

Abstract
The tricarboxylic acid cycle NAD+-specific isocitrate dehydrogenase (IDH) of Saccharomyces cerevisiae is an octameric enzyme composed of four heterodimers of regulatory IDH1 and catalytic IDH2 subunits. Recent structural analyses revealed the close proximity of Cys-150 residues from IDH2 in adjacent heterodimers, and features of the structure for the ligand-free enzyme suggested that formation of a disulfide bond between these residues might stabilize an inactive form of the enzyme. We constructed two mutant forms of IDH, one containing a C150S substitution in IDH2 and the other containing C56S/C242S substitutions in IDH2 leaving Cys-150 as the sole cysteine residue. Treatment of the affinity-purified enzymes with diamide resulted in the formation of disulfide bonds and in decreased activities for the wild-type and C56S/C242S enzymes. Both effects were reversible by the addition of dithiothreitol. Diamide had no effect on the C150S mutant enzyme, suggesting that Cys-150 is essential for the formation of a disulfide bond that inhibits IDH activity. Diamide-induced formation of the Cys-150 disulfide bond was also observed in vivo for yeast transformants expressing the wild-type or C56S/C242S enzymes but not for a transformant expressing the C150S enzyme. Finally, natural formation of the Cys-150 disulfide bond with a concomitant decrease in cellular IDH activity was observed during the stationary phase for the parental strain and for transformants expressing wild-type or C56S/C242S enzymes but not for a transformant expressing the C150S enzyme. A reduction in viability for the latter strain suggests that a decrease in IDH activity is important for metabolic changes in stationary phase cells.
The oxidative decarboxylation of isocitrate to form α-keto-glutarate catalyzed by mitochondrial NAD+-specific isocitrate dehydrogenase (IDH1) is an essentially irreversible reaction (1). On the basis of strong allosteric activation by AMP (2) and inhibition by NADH (1), this tricarboxylic acid cycle enzyme from Saccharomyces cerevisiae is proposed to be a key regulator of oxidative metabolism in response to cellular energy levels (2). Thus, when cellular levels of ATP and NADH are relatively high, flux through the tricarboxylic acid cycle would be reduced due to low levels of IDH activity, and citrate and isocitrate could be diverted into biosynthetic pathways.
Yeast IDH is an octameric enzyme composed of four IDH1 and four IDH2 subunits (3). Yeast mutants lacking either or both IDH subunits fail to grow with acetate as a carbon source (3), a growth phenotype shared with several other tricarboxylic acid cycle mutants (4–6). The IDH1 and IDH2 subunits have 42% sequence identity (7, 8), and results of targeted mutagenesis studies (9–14) suggested that (a) the basic functional unit of the enzyme is a heterodimer, (b) catalytic isocitrate; Mg2+- and NAD+-binding sites are primarily composed of residues from IDH2, (c) homologous sites for cooperative binding of isocitrate and for allosteric binding of AMP are primarily contained in IDH1, and (d) a few residues from the other subunit in each heterodimer contribute to ligand binding sites, providing a basis for communication between regulatory IDH1 and catalytic IDH2 subunits.
These inferences from mutagenesis studies were confirmed by our recent description of structures for yeast IDH (15) determined for enzyme crystallized in the absence of bound ligand, in the presence of the analogue citrate, or in the presence of citrate plus AMP2. IDH is organized with two heterodimers of IDH1 and IDH2 forming a heterotetramer, and with two heterotetramers forming the octameric holoenzyme (Figure 1). Within each heterodimer, distinct regulatory and catalytic sites are positioned near the interfaces between IDH1 and IDH2 subunits, and these sites contain many of the residues predicted by the mutagenesis studies summarized above. Differences between the structures for ligand-free and ligand-bound enzymes2 provided evidence for conformational changes related to allosteric mechanisms.
Figure 1.
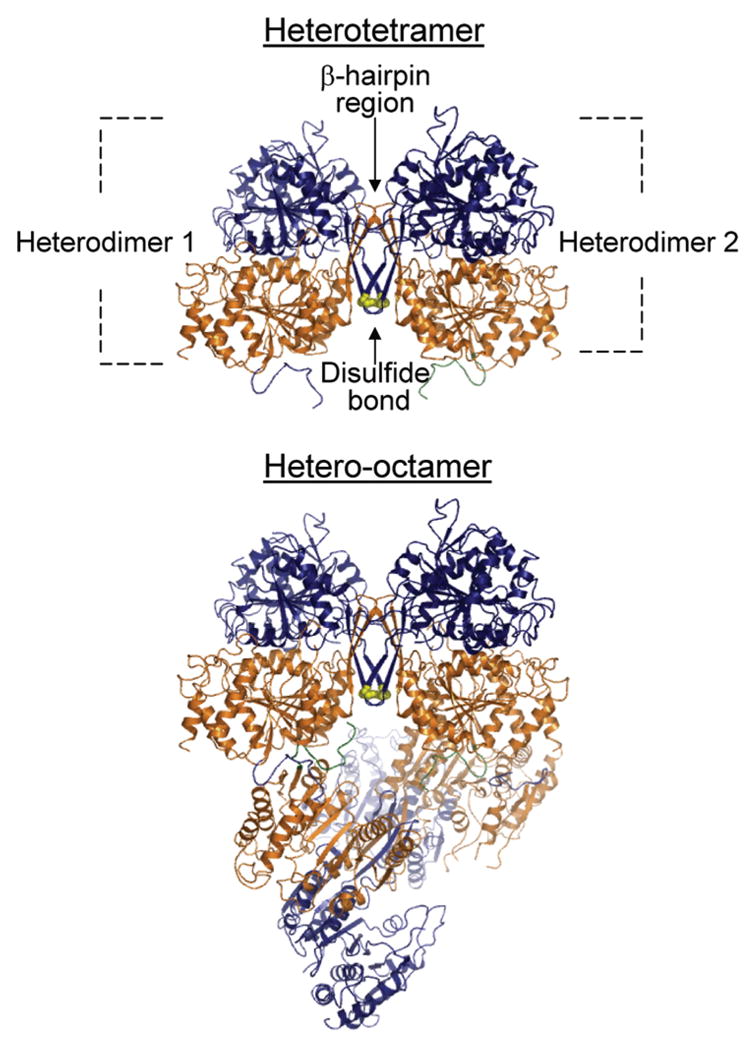
Organization of IDH1 and IDH2 subunits in the octameric holoenzyme. Shown is a depiction of the ligand-free structure determined for yeast IDH (15). Two heterodimers of IDH1 (orange) and IDH2 (navy) subunits form a heterotetramer primarily through interactions within a β-hairpin region, and two heterotetramers form the octamer primarily through contacts between IDH1 subunits in adjacent tetramers. The heterotetramers are offset from pseudo-222 symmetry by ~22°. IDH1 subunits form the internal core of the octamer with IDH2 subunits on the exterior. Cys-150 residues from IDH2 subunits in adjacent heterodimers are shown in yellow. Differences in environments around the Cys-150 residues in ligand-free and ligand-bound structures are presented and discussed in detail in ref 15.
One unexpected finding from structural analysis was the close proximity of Cys-150 residues from adjacent IDH2 subunits at the heterodimer interface in each tetramer (Figure 1), suggesting the possibility for the formation of a disulfide bond between Cys-150 side chains. For the ligand-bound structure, the proximity of the amino terminus and of basic amino acid residues of an IDH1 subunit from the other tetramer was proposed to stabilize a thiolate and disfavor disulfide bond formation. In the ligand-free structure, the amino terminus of the IDH1 subunit from the other tetramer is disordered, and the IDH2 Cys-150 residues are surrounded by apolar residues proposed to favor disulfide bond formation. If formed, this bond was predicted to stabilize an inactive form of IDH since, in the ligand-free structure of the enzyme, regulatory ligand-binding sites are largely inaccessible to solvent, and positions of residues in the catalytic isocitrate-binding site (particularly of IDH2 Tyr-142 of IDH2, a residue believed to be important for substrate binding) (12, 14) differ from those in the ligand-bound structure. Thus, the disulfide bond could represent some mechanism for reversible control of catalytic activity. In the current study, we examined the potential for the formation of a disulfide bond between IDH2 Cys-150 residues using the affinity-purified wild-type enzyme along with mutant enzymes containing either a serine replacement for the IDH2 Cys-150 residue or serine replacements for all cysteine residues other than Cys-150.
There is a well-characterized precedent for reversible down-regulation of isocitrate dehydrogenase activity in Escherichia coli. The E. coli enzyme (ICD) is an NADP+-specific homodimer and is not allosterically regulated (16). However, in bacterial cells shifted to cultivation medium with acetate as the carbon source, ~80% of the ICD molecules are rapidly inactivated by phosphorylation of Ser-113, a residue in the catalytic site important for substrate binding (17, 18). This modification results in a redirection of some isocitrate into the biosynthetic glyoxylate cycle (19–21) which converts acetyl CoA into four-carbon metabolites suitable for gluconeogenesis. The unmodified ICD molecules support oxidative energy production and provide NADPH for biosynthetic pathways. A specific kinase/phosphatase controls the reversible modification of ICD (22).
We hypothesized that formation of a disulfide bond in yeast IDH might represent an analogous mechanism for reversible regulation of catalytic activity. In the current study, we investigated the formation of the Cys-150 disulfide bond in vivo in cells treated with an exogenous oxidizing agent and sought to identify a physiological growth condition that might be associated with the formation of this bond.
EXPERIMENTAL PROCEDURES
Yeast Strains and Growth Conditions
The haploid parental strain of S. cerevisiae used in these studies was MMY011 (MATα ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1) (23). Wild-type and mutant forms of IDH were expressed in an idh1Δ idh2Δ yeast mutant constructed as previously described by incorporating deletion/disruption mutations (idh1Δ::kanMX4 idh2Δ::HIS3) in chromosomal IDH1 and IDH2 loci of the parental strain (10). Transformant strains were selected and maintained on plates containing defined YNB minimal medium (0.17% yeast nitrogen base and 0.5% ammonium sulfate at pH 6.5) containing 2% glucose and all nutrients needed to satisfy auxotrophic requirements for growth. In some experiments, parental and transformant strains were cultivated using or transferred to rich YP medium (1 % yeast extract and 2% Bacto-peptone) containing glucose, ethanol, glycerol, or sodium acetate added to 2% as the carbon source.
Mutagenesis, Expression, and Purification
Mutations were constructed using the Quickchange site-directed mutagenesis method (Stratagene). For expression in E. coli, a previously described plasmid (pET-15bIDH1His/IDH2) (15) carrying both IDH genes and codons for a histidine tag on the 3′-end of the IDH1 coding sequence was used as the template for mutagenesis. Mutagenesis was conducted to introduce a serine codon for the Cys-150 codon in the IDH2 gene (generating a plasmid designated pET-15bIDH1His/IDH2c150S). For expression of the C56S/C242S enzyme, the codon replacement for Cys-242 in the IDH2 gene was introduced initially, and the resulting plasmid was used for subsequent replacement of the codon for Cys-56 in IDH2 (generating a plasmid designated pET-15bIDH1His/IDH2C56S/C242S). Mutations were confirmed by DNA sequence analysis. For expression and purification of wild-type and mutant forms of IDH, plasmids were transformed into E. coli BL21-Gold (DE3) competent cells (Stratagene). Cells were grown to an optical density at A600nm = 0.6–0.8, and expression was induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside for 6 h at 30°C. Purification of enzymes from harvested cells using Ni2+-nitrilotriacetic acid (NTA) chromatography was performed as previously described (10). Concentrations of purified enzymes were determined by measuring A280 nm and using a molar extinction coefficient of 168,810 M−1 cm−1 (24). Purified protein samples were stored at −20 °C in the elution buffer (50 mM sodium phosphate at pH 8.0, 200 mM imidazole, and 300 mM sodium chloride) containing 15–25% glycerol.
For expression in yeast, a pRS316-IDH1/IDH2 plasmid (10) carrying both IDH genes was used for mutagenesis conducted as described above. The original pRS316 plasmid is a centromere-based plasmid designed for single-copy expression in yeast (25), and in the pRS316-IDH1/IDH2 plasmid, each IDH gene contains its authentic chromosomal promoter and mitochondrial targeting sequence. DNA sequence analysis was used to confirm mutagenesis, and the plasmids were transformed into the idh1Δidh2Δ mutant yeast strain using a lithium acetate procedure (26).
Kinetic Analyses and Electrophoretic Methods
IDH specific activity (units/mg protein, with 1 unit = 1 μmol NADH/min at 24 °C) was routinely measured in 1 mL assays containing 40 mM Tris-HCl (pH 7.4), 1 mM D-isocitrate, 4 mM MgCl2, and 0.5 mM NAD+. Apparent S0.5 values for isocitrate and corresponding Hill coefficients were determined in the absence of AMP using concentrations of D-isocitrate ranging from 0 to 5 mM or in the presence of 100 μM AMP using concentrations of D-isocitrate ranging from 0 to 1 mM. Apparent S0.5 values for NAD+ were determined using NAD+ concentrations ranging from 0 to 1.4 mM. Apparent Vmax values are the maxima1 rates obtained per mg of purified enzyme. Diamide from fresh 100 mM stock solutions in water was added as described in the text to determine effects on specific activity and electrophoretic properties.
Yeast cellular protein extracts were prepared by glass bead lysis as previously described (4) or, following the precipitation of cells with 25% cold trichloroacetic acid (TCA), as described by Delaunay et al. (27). To monitor disulfide bond formation in IDH, TCA precipitates were resuspended and lysed in a buffer containing 75 mM iodoacetamide as described in ref 27. Cellular protein concentrations were determined using the Bradford method (28).
Nonreducing gel electrophoresis was conducted using 10% or 11% polyacrylamide gels made according to Laemmli (29) but using a sample loading buffer (50 mM Tris-HCl at pH 6.8, 3% sodium dodecyl sulfate, and 2 mM EDTA) lacking β-mercaptoethanol. For reducing gel electrophoresis, the sample loading buffer also contained 0.35 M β-mercaptoethanol. Following electrophoresis, gels loaded with purified enzymes were stained with SYPRO Ruby Protein Gel Stain (Molecular Probes) or Coomassie blue. Gels loaded with cellular protein extracts were analyzed using an antiserum for the IDH holoenzyme that detects both subunits (diluted 1:5000) (3) or an antiserum specific for the yeast IDH1 subunit (diluted 1:5000) (30). Densitometry was performed using ImageJ software.
Mass Spectrometry Methods
To analyze the mass of the ~123 kDa electrophoretic form of IDH2 obtained by treatment with diamide, the C56S/C242S mutant enzyme (10 μg) was incubated with 50 mM diamide for 2 h at 24 °C. Nonreducing electrophoresis of a sample of treated enzyme indicated the presence of a ~38 kDa band (IDH1) and of the ~123 kDa band (disulfide bonded form of IDH2). For mass Spectrometry, the diamide-treated C56S/C242S enzyme was desalted using a C4 ZipTip column (Millipore) and infused into a ThermoFinnigan Quantum triple quadrapole mass spectrometer set in positive ion mode. Resulting spectra indicated peaks with masses of 38,728 (IDH1) and of 75,542. The latter is consistent with the mass of a dimer of IDH2.
RESULTS
Construction and Analysis of Mutant Forms of IDH
Yeast IDH contains three cysteine residues at residue positions 56, 150, and 242 in the IDH2 subunit and no cysteines in the IDH1 subunit. To investigate the possible formation of disulfide bonds, particularly between Cys-150 residues of IDH2 subunits which are in close proximity in adjacent dimers of each heterotetramer (Figure 1) (15), we constructed a mutant form of the enzyme containing a C150S substitution in IDH2. We also constructed a mutant form of the enzyme (with C56S and C242S substitutions in IDH2) that contains only one cysteine at residue position 150 in IDH2. The wild-type and mutant enzymes were expressed in E. coli and affinity-purified as described under Experimental Procedures using Ni2+-NTA chromatography based on the presence of a histidine tag on the carboxyl terminus of the IDH1 subunit. The relative purities of parental (lanes 1), C56S/C242S (lanes 2), and C150S (lanes 3) enzymes are illustrated in Figure 2A. Electrophoresis conducted under reducing conditions (plus β-mercaptoethanol) produced the expected patterns of migration for IDH1 and IDH2 subunits based on their molecular sizes (respective Mrs = 38,001 and 37,755) (7, 8), as well as evidence for some minor contaminant bands in the affinity purified enzyme samples. Electrophoresis conducted under nonreducing conditions (Figure 2A, minus β-mercaptoethanol) produced some evidence for additional slowly migrating species, in particular an ~123 kDa band in the C56S/C242S enzyme sample. We also examined a sample of wild-type IDH that had previously been extensively purified using multiple chromatography steps for crystallization studies (15) and that had been stored for several months at −20 °C (Figure 2B). Electrophoresis under reducing conditions produced the expected patterns of migration for IDH1 and IDH2 subunits and no evidence for contaminating polypeptides. With electrophoresis of the same enzyme preparation under nonreducing conditions, the IDH1 subunit migrated as expected on the basis of size, but most of the IDH2 subunit appeared as several more slowly migrating species, the slowest with an apparent molecular size of ~123 kDa. Thus, oxidation of the wild-type enzyme, which appears to involve disulfide-bond formation in the IDH2 subunit, is exacerbated by long-term storage.
Figure 2.

Affinity-purified enzymes. (A) Samples (1.6 μg ea) of affinity-purified wild-type enzyme (lanes 1) and C56S/C242S and C150S mutant enzymes (lanes 2 and 3, respectively) were electrophoresed under reducing (plus β-mercaptoethanol = β-ME) or nonreducing (minus β-ME) conditions. Positions of molecular size standards are indicated on the right of the figure. (B) Samples (0.5 μg ea) of native wild-type IDH previously purified for crystallography experiments (15) and stored for ~6 months at −20 °C were electrophoresed under reducing or nonreducing conditions as indicated. The specific activity of the enzyme preparation was ~50% lower than that measured immediately after purification. The gels were stained with SYPRO Ruby.
Kinetic Analyses
Kinetic parameters for affinity-purified wild-type, C56S/C242S, and C150S enzymes (i.e., those from Figure 2A) were examined using assays performed with various concentrations of isocitrate and in the absence or presence of the allosteric activator AMP. Representative results are illustrated in Figure 3, and cumulative results are presented in Table 1. These indicate that the C56S/C242S substitutions had little effect on apparent Vmax values, while apparent Vmax values obtained for the C150S enzyme were ~27% higher than those obtained for the wild-type enzyme. The C150S enzyme also exhibited a significant ~30% reduction in Hill coefficient values relative to the similar values measured for wild-type and C56S/C242S enzymes. Also, while both mutant enzymes exhibited increases in apparent S0.5 values for isocitrate relative to those for the wild-type enzyme, all three enzymes exhibited similar properties of AMP activation, i.e., increases of ~5- to 7-fold in apparent affinity for isocitrate in the presence versus in the absence of AMP. We also examined kinetic properties using NAD+ saturation curves (Table 1) and found that apparent Vmax values, relative to that for the wild-type enzyme, were somewhat higher (~14%) for the C150S enzyme and somewhat lower (~12%) for the C56S/C242S enzyme. The apparent affinity for NAD+ was similar for all three enzymes. (Hill coefficients for the three enzymes were also ~1.2, reflecting the absence of cooperativity with respect to NAD+ (1, 13).) Overall, these results suggest that the C56S/C242S and C150S mutant enzymes retain substantial catalytic activity, and as described below, we also found that the two mutant enzymes can functionally substitute for yeast IDH in vivo.
Figure 3.

Kinetic analyses of wild-type and mutant forms of IDH. Isocitrate saturation velocity experiments were conducted in the absence or presence of 100 μM. AMP as indicated using affinity purified wild-type (●), C56S/C242S (○), and C150S (▼;) enzymes. Note the differences in scales for [D-isocitrate].
Table 1.
Kinetic Properties of Wild-Type and Mutant Forms of IDH
| isocitratea |
NAD+b |
||||
|---|---|---|---|---|---|
| enzyme | Vmax (U/mg) (−AMP/+AMP) | S0.5 (mM) (−AMP/+AMP) | Hill coef. (−AMP/+ AMP) | Vmax(U/mg) | S0.5 (mM) |
| wild-type | 30.5/31.8 (±1.0/1.6) | 0.57/0.10 (±0.04/0.01) | 3.6/3.5 (±0.1/0.3) | 35.0 | 0.12 |
| C56S/C242S | 29.8/31.7 (±1.5/2.1) | 1.11/0.16 (±0.02/0.04) | 3.6/3.1 (±0.1/0) | 30.7 | 0.08 |
| C150S | 38.7/40.3 (±1.4/2.5) | 0.85/0.19 (±0.01/0.02) | 2.5/2.5 (±0.4/0.3) | 39.9 | 0.10 |
Kinetic properties were determined as described under Experimental Procedures. Properties with respect to isocitrate were determined in the absence or presence of 100μM AMP, and values represent three independent measurements (±standard deviations).
Values for kinetic properties with respect to NAD+ represent averages of two independent measurements.
The kinetic properties of the C150S enzyme with respect to isocitrate suggested that the presence of the IDH2 Cys-150 residue (and perhaps the potential for disulfide bond formation) may contribute to cooperativity and may also limit maximal IDH activity. Since the lower Vmax value measured for the wild-type enzyme could be due to the presence of some disulfide bonds in the purified enzyme (Figure 2A), we analyzed kinetic parameters for the wild-type enzyme in the presence of dithiothreitol. Relative to kinetic properties for the untreated wild-type enzyme (Table 1), the addition of 0.5 mM dithiothreitol (or of 2 mM dithiothreitol) to assays produced an increase in apparent Vmax values of 30% in the absence of AMP and of 26% in the presence of 100 μM AMP, as well as a ~37% decrease in cooperativity under both conditions. AMP activation was largely unchanged by the presence of dithiothreitol. Similar but less dramatic effects were obtained for the C56S/C242S enzyme, but the addition of dithiothreitol had no effect on kinetic properties of the C150S enzyme. Thus, the effects of a disulfide reducing reagent on kinetic properties of the wild-type enzyme are quite similar to those obtained by replacement of Cys-150 with a serine residue. These data suggest that oxidation and disulfide bond formation in the isolated wild-type enzyme may reduce catalytic activity. However, the similar effects of dithiothreitol and of the C150S residue substitution on cooperativity suggest that Cys-150 disulfide bonds may affect this kinetic property.
Diamide-Induced Formation of Disulfide Bonds in Purified IDH
To examine disulfide bond formation in IDH, aliquots of the purified wild-type enzyme were incubated for 2 h with increasing amounts of diamide, an oxidizing agent used to induce disulfide bond formation in vitro and in vivo (31). Following incubation, protein samples were electrophoresed using nonreducing conditions. As shown in Figure 4A, the IDH1 and IDH2 subunits in untreated samples of the wild-type enzyme (lane 0) migrated essentially as expected on the basis of molecular sizes for each monomer. Following incubation with increasing concentrations of diamide, levels of the IDH2 monomer in the wild-type enzyme decreased, concomitant with the appearance of several more slowly migrating species, the slowest with an apparent size of ~123 kDa. Mass spectrometry was used to confirm that IDH2 is the major component in the three most slowly migrating species in samples treated with 5 mM or 10 mM diamide. As indicated below each lane (and as graphed in Figure 4C, ●), the specific activity of the wild-type enzyme decreased with increasing diamide concentrations. Also, as shown in Figure 4B, for the wild-type enzyme incubated with an intermediate concentration (0.5 mM) of diamide, the decrease in activity was time dependent. Higher concentrations of diamide produced more rapid decreases in activity (data not shown).
Figure 4.

Effects of diamide on electrophoretic properties of wild-type and mutant forms of IDH. (A) Samples (1 μM in 50 mM Tris-HCl at pH 7.4) of affinity-purified wild-type IDH were incubated with the indicated concentrations of diamide for 2 h on ice prior to nonreducing electrophoresis of 1.6 μg samples. IDH specific activity, expressed as the percent of the maximum activity measured, is indicated below each lane. (B) Decrease with time in specific activity (expressed as percent of the maximum activity measured) for wild-type IDH incubated with 0.5 mM diamide. (C) IDH specific activities were determined for wild-type (●), C56S/C242S (○), and C150S (▼;) enzymes following 2 h incubations with the indicated concentrations of diamide. Activities are expressed as the percent of the maximum activity measured. (D) Samples (1 μM in 50 mM Tris-HCl at pH 7.4) of affinity-purified C56S/C242S and C150S enzymes were incubated with the indicated concentrations of diamide for 2 h on ice prior to nonreducing electrophoresis of 1.6 μg samples. The gels were stained with SYPRO Ruby.
To determine the relevance of IDH2 Cys-150 on diamide-induced formation of disulfide bonds and on the associated decrease in activity, we similarly examined properties of affinity-purified C56S/C242S and C150S enzymes following incubations with diamide. Incubation with increasing concentrations of diamide (Figure 4C) was found to decrease the specific activity of the C56S/C242S enzyme (○) to levels similar to those observed for the wild-type enzyme (●), but incubation with diamide had little effect on the specific activity of the C150S enzyme (▼;). Nonreducing gel electrophoresis was used to examine the mutant enzymes following incubations with higher concentrations of diamide (Figure 4D). For the C56S/C242S enzyme, relative to the untreated enzyme (lane 0), incubation with 2.5 mM, 5 mM, or 10 mM diamide produced a decrease in levels of the IDH2 monomer and a concomitant increase in levels of the ~123 kDa species. Thus, diamide-induced oxidation apparently produced a disulfide bond between Cys-150 residues in the C56S/C242S enzyme resulting in the formation of the ~123 kDa species. In contrast, incubation of the C150S mutant enzyme with similar concentrations of diamide did not alter electrophoretic properties of the monomeric IDH2 subunit (Figure 4D). In addition, results for the C150S enzyme suggest that the formation of most of the other slowly migrating species observed for the diamide-treated wild-type enzyme (Figure 4A) is also dependent on Cys-150. Since diamide-induced reductions in activity correlated with the formation of higher molecular weight forms of IDH2 in both enzymes containing Cys-150, it appears that disulfide bond formation involving only Cys-150 residues (in the C56S/C242S enzyme) or involving Cys-150 and other cysteine residues in IDH2 (in the wild-type enzyme) stabilizes an inactive form of IDH. As discussed below, the disulfide bond involving only Cys-150 residues is the predominant bond formed in vivo, suggesting that other disulfide bonds formed in the purified wild-type enzyme during auto-oxidation (e.g., as in Figure 2B) or following incubation with diamide (Figure 4A) are a function of the purity of the enzyme.
On the basis of these observations, we conclude that the ~123 kDa form of IDH2 represents a homodimer linked by a disulfide bond between Cys-150 residues from adjacent dimers in the heterotetramer. However, since the electrophoretic migration of this species under nonreducing conditions is slower than would be expected for a linear polypeptide of the same size (i.e., ~76 kDa), we used mass spectrometry as described under Experimental Procedures to examine the C56S/C242S enzyme following treatment with 50 mM diamide. Masses were consistent with the presence of the monomeric form of the IDH1 subunit and of a dimeric form of the IDH2 subunit. Thus, the electrophoretic ~123 kDa form of IDH2 represents a dimer of IDH2 linked by a Cys-150 disulfide bond. We note that similar aberrant electrophoretic properties have been reported for standard proteins containing artificially induced cross-links (32).
As shown in Figure 1 of Supporting Information, diamide-induced formation of the IDH2 Cys-150 disulfide bond was also confirmed by chemical cleavage using 2-nitro-5-thiocyanobenzoate, a reagent specific for free cysteine residues (33).
Reduction of Disulfide Bonds in IDH
The reversible nature of diamide-induced formation of high molecular weight forms of IDH2 in wild-type and mutant enzymes was investigated using dithiothreitol. As shown in Figure 5 and as described above, relative to untreated enzymes (left lane in each panel), the appearance of multiple electrophoretic forms of IDH2 in the wild-type enzyme and of the ~123 kDa form of IDH2 in the C56S/C242S enzyme, along with concomitant reduction in levels of the monomeric form of the IDH2 subunit for both enzymes, were observed following incubation for 2 h with 5 mM diamide (central lane in each panel). These effects on the electrophoretic mobility of IDH2 for both the wild-type and C56S/C242S enzyme were reversed by subsequent addition and incubation for 30 min with 50 mM dithiothreitol (right lane in each panel). Also, as indicated below each lane in Figure 5, the diamide-induced formation of more slowly migrating forms of IDH2 for the wild-type and C56S/C242S enzymes correlated with substantial reductions in specific activities (~5-fold and 10-fold, respectively), whereas subsequent incubation with dithiothreitol restored activities to levels observed for the untreated enzymes. Incubation of the C150S enzyme with diamide and subsequently with dithiothreitol produced little change in electrophoretic properties or in specific activity, indicating that effects of both reagents are dependent on the presence of IDH2 Cys-150.
Figure 5.

Effects of dithiothreitol on disulfide bond formation in IDH. Samples (1 μM in 50 mM Tris-HCl at pH 7.4) of affinity-purified wild-type, C56S/C242S, and C150S enzymes as indicated were untreated (left lane in each panel), were incubated on ice with 5 mM diamide for 2 h (central lane in each panel), or were similarly incubated with diamide prior to the addition of 50 mM dithiothreitol and further incubation on ice for 30 min (right lane in each panel). Nonreducing electrophoresis was conducted using samples containing 1.6μg enzyme, and the gels were stained with SYPRO Ruby. IDH specific activity, expressed as the percent of that measured for the untreated enzyme, is indicated below each lane.
Diamide-Induced Formation of an IDH2 Disulfide Bond in Vivo
To examine the formation of disulfide bonds in IDH2 in vivo, we constructed single copy plasmids for the expression of wild-type, C56S/C242S, and C150S enzymes in an idh1Δidh2Δ yeast strain lacking the endogenous mitochondrial enzyme. All three enzymes were found to be expressed to similar levels in extracts from transformant strains and to levels approximately equal to those of IDH in the parental strain, as illustrated in the experimental data presented below. Cellular IDH specific activities were also similar for the parental strain and all transformants (0.3–0.5 units/mg cellular protein relative to no detectable activity for the idh1Δidh2Δ strain). Finally, we found that all transformant strains grew as well as the parental strain on YP plates with acetate as the carbon source, whereas the idh1Δidh2Δ yeast strain failed to grow under this condition. This indicated functional complementation for IDH by the C56S/C242S and C150S enzymes in vivo, as expected on the basis of the robust catalytic activities of the purified enzymes (Table 1).
To test in vivo effects of diamide, the transformant strains were grown in rich medium with galactose, a nonrepressing fermentable carbon source that produces significant levels of IDH expression (34). Cells were harvested at various times following the addition of 2 mM diamide to logarithmically growing cultures. IDH was analyzed using nonreducing gel electrophoresis and immunoblots with an antiserum for the holoenzyme (3). As shown in Figure 6, low levels of the ~123 kDa form of IDH2 were detected immediately after diamide addition to strains expressing the wild-type or C56S/C242S enzymes (lanes 0). Levels of the ~123 kDa form of IDH2 increased in these strains ~5-fold within ~30 min then remained constant over a 5 h period of incubation. No high-molecular weight form of IDH2 was seen in samples from untreated control cultures of these strains (ctl lanes) after 5 h of growth. There was no evidence for formation of the ~123 kDa form of IDH2 in extracts from the transformant expressing the C150S mutant enzyme at any time following the addition of diamide, indicating that Cys-150 is essential for disulfide bond formation in vivo.
Figure 6.

Effects of diamide on IDH in vivo. Yeast idh1Δidh2Δ transformants expressing the wild-type enzyme or the indicated mutant forms of IDH were grown in YP galactose medium to culture A600 nm values of ~2. Cells were harvested immediately after (times 0) and at the times indicated following the addition of 2 mM diamide to the cultures. Cells from control cultures (ctl lanes) containing no diamide were harvested after 5 h of growth. Cellular protein extracts were obtained by glass bead lysis, and ~5μg samples were electrophoresed under nonreducing conditions. Immunoblot analysis was conducted using an antiserum for the IDH holoenzyme (3). Positions of molecular size standards are indicated on the right of the figure.
It is notable that the ~123 kDa form of IDH2 was the primary slowly migrating species of IDH2 detected in extracts from the diamide-treated transformant expressing the wild-type enzyme (Figure 6). This suggests, as stated above, that the formation of disulfide bonds other than those between Cys-150 residues in the affinity-purified wild-type enzyme (e.g., as in Figures 2B and 4A) may be due to the highly purified nature of the enzyme used in in vitro assays. It should also be noted that, while the untreated control cultures continued to grow over the 5 h period of this experiment (Figure 6), with A600 nm values increasing ~2.5-fold, the diamide-treated cultures exhibited little change in A600 nm values. This is likely due to detrimental pleiotropic effects of this oxidizing agent (31). Obviously, the lack of growth was not specifically due to the modification of IDH2 since the C150S transformant, which exhibited no change in electrophoretic mobility of IDH2, also failed to grow. While these are clearly not physiological conditions, our results do show that IDH2 can be induced to form a disulfide bond in vivo, and this bond involves only Cys-150 residues since other cysteines are absent in the C56S/C242S enzyme.
Natural Formation of an IDH2 Disulfide Bond in Vivo
In order to identify a physiological condition that might correlate with the formation of the Cys-150 disulfide bond in vivo, we used immunoblots to examine IDH in extracts from cells cultivated under a variety of growth conditions. These included logarithmic growth with various carbon sources and various shifts of logarithmically growing cells from a medium with one carbon source to another. None of these conditions provided any evidence for modification of IDH in vivo. In another experiment originally designed to test possible analogies with the regulation of ICD in E. coli, i.e., rapid covalent modification and inactivation following a shift of bacterial cells to medium with acetate as the carbon source (17, 18), we examined IDH in the parental yeast strain at multiple time points following a shift of cells growing logarithmically in YP glucose medium to YP acetate medium. With immunoblots, we found no evidence for modification of IDH at early time points following the shift to acetate medium (data not shown). Instead, cellular levels of IDH increased ~3-fold within 12 h following the shift, consistent with previous observations that levels of IDH are higher in yeast cells grown with a nonfermentable carbon source than in cells grown with glucose (35). Thus, unlike E. coli ICD, yeast IDH is not modified during this carbon source shift. However, as shown in Figure 7A (left panel), using the antiserum for the IDH holoenzyme, we did find evidence for the ~123 kDa electrophoretic form of IDH2 in cellular extracts at later time points (48 and 60 h) after acetate-grown cells had entered stationary phase, i.e., at times when culture A600 nm values ceased to increase (Figure 7B, ●). To establish that the ~123 kDa form of IDH2 contained only that subunit, we performed an immunoblot using an antiserum specific for IDH1 (30) with the same cellular extracts. As shown in Figure 7A (right panel), only the monomeric form of IDH1 was present in all samples including those from stationary phase cells at 48 and 60 h time points. (There is no comparable antiserum specific for IDH2.)
Figure 7.

Formation of a disulfide bond in IDH in stationary phase cells grown with acetate as the carbon source. (A) The parental yeast strain was cultivated using YP acetate medium, and protein extracts (~5μg ea) from cells harvested by trichloroacetic acid precipitation at the indicated times were electrophoresed on two gels under nonreducing conditions. One gel (left panel) was used for immunoblot analysis with an antiserum for the IDH holoenzyme (3), and the other gel (right panel) was used for immunoblot analysis with an antiserum specific for the IDH1 subunit (30). (B) Growth of cultures of the parental yeast strain (●) and of transformants expressing the wild-type (○), C56S/C242S (▼;), or C150S (∇) enzymes in YP acetate medium was monitored as A600 nm. (C) Protein samples (~5μg ea) from YP acetate cultures of transformants expressing the wild-type or mutant IDH enzymes as indicated were electrophoresed under nonreducing conditions, and the gels were used for immunoblot analysis with an antiserum for the IDH holoenzyme.
To verify that formation of the ~123 kDa form of IDH2 in cells grown to stationary phase is due to Cys-150, we grew the yeast transformant strains expressing the wild-type, C56S/C242S, or C150S enzymes using YP acetate medium. As shown in Figure 7B, the transformant strains exhibited logarithmic growth rates (to ~24 h) and transitions to stationary phase comparable to those observed for the parental strain. Extracts from the transformants were used for immunoblot analyses (Figure 7C). Relative to starting levels (0 h), levels of IDH subunits increased 3- to 5-fold in extracts from the transformant strains by 12 h time points, comparable to the increase in levels observed for the parental strain (Figure 7A). At 48 and 60 h time points, the ~123 kDa form of IDH2 was observed in extracts from transformants expressing the wild-type enzyme or the C56S/C242S mutant enzyme, but not in extracts from the transformant expressing the C150S mutant enzyme (Figure 7C). Thus, formation of the ~123 kDa form of IDH2 in extracts from stationary phase cells depends on the presence of the IDH2 Cys-150 residue.
IDH specific activities were measured using cellular extracts taken at the time points indicated in Figure 7A and C. For the parental strain and for transformants expressing the wild-type or C56S/C242S mutant enzymes, IDH specific activities were found to be highest in the 24 h cell extract samples, and activities decreased from these peaks by 25–45% in the 60 h samples. This suggests that formation of the Cys-150 disulfide bond in IDH reduces catalytic activity in vivo. Furthermore, for the transformant expressing the C150S enzyme, IDH specific activity in the 24 h sample was comparable to those determined for other strains at that time point, but the specific activity increased from this value by 33% in the 60 h sample. Thus, the presence of the Cys-150 residue (in wild-type and C56S/C242S transformants) also appears to prevent the increase in IDH activity observed in stationary phase samples from the transformant expressing the C150S enzyme.
Since acetate is a poor nonfermentable carbon source, producing less than two culture doublings after two days (Figure 7B), we also examined the formation of the Cys-150 disulfide bond and IDH activity in strains grown with ethanol, a carbon source permissive for the growth of strains lacking IDH (6). For the strain expressing the wild-type enzyme (Figure 8B, upper panel, ●), growth with ethanol produced a ~90-fold increase in viable cell numbers over four days as the culture reached stationary phase. Levels of IDH subunits increased ~5-fold by day 1 (Figure 8A), concomitant with a ~ 11-fold increase in cellular activity (Figure 8B, lower panel, ●). The ~123 kDa form of IDH2 appeared in extracts by day 2, and levels increased to day 4 (Figure 8A), concomitant with a ~50% decrease in cellular IDH activity (Figure 8B, lower panel, ●). The strain expressing the C56S/C242S mutant enzyme (Figure 8B, upper panel, ○) exhibited growth patterns essentially identical with those of the strain expressing the wild-type enzyme. Appearance and levels of the ~123 kDa species was also similar for the C56S/C242S mutant strain (data not shown), as was the ~40% decrease in cellular IDH activity from a peak at day 2 to day 4 (Figure 8B, lower panel, ○). For the strain expressing the C150S mutant enzyme, which cannot form the IDH2 disulfide bond, viable cell numbers (Figure 8B, upper panel, ▼;) actually exceeded those of wild-type and C56S/C242S strains by ~20% on day 2 in ethanol medium, then decreased to levels ~25% lower than those of the other strains on day 4. Also, unlike the other strains, cellular IDH activity for the C150S strain remained elevated after four days in ethanol medium (Figure 8B, lower panel, ▼;). These results suggest that the formation of the Cys-150 disulfide bond and reduction in IDH cellular activity may confer an advantage to cells in terms of viability during adaptation to stationary phase growth under these conditions.
Figure 8.

Formation of a disulfide bond in IDH in stationary phase cells grown with ethanol as the carbon source. (A) Protein samples (~5μg ea) taken at indicated times during growth in YP ethanol medium of the yeast strain expressing wild-type IDH were electrophoresed under nonreducing conditions. The gel was used for immunoblot analysis with an antiserum for the IDH holoenzyme. (B) Samples of cultures of yeast strains expressing wild-type (●), C56S/C242S (○), or C150S (▼;) enzymes grown in YP ethanol medium were taken at indicated times to assess cellular viability (by plating dilutions on YP glucose plates and counting colonies after 3–5 days of growth at 30 °C) (top panel) or to determine cellular levels of IDH activity as described under Experimental Procedures (bottom panel). Cell numbers are normalized relative to numbers at time 0 (set at 1). IDH activity is expressed relative to mg cellular protein.
The ~50% decrease in cellular IDH activity in yeast cells expressing the wild-type or C56S/C242S enzymes and grown to stationary phase with acetate or ethanol as the carbon source was associated with only a 20–25% decrease in immunochemical levels of the monomeric form of the IDH2 as the ~123 kDa IDH2 species appeared (as estimated with densitometric analysis of immunoblots of lower exposure times than those illustrated in Figures 7 and 8). Thus, while quantification of protein and activity levels in cellular extracts is inexact, these estimates suggest that formation of only one of the two potential Cys-150 disulfide bonds in the octameric enzyme may be sufficient to substantially reduce activity of the enzyme in vivo.
DISCUSSION
We have demonstrated that a disulfide bond can form in yeast IDH. The ~123 kDa electrophoretic form of IDH2 containing this bond was observed following treatment of newly purified enzyme using diamide and in extracts from yeast cells treated with diamide or simply grown to stationary phase. Formation of the disulfide bond correlated with a decrease in IDH activity. Use of mutant enzymes demonstrated that the formation of the bond is dependent on the presence of the IDH2 Cys-150 residue and that the bond can form in the absence of any other cysteine residue in the IDH holoenzyme. Diamide-induced formation of disulfide bonds and diminished activity for the purified wild-type and C56S/C242S enzymes were reversible by treatment with dithiothreitol.
The decrease in cellular IDH activity associated with the appearance of the Cys-150 disulfide bond in extracts from parental yeast cells grown to stationary phase suggests that down-regulation of IDH activity may be related to changes in metabolism that occur during this time. In other studies, we have found that loss of IDH correlates with an accumulation of high cellular levels of citrate and isocitrate (34) and in a diminished capacity for reduction of NAD+ (36). Presumably, the ~50% reduction in IDH activity in stationary phase cells would have similar though less dramatic effects. If so, then the excess citrate and isocitrate could be transported into the cytosol for use in biosynthetic glyoxylate and gluconeogenic pathways. As in bacteria, the glyoxylate cycle in yeast permits net assimilation of two-carbon precursors (acetyl CoA) into four-carbon metabolites (succinate or malate). In addition to the provision of four-carbon metabolites, the isocitrate accumulated because of the down-regulation of IDH activity could be used as a substrate for mitochondrial or cytosolic NADP+-specific isocitrate dehydrogenases, thus providing the NADPH needed for biosynthetic and/or antioxidant functions. Consistent with these predictions, yeast genes encoding glyoxylate cycle enzymes and the IDP2 gene encoding cytosolic NADP+-specific isocitrate dehydrogenase are up-regulated during the diauxic shift and transition into stationary phase (37–39). If these speculations prove correct, then the different covalent mechanisms for decreasing activities of yeast IDH by disulfide bond formation and of E. coli ICD by phosphorylation could have similar metabolic effects. In addition, the reduced viability of the yeast strain expressing the C150S enzyme (Figure 8) suggests that the decrease in IDH activity is important for metabolic changes that occur in stationary phase.
The Cys-150 residue of IDH2 is relatively unique among catalytic subunits of eukaryotic homologues including those from Caenorhabiditis elegans, Drosophila melanogaster, and Homo sapiens. Even among other yeasts for which genomic sequences are available, only members of very closely related species in a group denoted Saccharomyces sensu stricto (40, 41)3 contain cysteines in residue positions equivalent to that of IDH2 Cys-150 of S. cerevisiae. This group is metabolically unique compared to other yeasts because its members are able to ferment glucose to produce ethanol even in the presence of oxygen, accounting for the exceptional commercial exploitation of this group of organisms. It is tempting to speculate that the reversible formation of the Cys-150 disulfide bond could also be a mechanism for sensing oxygen levels and could control this unique metabolic property. However, preliminary experiments have produced no evidence for changes in the redox status of Cys-150 as a function of hypoxic growth conditions.
Finally, the formation of a reversible disulfide bond involving IDH2 C-150 was also recently reported in a proteomic analysis of yeast cells treated with a concentration of hydrogen peroxide sufficient to produce a 5-fold decrease in culture viability (42). Thus, reduction of IDH activity may additionally be related to redox stress responses.
Mammalian IDH enzymes are also octameric but contain four catalytic α subunits and two each of regulatory β and γ subunits (43–45). The human enzyme contains numerous cysteine residues but none in positions comparable to that of yeast IDH2 Cys-150. Given the absence of a glyoxylate cycle in mammalian cells, a potential role for covalent modification of IDH is unclear. Also, in terms of contributions to and/or regulation of flux through the tricarboxylic acid cycle, the role of human IDH has recently been questioned as a result of studies of families with hereditary retinitis pigmentosa (46). Affected individuals were found to be homozygous for a loss-of-function mutation in the β subunit of IDH but, other than retinal problems, exhibited no other health problems. Thus, the human mitochondrial NADP+-specific enzyme was speculated to play a crucial role in support of oxidative metabolism. This is clearly not the case for S. cerevisiae since yeast mutants lacking IDH have growth phenotypes similar to those of mutants lacking several other tricarboxylic acid cycle enzymes (4–6), despite the presence of substantial levels of the mitochondrial NADP+-specific enzyme (35). Also yeast mutants lacking the latter enzyme demonstrate no growth or respiratory defects (47).
While disulfide bond formation in purified yeast IDH can be induced by diamide and the bonds subsequently reduced by dithiothreitol, the mechanisms for oxidation and reduction of the Cys-150 disulfide bond in vivo are unknown. One well-characterized disulfide relay system in the intermembrane space of yeast mitochondria (48) has been shown to be important for maturation, e.g., disulfide bond formation and copper acquisition, of several proteins in this compartment including components of cytochromes (48) as well as superoxide dismutase (SOD1) and its copper chaperone (CCS1) (49). However, it seems unlikely that this system would contribute to oxidation/reduction of disulfide bonds in a soluble mitochondrial matrix enzyme. In fact, other than IDH as described here, there have been few reports of soluble matrix enzymes containing disulfide bonds. One reported example is human mitochondrial presequence peptidase. This enzyme contains cysteine residues that form a disulfide bond upon oxidation resulting in inhibition of activity (50). However, the yeast homologue of the presequence peptidase is reported to be localized in the intermembrane space (51), suggesting that oxidation/reduction of thiols in this enzyme might be regulated by the disulfide relay system described above.
Yeast mitochondria do contain a mitochondrial thioredoxin system comprising a thioredoxin (TRX3) and a thioredoxin reductase (TRR2) (52), and a glutathione reductase (GLR1) is reported to be localized in both cytosolic and mitochondrial compartments (53). It will be important to test potential contributions of these systems to the redox state of Cys-150 residues in IDH. Given that we observe the oxidation of purified wild-type IDH after long-term storage, it is also possible that the state of the IDH2 Cys-150 side chain may directly reflect the redox status of the mitochondrial matrix.
Similar kinetic parameters with respect to isocitrate measured for the C150S mutant enzyme and the wild-type enzyme in the presence of dithiothreitol suggested that some of the Cys-150 residues in the purified wild-type enzyme may be oxidized and that addition of the reducing reagent results in higher velocities than those observed for the untreated enzyme. However, decreases in cooperativity observed for the C150S enzyme and for the dithiothreitol-treated wild-type enzyme suggest that the capacity for the formation of the disulfide bond may be important for communication among isocitrate-binding sites. We are particularly interested in this possibility because, although the existence of allosteric disulfide bonds has been predicted (54), there are currently few examples. We are also interested in the possible relevance of the disulfide bond to the unusual property of half-site ligand binding by yeast IDH (12,13, 55), i.e., that ligand-binding assays measure only half of the sites predicted for each ligand of the enzyme on the basis of the structure determined for IDH. We will use mutant enzymes constructed in these studies to investigate these possibilities.
Supplementary Material
Acknowledgments
We thank Kevin W. Hakala and Dr. Susan T. Weintraub of the Center for Mass Spectrometry at the UTHSCSA for protein analyses. We also thank Sondra L. Anderson for development of methods and editorial comments, and Dr. Mark T. McCammon for providing the antiserum specific for the IDH1 subunit.
Footnotes
Abbreviations: IDH, mitochondrial NAD+-specific isocitrate dehydrogenase; ICD, Escherichia coli NADP+-specific isocitrate dehydrogenase; NTA, nitrilotriacetic acid; β-ME, β-mercaptoethanol; NTCB, 2-nitro-5-thiocyanobenzoate.
Respective accession codes in the Protein Data Bank are 3BLX, 3BLV, and 3BLW. Structures obtained in the presence of citrate (3BLV) or of citrate plus AMP (3BLW) were similar, and the former was used for most comparisons with the ligand-free structure (3BLX).
The senso stricto group includes, in addition to the bakers’ yeast S. cerevisiae, the brewers’ yeast S. partorianus/carlbergensis as well as S. paradoxus and S. mikatae (40, 41). The IDH2 subunits from this group share 97–100% sequence identities, and all have a cysteine at the residue position equivalent to Cys-150 in the S. cerevisiae enzyme. The sensu stricto group does not include other yeast species such as S. kluyveri, Candida glabrata, or Kluyveromyces lactis. Despite overall sequence identities of 86–88% with S. cerevisiae IDH2, none of the subunits from the latter species contains a cysteine at the residue position analogous to Cys-150.
This work was supported by NIH grants GM051265 and AG017477 (to L.M-H.).
SUPPORTING INFORMATION AVAILABLE
Figure 1 shows predicted patterns and experimental results of chemical cleavage to map disulfide bonds in IDH2 subunits from wild-type and mutant forms of IDH prior to and following treatment with diamide. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Barnes LD, McGuire JJ, Atkinson DE. Yeast diphosphopyridine nucleotide specific isocitrate dehydrogenase. Regulation of activity and unidirectional catalysis. Biochemistry. 1972;11:4322–4329. doi: 10.1021/bi00773a019. [DOI] [PubMed] [Google Scholar]
- 2.Hathaway JA, Atkinson DE. The effect of adenylic acid on yeast nicotinamide adenine dinueleotide isocitrate dehydrogenase, a possible control mechanism. J Biol Chem. 1963;238:2875–2881. [PubMed] [Google Scholar]
- 3.Keys DA, McAlister-Henn L. Subunit structure, expression, and function of NAD(H)-specific isocitrate dehydrogenase in Saccharomyces cerevisiae. J Bacteriol. 1990;172:4280–4287. doi: 10.1128/jb.172.8.4280-4287.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson LM, Sutherland P, Steffan JS, McAlister-Henn L. Gene sequence and primary structure of mitochondrial malate dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 1988;27:8393–8400. doi: 10.1021/bi00422a015. [DOI] [PubMed] [Google Scholar]
- 5.Kim KS, Rosenkrantz MS, Guarente L. Saccharomyces cerevisiae contains two functional citrate synthase genes. Mol Cell Biol. 1986;6:1936–1942. doi: 10.1128/mcb.6.6.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCammon MT. Mutants of Saccharomyces cerevisiae with defects in acetate metabolism: isolation and characterization of Acn- mutants. Genetics. 1996;144:57–69. doi: 10.1093/genetics/144.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cupp JR, McAlister-Henn L. NAD+-dependent isocitrate dehydrogenase. Cloning, nucleotide sequence, and disruption of the IDH2 gene from Saccharomyces cerevisiae. J Biol Chem. 1991;266:22199–22205. [PubMed] [Google Scholar]
- 8.Cupp JR, McAlister-Henn L. Cloning and characterization of the gone encoding the IDH1 subunit of NAD+-dependent isocitrate dehydrogenase from Saccharomyces cerevisiae. J Biol Chem. 1992;267:16417–16423. [PubMed] [Google Scholar]
- 9.Cupp JR, McAlister-Henn L. Kinetic analysis of NAD+-isocitrate dehydrogenase with altered isocitrate binding sites: contribution of IDH1 and IDH2 subunits to regulation and catalysis. Biochemistry. 1993;32:9323–9328. doi: 10.1021/bi00087a010. [DOI] [PubMed] [Google Scholar]
- 10.Zhao WN, McAlister-Henn L. Affinity purification and kinetic analysis of mutant forms of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 1997;272:21811–21817. doi: 10.1074/jbc.272.35.21811. [DOI] [PubMed] [Google Scholar]
- 11.Panisko EA, McAlister-Henn L. Subunit interactions of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2001;276:1204–1210. doi: 10.1074/jbc.M005056200. [DOI] [PubMed] [Google Scholar]
- 12.Lin AP, McAlister-Henn L. isocitrate binding at two functionally distinct sites in yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2002;277:22475–22483. doi: 10.1074/jbc.M202534200. [DOI] [PubMed] [Google Scholar]
- 13.Lin AP, McAlister-Henn L. Homologous binding sites in yeast isocitrate dehydrogenase for cofactor (NAD+) and allosteric activator (AMP) J Biol Chem. 2003;278:12864–12872. doi: 10.1074/jbc.M300154200. [DOI] [PubMed] [Google Scholar]
- 14.Lin AP, McCammon MT, McAlister-Henn L. Kinetic and physiological effects of alterations in homologous isocitrate-binding sites of yeast NAD+-specific isocitrate dehydrogenase. Biochemistry. 2001;40:14291–14301. doi: 10.1021/bi0111707. [DOI] [PubMed] [Google Scholar]
- 15.Taylor AB, Hu G, Hart PJ, McAlister-Henn L. Allosteric motions in structures of yeast NAD+-specific isocitrate dehydrogenase. J Biol Chem. 2008;283:10872–10880. doi: 10.1074/jbc.M708719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke WF, Johanson RA, Reeves HC. NADP+-specific isocitrate dehydrogenase of Escherichia coli. II Subunit structure. Biochim Biophys Acta. 1974;351:333–340. doi: 10.1016/0005-2795(74)90196-2. [DOI] [PubMed] [Google Scholar]
- 17.Thorsness PE, Koshland DE., Jr Inactivation of isocitrate dehydrogenase by phosphorylation is mediated by the negative charge of the phosphate. J Biol Chem. 1987;262:10422–10425. [PubMed] [Google Scholar]
- 18.Dean AM, Lee MH, Koshland DE., Jr Phosphorylation inactivates Escherichia coli isocitrate dehydrogenase by preventing isocitrate binding. J Biol Chem. 1989;264:20482–20486. [PubMed] [Google Scholar]
- 19.LaPorte DC, Walsh K, Koshland DE., Jr The branch point effect. Ultrasensitivity and subsensitivity to metabolic control. J Biol Chem. 1984;259:14068–14075. [PubMed] [Google Scholar]
- 20.Walsh K, Koshland DE., Jr Determination of flux through the branch point of two metabolic cycles. The tricarboxylic acid cycle and the glyoxylate shunt. J Biol Chem. 1984;259:9646–9654. [PubMed] [Google Scholar]
- 21.Walsh K, Koshland DE., Jr Branch point control by the phosphorylation state of isocitrate dehydrogenase. A quantitative examination of fluxes during a regulatory transition. J Biol Chem. 1985;260:8430–8437. [PubMed] [Google Scholar]
- 22.LaPorte DC. The isocitrate dehydrogenase phosphorylation cycle: regulation and enzymology. J Cell Biochem. 1993;51:14–18. doi: 10.1002/jcb.240510104. [DOI] [PubMed] [Google Scholar]
- 23.McCammon MT, Veenhuis M, Trapp SB, Goodman JM. Association of glyoxylate and beta-oxidation enzymes with peroxisomes of Saccharomyces cerevisiae. J Bacteriol. 1990;172:5816–5827. doi: 10.1128/jb.172.10.5816-5827.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.St Gietz D, Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delaunay A, Isnard AD, Toledano MB. H2O2 sensing through oxidation of the Yap1 transcription factor. Embo J. 2000;19:5157–5166. doi: 10.1093/emboj/19.19.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Gadde DM, Yang E, McCammon MT. An unassembled subunit of NAD+-dependent isocitrate dehydrogenase is insoluble and covalently modified. Arch Biochem Biophys. 1998;354:102–110. doi: 10.1006/abbi.1998.0677. [DOI] [PubMed] [Google Scholar]
- 31.Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- 32.Steele JCH, Nielsen TB. Evaluation of cross-lined polypeptides in SDS gel electrophoresis. Anal Biochem. 1978;84:218–224. doi: 10.1016/0003-2697(78)90502-x. [DOI] [PubMed] [Google Scholar]
- 33.Smith BJ. Chemical cleavage of polypeptides. Methods Mol Biol. 2003;211:63–82. doi: 10.1385/1-59259-342-9:63. [DOI] [PubMed] [Google Scholar]
- 34.Lin AP, Hakala KW, Weintraub ST, McAlister-Henn L. Suppression of metabolic defects of yeast isocitrate dehydrogenase and aconitase mutants by loss of citrate synthase. Arch Biochem Biophys. 2008;474:205–212. doi: 10.1016/j.abb.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haselbeck RJ, McAlister-Henn L. Function and expression of yeast mitochondrial NAD- and NADP-specific isocitrate dehydrogenases. J Biol Chem. 1993;268:12116–12122. [PubMed] [Google Scholar]
- 36.Minard KI, McAlisier-Henn L. Redox responses in yeast to acetate as the carbon source. Arch Biochem Biophys. 2008;483:136–143. doi: 10.1016/j.abb.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasek AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 39.Minard KI, McAlister-Henn L. Sources of NADPH in yeast vary with carbon source. J Biol Chem. 2005;280:39890–39896. doi: 10.1074/jbc.M509461200. [DOI] [PubMed] [Google Scholar]
- 40.Piskur J, Langkjaer RB. Yeast genome sequencing: the power of comparative genomics. Mol Microbiol. 2004;53:381–389. doi: 10.1111/j.1365-2958.2004.04182.x. [DOI] [PubMed] [Google Scholar]
- 41.Dujon B. Hemiascomycetous yeasts at the forefront of comparative genomics. Curr Opin Genet Dev. 2005;15:614–620. doi: 10.1016/j.gde.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 42.McDonagh B, Ogueta S, Lasarte G, Padilla CA, Barcena JA. Shotgun redox proteomics identities specifically modified cysteines in key metabolic enzymes under oxidative stress in Saccharomyces cerevisiae. J Proteomics. 2009;72:677–689. doi: 10.1016/j.jprot.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 43.Ramachandran N, Colman RF. Chemical characterization of distinct subunits of pig heart DPN-specific isocitrate dehydrogenase. J Biol Chem. 1980;255:8859–8864. [PubMed] [Google Scholar]
- 44.Soundar S, Park JH, Huh TL, Colman RF. Evaluation by mutagenesis of the importance of 3 arginines in alpha, beta, and gamma subunits of human NAD-dependent isocitrate dehydrogenase. J Biol Chem. 2003;278:52146–52153. doi: 10.1074/jbc.M306178200. [DOI] [PubMed] [Google Scholar]
- 45.Kim YO, Oh IU, Park HS, Jeng J, Song BJ, Huh TL. Characterization of a cDNA clone for human NAD+-specific isocitrate dehydrogenase alpha-subunit and structural comparison with its isoenzymes from different species. Biochem J. 1995;308:63–68. doi: 10.1042/bj3080063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008;40:1230–1234. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haselbeck RJ, McAlister-Henn L. Isolation, nucleotide sequence, and disruption of the Saccharomyces cerevisiae gene encoding mitochondrial NADP(H)-specific isocitrate dehydrogenase. J Biol Chem. 1991;266:2339–2345. [PubMed] [Google Scholar]
- 48.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 49.Reddehase S, Grumbt B, Neupert W, Hell K. The disulfide relay system of mitochondria is required for the biogenesis of mitochondrial Ccs1 and Sod1. J Mol Biol. 2008;385:331–338. doi: 10.1016/j.jmb.2008.10.088. [DOI] [PubMed] [Google Scholar]
- 50.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, Eneqvist T, Tjernberg L, Ankarcrona M, Glaser E. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006;281:29096–29104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 51.Kambacheld M, Augustin S, Tatsuta T, Muller S, Langer T. Role of the novel metallopeptidase Mop112 and saccharolysin for the complete degradation of proteins residing in different sub-compartments of mitochondria. J Biol Chem. 2005;280:20132–20139. doi: 10.1074/jbc.M500398200. [DOI] [PubMed] [Google Scholar]
- 52.Pedrajas JR, Kosmidou E, Miranda-Vizuete A, Gustafsson JA, Wright AP, Spyrou G. Identification and functional characterization of a novel mitochondrial thioredoxin system in Saccharomyces cerevisiae. J Biol Chem. 1999;274:6366–6373. doi: 10.1074/jbc.274.10.6366. [DOI] [PubMed] [Google Scholar]
- 53.Outten CE, Culotta VC. Alternative start sites in the Saccharomyces cerevisiae GLR1 gene are responsible for mitochondrial and cytosolic isoforms of glutathione reductase. J Biol Chem. 2004;279:7785–7791. doi: 10.1074/jbc.M312421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt B, Ho L, Hogg PJ. Allosteric disulfide bonds. Biochemistry. 2006;45:7429–7433. doi: 10.1021/bi0603064. [DOI] [PubMed] [Google Scholar]
- 55.Kuehn GD, Barnes LD, Atkinson DE. Yeast diphosphopyridine nucleotide specific isocitrate dehydrogenase. Binding of ligands. Biochemistry. 1971;10:3945–3951. doi: 10.1021/bi00797a023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.