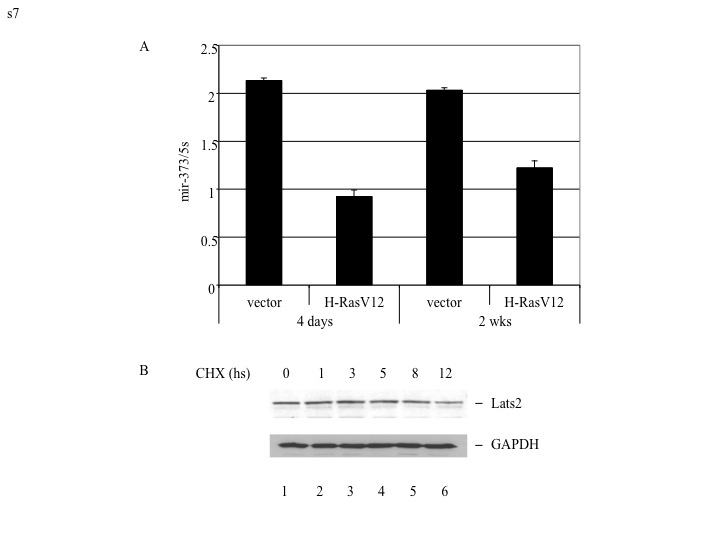
How is the Lats2 response dampened in cells that have evaded the selective pressure imposed by persistent H-RasV12 expression? We observed no significant difference in miR-373 expression between short and long-term H-RasV12 expressors (Supplementary Fig S7A), nor was there a change in Lats2 protein half-life, which remained approximately 8 hours (Supplementary Fig S7B). In contrast, Lats2 transcription was markedly downregulated in the “escaper” cells, as evident from the quantitative analysis of mature as well as precursor intronic Lats2 mRNA (Fig 5B). Numbing of the Lats2-p53 axis by long-term H-RasV12 activation could be confirmed also at the protein level (Fig 5C). Furthermore, diminished Lats2 expression resulted in reduced levels of p53 and p21 (Fig 5C, compare lane 2 and 4). Remarkably, once cells had switched off the Lats2 axis, even reintroduction of extra H-RasV12 failed to upregulate Lats2 again, despite a clear increase in H-Ras protein (Fig 5D, compare lane 4 and 6). Likewise, Abulaiti et al. (2006) reported that chronic overexpression of Ras in cultured rat thyroid cells selects for the emergence of survivors that have switched off the apoptotic response to excess Ras; such cells were shown to display compromised p53 activity and to have become refractory to a second challenge with activated Ras.
Promoter hypermethylation plays a significant role in gene inactivation in various human cancers (reviewed in Esteller 2008). Moreover, Lats2 promoter hypermethylation in tumors has been documented (Jiang et al. 2006; Shema et al. 2008). We therefore asked whether elevated Lats2 promoter hypermethylation might underlie also the downregulation of Lats2 transcription in this in vitro “tumor progression” model. To this end, we performed methylation-specific qPCR analysis on genomic DNA. As seen in figure 5D, selection for survival with chronic expression of H-RasV12 resulted in a pronounced increase of Lats2 promoter hypermethylation. This suggests that Lats2 promoter hypermethylation enables the emergence of transformed cells harboring activated oncogenic H-Ras.
If the silencing of Lats2 expression contributes to the establishment of the H-Ras-driven transformed phenotype, it is conceivable that coexpression of ectopic Lats2 might attenuate this phenotype. One characteristic often associated with oncogenic H-Ras is acquisition of anchorage-independent growth, manifested experimentally as an ability to form colonies in soft agar. Importantly, overexpression of Lats2 dramatically decreased the ability of H-RasV12 cells to form agar colonies (Fig 6). Notably, knockdown of p53 in conjunction with Lats2 overexpression restored the ability of H-RasV12 expressing cells to form colonies (Fig. 6A, right panels), indicating that inhibition of anchorage-independent cell growth by Lats2 is p53-dependent.
Figure 6. Overexpression of Lats2 prevents soft agar colony formation in a p53-dependent manner and triggers apoptosis.
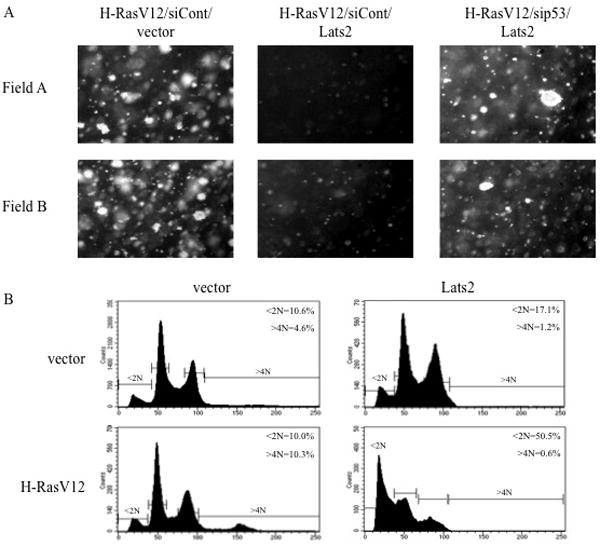
(A) WI-38 cells were infected with various combinations of recombinant retroviruses encoding H-RasV12, Myc-tagged Lats2 (Lats2), p53 shRNA (sip53), mouse-specific PUMA shRNA (siCont) or empty retrovirus (vector). Following selection for two weeks under hygromycin and blasticidin, equal amounts of cells were subjected in duplicate to a soft agar colony formation assay. Four weeks after plating, microcolonies were photographed. Two representative fields are shown for each condition. (B) WI-38 cells were infected with vector only or H-RasV12. Following three weeks of hygromycin selection, cells were infected with vector only or Myc-tagged Lats2 (Lats2) and subjected to blasticidin selection. Cells were harvested 4 days after the second infection, fixed, stained with PI and subjected to FACS analysis.
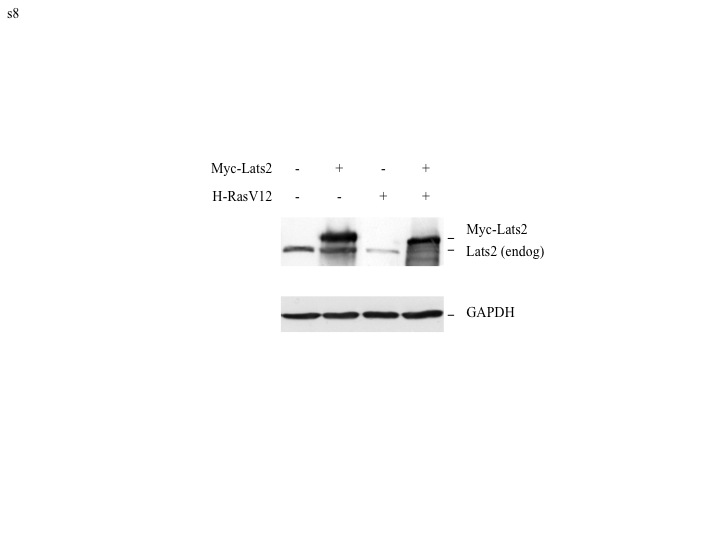
In Fig. 6A, ectopic Lats2 was co-introduced together with H-RasV12 infection, before selection of H-RasV12 “escapers” took place. Since “escapers” presumably underwent strong selective pressure to subvert the Lats2 response, subsequent reintroduction of ectopic Lats2 might be expected to exert even more profound inhibitory effects on those cells. Indeed, whereas overexpression of ectopic Lats2 led to only a modest increase in apoptosis (10.6% to 17.1%) in control (vector) cells (Fig. 6B, upper panels), it had a dramatic effect on the “escapers”, massively inducing apoptosis (10.0% to 50.5%, lower panels), even though the extent of Lats2 overexpression was comparable in the vector and H-Ras “escapers” (Supplementary Fig S8). Moreover, Lats2 expression almost completely eliminated the polyploid subpopulation, suggesting that these cells were preferentially shunted to apoptosis. Thus, restoration of high Lats2 levels in the “escapers” can effectively reverse the outcome of the long-term selection process and curtail the cancer-promoting consequences of chronic H-RasV12 expression.
Discussion
H-Ras is among the most frequently mutated genes in cancer. Intrinsically, H-Ras activation delivers potent oncogenic signals and contributes richly to deregulation of cell proliferation and establishment of transformed features. Luckily, cellular transformation is a relatively rare event. This is thanks to the action of checkpoint mechanisms that eliminate potentially dangerous cells from the replicative pool by shunting them to cell cycle arrest, apoptosis or senescence. Hence, tumor cells must cope with heavy selective pressure to undergo adaptations that allow them to neutralize or bypass these barriers.
The observations described in the present report provide additional insights into the mechanisms that underlie the H-Ras-triggered protective checkpoint response as well as the escape of mutant H-Ras-expressing cells from this response en route to becoming fully transformed. Our findings are consistent with a model wherein activation of oncogenic H-RasV12 results in a stress that is sensed by the ATR-Chk1 checkpoint pathway. Activation of ATR and Chk1 leads to nuclear accumulation and stabilization of Lats2 protein. Lats2 subsequently mediates the p53 response to H-Ras oncogene activation, directing cells towards an antiproliferative transcriptional program as well as inducing p53-dependent transcription of the Lats2 gene itself. Thus, the Lats2-p53 tumor suppressor positive feedback pathway acts to combat the potentially harmful outcome of oncogenic activation. However, rare cells might escape this checkpoint block by inactivating Lats2, as well as other components of the pathway, such as p53. In the case of Lats2, such bypass can be achieved epigenetically by promoter hypermethylation and subsequent quenching of Lats2 gene expression.
At which point in the history of the cells does the Lats2 promoter become hypermethylated? A number of studies suggest that oncogenic Ras can induce the expression of several DNA methyltransferases (Pruitt et al. 2005; Chang et al. 2006), hence, it is possible that the silencing of Lats2 occurs only after H-RasV12 introduction. Yet, given the short duration of the experiment shown in Fig. 5D and the fact that the WI-38/hTERTfast cells underwent extensive passaging in vitro (Milyavsky et al. 2003) prior to H-RasV12 infection, it appears more likely that the stress response elicited through activation of the ATR-Chk1-Lats2-p53 pathway selects for cells with preexisting Lats2 promoter hypermethylation, rather than inducing multiple independent de novo methylation events. In fact, the extent of hypermethylation in the control WI-38/hTERTfast cells (Fig. 5D, vector), albeit low, is already significantly higher than in earlier passage WI-38 cells (data not shown). This scenario is reminiscent of observations made in human breast tissue, where preexistent silencing of the INK4A/ARF tumor suppressor locus by promoter hypermethylation in seemingly normal tissue might provide the setting for the emergence of tumor initiating cells upon subsequent oncogene activation (Bean et al. 2007). Thus, it is tempting to speculate that, likewise, cells with silenced Lats2 may also be present sporadically in normal tissue, offering ready soil for oncogene-driven transformation and tumor development. Notably, loss of p16(INK4A) expression, which took place in the course of WI-38/hTERTfast cell propagation in culture (Milyavsky et al. 2003), may have accelerated DNA methylation (Reynolds et al. 2006), including that of the Lats2 promoter, thus generating a larger pool of cells to select from in response to H-RasV12-induced stress. Furthermore, by creating centrosome dysfunction (McDermott et al. 2006), prior loss of p16 might facilitate Lats2 activation through chronic mitotic apparatus stress (Aylon et al. 2006). This may account for the occasional activation of Lats2 prior to H-RasV12 overexpression (Fig. 2B, upper left).
It still remains to be determined exactly how activation of the Ras-ATR-Chk1 axis leads to Lats2 induction. The data presented in this study implies that this induction is driven at multiple levels. On the one hand, there is an increase in the rate of Lats2 gene transcription, which is dependent on p53 (Fig. 3D); this is reminiscent of the response to mitotic apparatus stress, where p53 is also required for induction of Lats2 gene expression (Aylon et al. 2006). On the other hand, the Ras-ATR-Chk1 axis alters the stability and subcellular localization of the Lats2 protein. Activated H-Ras triggers the departure of Lats2 from centrosomes and its subsequent translocation into the nucleus; whereas the former is not dependent on ATR and Chk1, the latter is. Interestingly, the increased stability of Lats2 protein appears to be correlated with its nuclear translocation, since treatment with compounds such as caffeine and UCN-01, which prevent Lats2 nuclear translocation, also abolish the increase in steady state Lats2 levels. These events might be regulated, at least in part, by putative covalent modifications on the Lats2 protein. Such modification might differ between the different states of Lats2: centrosomal (non-stressed), non-centrosomal cytoplasmic (activated H-Ras together with ATR-Chk1 pathway inhibition, or nuclear (activated H-Ras and functional ATR-Chk1 pathway). Perhaps relevant to this notion is the observation by Toji et al. (2004) that preventing the phosphorylation of Lats2 by Aurora A kinase releases Lats2 from the centrosome, but is not sufficient for nuclear accumulation. We propose that generation of mitotic stress, either by treatment with nocodazole (Aylon et al. 2006) or oncogene activation (this work) is necessary for Lats2 nuclear import.
Our data predict that inactivation of the Lats2 tumor suppressor might facilitate tumor progression. Indeed, this conjecture is further supported by the analysis of human tumors. Thus, in breast cancers, decreased Lats2 expression is linked to biologically aggressive phenotypes, such as large tumor size, high lymph node metastasis and estrogen- and progresterone-receptor negativity (Takahashi et al. 2005). Similarly, in astrocytoma, hypermethylation of the Lats2 promoter was detected in over 70% of the cases and associated with higher tumor grade and increased overall malignancy (Jiang et al. 2006). Moreover, treatment of astrocytoma cell lines with a DNA methyltransferase inhibitor restored Lats2 levels and caused apoptosis, indicating that silencing of Lats2 is an important step in tumor cell resistance to apoptosis (Jiang et al. 2006). Downregulation of Lats2 via promoter hypermethylation is also associated with poor prognosis in acute lymphoblastic leukemia (Jimenez-Velasco et al. 2005). Furthermore, 50% of non-small cell lung carcinoma tumors exhibit decreased Lats2 expression (Strazisar et al. 2008). Interestingly, diminished Lats2 levels are highly correlated with lower expression of the p53 target gene Mdm2 (Strazisar et al. 2008), supporting the notion that downregulation of Lats2 might negatively affect p53 transcriptional activity. Together, these studies are consistent with the view that evasion of the Lats2-p53 tumor suppressor pathway is an important contributor to tumor progression. Reconstitution of Lats2 expression, for instance by erasure of Lats2 promoter hypermethylation, might thus qualify as a potential means to combat tumors in which Lats2 has been silenced but the p53 gene remains intact.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Doron Ginsberg, Reuven Agami and Scott Lowe for the generous gift or plasmids and the following people for other help: Lior Golomb (β-gal staining), Efrat Shema (migration assays) and Sylvia Wilder (excellent technical help). Supported in part by grant R37 CA40099 from the National Cancer Institute, EC FP6 grant LSHC-CT-2004-503576, FP7 funding (ONCOMIRS, agreement 201102), the Dr. Miriam and Sheldon Adelson Medical Research Foundation, and the Yad Abraham Center for Cancer Diagnosis and Therapy. The EC is not liable for any use that may be made of the information contained herein.
References
- Abulaiti A, Fikaris AJ, Tsygankova OM, Meinkoth JL. Ras induces chromosome instability and abrogation of the DNA damage response. Cancer Res. 2006;66(21):10505–10512. doi: 10.1158/0008-5472.CAN-06-2351. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20(19):2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26(56):7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bean GR, Bryson AD, Pilie PG, Goldenberg V, Baker JC, Jr, Ibarra C, et al. Morphologically normal-appearing mammary epithelial cells obtained from high-risk women exhibit methylation silencing of INK4a/ARF. Clin Cancer Res. 2007;13(22 Pt 1):6834–6841. doi: 10.1158/1078-0432.CCR-07-0407. [DOI] [PubMed] [Google Scholar]
- Burri N, Shaw P, Bouzourene H, Sordat I, Sordat B, Gillet M, et al. Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab Invest. 2001;81(2):217–229. doi: 10.1038/labinvest.3780230. [DOI] [PubMed] [Google Scholar]
- Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res. 2006;66(17):8413–8420. doi: 10.1158/0008-5472.CAN-06-0685. [DOI] [PubMed] [Google Scholar]
- Denko N, Stringer J, Wani M, Stambrook P. Mitotic and post mitotic consequences of genomic instability induced by oncogenic Ha-ras. Somat Cell Mol Genet. 1995;21(4):241–253. doi: 10.1007/BF02255779. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Fikaris AJ, Lewis AE, Abulaiti A, Tsygankova OM, Meinkoth JL. Ras triggers ataxia-telangiectasia-mutated and Rad-3-related activation and apoptosis through sustained mitogenic signaling. J Biol Chem. 2006;281(46):34759–34767. doi: 10.1074/jbc.M606737200. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem. 2003;278(27):25207–25217. doi: 10.1074/jbc.M300070200. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Li X, Hu J, Zhou W, Jiang Y, Li G, et al. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci Res. 2006;56(4):450–458. doi: 10.1016/j.neures.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Jimenez-Velasco A, Roman-Gomez J, Agirre X, Barrios M, Navarro G, Vazquez I, et al. Downregulation of the large tumor suppressor 2 (LATS2/KPM) gene is associated with poor prognosis in acute lymphoblastic leukemia. Leukemia. 2005;19(12):2347–2350. doi: 10.1038/sj.leu.2403974. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27(2):662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Pei J, Xia H, Ke H, Wang H, Tao W. Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene. 2003;22(28):4398–4405. doi: 10.1038/sj.onc.1206603. [DOI] [PubMed] [Google Scholar]
- Lowe SW. Activation of p53 by oncogenes. Endocr Relat Cancer. 1999;6(1):45–48. doi: 10.1677/erc.0.0060045. [DOI] [PubMed] [Google Scholar]
- Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21(1):43–48. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, Tlsty TD. p16(INK4a) prevents centrosome dysfunction and genomic instability in primary cells. PLoS Biol. 2006;4(3):e51. doi: 10.1371/journal.pbio.0040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milyavsky M, Shats I, Erez N, Tang X, Senderovich S, Meerson A, et al. Prolonged culture of telomerase-immortalized human fibroblasts leads to a premalignant phenotype. Cancer Res. 2003;63(21):7147–7157. [PubMed] [Google Scholar]
- Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell. 2004;16(4):631–639. doi: 10.1016/j.molcel.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Phelps ED, Updike DL, Bullen EC, Grammas P, Howard EW. Transcriptional and posttranscriptional regulation of angiopoietin-2 expression mediated by IGF and PDGF in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;290(2):C352–361. doi: 10.1152/ajpcell.00050.2005. [DOI] [PubMed] [Google Scholar]
- Ponzio TA, Yue C, Gainer H. An intron-based real-time PCR method for measuring vasopressin gene transcription. J Neurosci Methods. 2007;164(1):149–154. doi: 10.1016/j.jneumeth.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K, Ulku AS, Frantz K, Rojas RJ, Muniz-Medina VM, Rangnekar VM, et al. Ras-mediated loss of the pro-apoptotic response protein Par-4 is mediated by DNA hypermethylation through Raf-independent and Raf-dependent signaling cascades in epithelial cells. J Biol Chem. 2005;280(24):23363–23370. doi: 10.1074/jbc.M503083200. [DOI] [PubMed] [Google Scholar]
- Reynolds PA, Sigaroudinia M, Zardo G, Wilson MB, Benton GM, Miller CJ, et al. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281(34):24790–24802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- Serrano M. The tumor suppressor protein p16INK4a. Exp Cell Res. 1997;237(1):7–13. doi: 10.1006/excr.1997.3824. [DOI] [PubMed] [Google Scholar]
- Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008;22(19):2664–2676. doi: 10.1101/gad.1703008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazisar M, Mlakar V, Glavac D. LATS2 tumour specific mutations and down-regulation of the gene in non-small cell carcinoma. Lung Cancer. 2008 doi: 10.1016/j.lungcan.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, et al. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res. 2005;11(4):1380–1385. doi: 10.1158/1078-0432.CCR-04-1773. [DOI] [PubMed] [Google Scholar]
- Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, et al. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells. 2004;9(5):383–397. doi: 10.1111/j.1356-9597.2004.00732.x. [DOI] [PubMed] [Google Scholar]
- Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7(12):979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124(6):1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Wang W, El-Deiry WS. Restoration of p53 to limit tumor growth. Curr Opin Oncol. 2008;20(1):90–96. doi: 10.1097/CCO.0b013e3282f31d6f. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuta N, Okada N, Ito A, Hosomi T, Nishihara S, Sasayama Y, et al. Lats2 is an essential mitotic regulator required for the coordination of cell division. J Biol Chem. 2007;282(26):19259–19271. doi: 10.1074/jbc.M608562200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.