

Abstract
Genome-wide associations between single-nucleotide polymorphisms and clinical traits were simultaneously conducted using penalized orthogonal-components regression. This method was developed to identify the genetic variants controlling phenotypes from a massive number of candidate variants. By investigating the association between all single-nucleotide polymorphisms to the phenotype of antibodies against cyclic citrullinated peptide using the rheumatoid arthritis data provided by Genetic Analysis Workshop 16, we identified genetic regions which may contribute to the pathogenesis of rheumatoid arthritis. Bioinformatic analysis of these genomic regions showed most of them harbor protein-coding gene(s).
Background
Most of the available statistical approaches for genome-wide association study (GWAS) have focused on studying one single-nucleotide polymorphism (SNP) at a time [1], thereby ignoring the multigenic nature of complex diseases [2,3] and the strong correlation between some SNPs due to linkage disquilibrium (i.e., some SNPs are inherited together in blocks of DNA). As pointed out by Waldron et al. [4], association studies using multiple SNPs have substantial advantages over those based on SNP associations. To capture the correlation between SNPs in regions of low recombination, haplotype-based methods that recognize the existence of linkage disequilibrium between genetic markers have been employed for multi-SNP analyses. However, such methods introduce additional problems, including the need to infer haplotypes, the impact of including (or excluding) rare haplotypes, and the need to define haplotype block boundaries. Even with haplotype-based association methods, high false-positive rates and a lack of reproducibility remain major concerns.
Because hopes remain high for the value of GWAS, other approaches need to be pursued that account for the correlation structure among SNPs. Here we have developed a method for GWAS that incorporates regression of a phenotype simultaneously on all available SNPs, i.e., considering a multiple linear regression,
where Y is the phenotypic value, Xj counts one of the two alleles at the jth SNP, and βj is the additive effect of that allele, j = 1,..., p. To account for the issues raised by large p, such as lack of independence between SNPs due to linkage disequilibrium, we will conduct the GWAS using the penalized orthogonal-components regression (POCRE) [5]. POCRE offers a fast and efficient way to identify significant SNPs simultaneously from a large number of candidates.
Methods
GWAS usually entails the collection of a massive amount of SNPs (i.e., large p) for only a small number of biological individuals (i.e., small n). Therefore, identifying the few genetic variants underlying disease risk is equivalent to the task of finding "a very few needles in a haystack", and poses a challenging statistical issue. Zhang et al. [5] recently described the POCRE approach, which sequentially constructs sparsely loaded orthogonal components with proper regularization. They demonstrate this approach works well when fitting regression models with n <<p data.
Let Y be an n-dimensional column phenotype vector, and X be an n × p genotype matrix, where n and p are the number of individuals and number of SNPs, respectively. Further assume both Y and X are centralized, and accordingly assume μ = 0 in Eq. (1). POCRE sequentially constructs orthogonal components 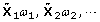 , where
, where 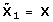 and
and 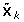 , k ≥ 2 is iteratively built to be orthogonal to
, k ≥ 2 is iteratively built to be orthogonal to 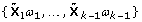 . The loading ωκ, k ≥ 1, is obtained as γ/||γ||, which minimizes
. The loading ωκ, k ≥ 1, is obtained as γ/||γ||, which minimizes
where gλ(γ) is a penalty function defined by a proper regularization on γ with tuning parameter λ.
When gλ(γ) ≡ 0, the optimal γ solving Eq. (2) is proportional to the leading eigenvector of 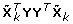 . Zhang et al. [5] employed empirical Bayes thresholding methods proposed by Johnstone and Silverman [6] to introduce proper penalty gλ(γ). Such penalty benefits estimating covariance between phenotype and genotypes, and provides adaptively sparse loadings of orthogonal components. The empirical Bayes implementation is also computationally efficient. The tuning parameter λ also accounts for possible dependence structure among different SNPs, and 10-fold cross-validation was employed to elicit its optimal values ranging from 0.8 to 1, i.e., considering candidate values λ ∈ {0.8, 0.82, 0.84, 0.86, 0.88, 0.9, 0.92, 0.94, 0.96, 0.98, 1}.
. Zhang et al. [5] employed empirical Bayes thresholding methods proposed by Johnstone and Silverman [6] to introduce proper penalty gλ(γ). Such penalty benefits estimating covariance between phenotype and genotypes, and provides adaptively sparse loadings of orthogonal components. The empirical Bayes implementation is also computationally efficient. The tuning parameter λ also accounts for possible dependence structure among different SNPs, and 10-fold cross-validation was employed to elicit its optimal values ranging from 0.8 to 1, i.e., considering candidate values λ ∈ {0.8, 0.82, 0.84, 0.86, 0.88, 0.9, 0.92, 0.94, 0.96, 0.98, 1}.
The sequential construction of the orthogonal components stops when the optimal γ solving Eq. (2) is zero, which implies 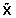 is hardly correlated to Y Then, regressing Y on the orthogonal components, i.e., , provides an estimate of β1,⋯, βp in Eq. (1). Because non-zero loadings in ωj, j = 1,2,⋯, are sparse, most of estimated β1,⋯, βp are therefore zero, reflecting the fact that most SNPs are insignificantly associated or are even completely uncorrelated to the phenotype of interest.
is hardly correlated to Y Then, regressing Y on the orthogonal components, i.e., , provides an estimate of β1,⋯, βp in Eq. (1). Because non-zero loadings in ωj, j = 1,2,⋯, are sparse, most of estimated β1,⋯, βp are therefore zero, reflecting the fact that most SNPs are insignificantly associated or are even completely uncorrelated to the phenotype of interest.
Results
Using our novel method (POCRE), the rheumatoid arthritis data from Genetic Analysis Workshop 16 were investigated for associations between SNPs and a serum biomarker for rheumatoid arthritis, i.e., antibodies anti-cyclic citrullinated peptide (anti-CCP). In this dataset, only 867 samples were positive for anti-CCP. The data set was preprocessed with the computer program PLINK [7] to control data quality, and with the computer program EIGENSTRAT [8] to control potential population structures. After preprocessing there were 490,613 SNPs remaining for the GWAS. POCRE was applied individually to each chromosome for the simultaneous association of the SNPs in that chromosome with the anti-CCP phenotype. The effects of the 10 principal components constructed by EIGENSTRAT were considered as covariates for the POCRE. Nonzero effects of SNPs were reported on seven chromosomes where the positive 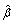 indicates that the minor allele will increase the level of anti-CCP (Table 1).
indicates that the minor allele will increase the level of anti-CCP (Table 1).
Table 1.
SNPs identified with nonzero coefficients in Eq. (1)
| SNP | 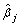 |
Chromosome | SNP Location (bp) | Gene | Gene Location (bp) |
|---|---|---|---|---|---|
| rs233492 | 149.7514 | 6p23 | 14,910,666 | JARID2a | 14,754,745-14,754,848 |
| rs17068819 | 109.0095 | 6q21 | 108,246,263 | SCML4 | 108,130,060-108,252,214 |
| rs922898 | 56.7294 | 6q25.1 | 149,155,750 | UST | 149,110,165-149,439,818 |
| rs6929401 | 83.7685 | 6q25.1 | 149,158,549 | UST | 149,110,165-149,439,818 |
| rs17087579 | 187.2452 | 6q25.1 | 149,886,810 | PPIL4 | 149,867,324-149,908,864 |
| rs11760836 | 358.7046 | 7p21.2 | 14,903,193 | DGKB | 14,151,199-14,909,359 |
| rs11029744 | 300.2939 | 11p14.2 | 26,909,033 | AC016450.10 | 26,972,204-26,975,206 |
| rs10861038 | 63.7626 | 12q23.3 | 102,455,524 | STAB2a | 102,505,181-102,684,635 |
| rs10507167 | 117.5381 | 12q23.3 | 102,490,212 | STAB2a | 102,505,181-102,684,635 |
| rs17055893 | 265.3688 | 13q13.3 | 36,961,628 | POSTNa | 37,034,779-37,070,874 |
| rs2322047 | 324.3976 | 17p12 | 11,556,512 | DNAH9 | 11,442,473-11,813,856 |
| rs9305833 | 102.7568 | 21q21.1 | 18,238,917 | CHODL | 18,195,451-18,561,561 |
aSNP does not reside in any gene. The nearest gene is shown.
Of the 12 SNPs identified to be associated with anti-CCP level, 5 SNPs are from chromosome 6, 2 SNPs from chromosome 12, and 1 SNP each is from chromosomes 7, 11, 13, 17, and 21, respectively. The location of each significant SNP was mapped to the human genome using the Ensembl database http://www.ensembl.org. Based on this analysis, eight of the SNPs were found to reside in seven genes (Table 1). For the other four SNPs, the nearest neighboring genes are listed in the table. None of these genes we identified have previously been linked to rheumatoid arthritis, but several of them encode proteins whose biological roles may be involved in the pathogenesis of this disease. For example, two SNPs (rs922898, rs6929401) that reside in the uronyl-2-sulfotransferase (UST) gene were identified by our method. UST is involved in the chondroitin 3 sulfate and glycan structure biosynthesis pathways and could contribute to optimal cartilage development or repair. PPIL4 is a member of cyclophilin family, a group of proteins crucial for protein folding and immunosuppression by cyclosporin A (CsA) [9]. Polymorphisms in this gene may influence development of inflammation during rheumatoid arthritis or the response of individuals with rheumatoid arthritis to treatment. DGKB encodes diacylglycerol kinase and participates in intracellular signalling processes via several pathways including the protein kinase C pathway [10]. Alteration in intracellular signalling could negatively influence inflammatory processes. Finally, CHODL encodes a type I transmembrane protein including a single carbohydrate recognition domain for the C-type lectins that can worsen inflammation [11].
Discussion
In addition to its diagnostic value for the general risk of rheumatoid arthritis, a high anti-CCP level is associated with a high risk of developing joint damage during the disease. As a result, an anti-CCP-test has been employed to monitor the progression of the disease. With our novel approach for GWAS, we identified several candidate SNPs associated with the level of anti-CCP in rheumatoid arthritis patients. Among the candidate genes identified, only PPIL4 is functionally related to the immune system. Further investigation will be necessary to define the potential roles that the other gene products play in rheumatoid arthritis. Due to the small sample size available for this study (867) and the large total number of SNPs measured (490,613), we expect some of the associations we identified to constitute false positives. Determining false detection rates will require development of additional procedures.
Conclusion
Our analyses using the newly developed method POCRE indicate that the genomic region 6q25.1 may harbor genes associated with anti-CCP level in rheumatoid arthritis patients. Further investigation is necessary to confirm this observation.
List of abbreviations used
anti-CCP: Anti-cyclic citrullinated peptide; GWAS: Genome-wide association study; POCRE: Penalized orthogonal-components regression; SNP: Single-nucleotide polymorphism.
Authors' contributions
YL and MZ designed the study, and YL performed the statistical analysis. MZ and DZ both conceived the study, and drafted the manuscript. LW and VP participated in the design of the study and preprocessing of the data. JCF participated in interpreting the statistical analysis results, reviewing and editing the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yanzhu Lin, Email: YL: lin43@purdue.edu.
Min Zhang, Email: MZ: minzhang@purdue.edu.
Libo Wang, Email: LW: wang220@purdue.edu.
Vitara Pungpapong, Email: VP: vpungpap@purdue.edu.
James C Fleet, Email: JCF: fleet@purdue.edu.
Dabao Zhang, Email: DZ: zhangdb@purdue.edu.
Acknowledgements
The Genetic Analysis Workshops are supported by NIH grant R01 GM031575 from the National Institute of General Medical Sciences. Support from the CCE project at the Oncological Sciences Center of Purdue University is gratefully acknowledged. This research was also partially supported by NSF grant IIS 0844945 to DZ, Purdue Research Foundation grant to MZ, and Purdue Discovery Park seed grant to MZ and DZ.
This article has been published as part of BMC Proceedings Volume 3 Supplement 7, 2009: Genetic Analysis Workshop 16. The full contents of the supplement are available online at http://www.biomedcentral.com/1753-6561/3?issue=S7.
References
- Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham P, Bader JS, Craig I, O'Dorrovan M, Owen M. DNA pooling: a tool for large-scale association studies. Nat Rev Genet. 2002;3:862–871. doi: 10.1038/nrg930. [DOI] [PubMed] [Google Scholar]
- Waldron ERB, Whittaker JC, Balding DJ. Fine mapping of disease genes via haplotype clustering. Genet Epidemiol. 2006;30:170–179. doi: 10.1002/gepi.20134. [DOI] [PubMed] [Google Scholar]
- Zhang D, Lin Y, Zhang M. Penalized orthogonal-components regression for large p small n Data. Electron J Stat. 2009;3:781–796. doi: 10.1214/09-EJS354. [DOI] [Google Scholar]
- Johnstone IM, Silverman BW. Needles and straw in haystacks: empirical Bayes estimates of possibly sparse sequences. Ann Stat. 2004;32:1594–1649. doi: 10.1214/009053604000000030. [DOI] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Baker PIW, Daly MJ, Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Wang P, Cardenas M, Cox G, Perfect J, Heitman J. Two cyclophilin A homologs with shared and distinct functions important for growth and Cryptococcus neoformans. EMBO Rep. 2001;2:511–518. doi: 10.1093/embo-reports/kve135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Kondo H. Molecular cloning and expression of a 90-kDa diacylglycerol kinase that predominantly localizes in neurons. Proc Natl Acad Sci USA. 1993;90:7598–7602. doi: 10.1073/pnas.90.16.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L, Smits P, Wauters J, Merregaert J. Molecular cloning and characterization of human chondrolection, a novel type I transmembrane protein homologous to C-type lections. Genomics. 2002;80:62–70. doi: 10.1006/geno.2002.6806. [DOI] [PubMed] [Google Scholar]