

Abstract

Monohalo and dihalo 1,3-thiazole derivatives can be efficiently and selectively prepared under mild conditions from 2-amino-1,3-thiazoles. Halogenations proceed easily in the presence of copper(I) or copper(II) chlorides, bromides, or iodides directly in solution or with supported copper halides.
1,3-Thiazole rings appear in many compounds that exhibit important biological and pharmacological activities. For example, these rings feature in all the potent epothilones1used against multidrug-resistant tumor cell lines. They are also found among pharmaceuticals for the treatment of type 2 diabetes,2 antibiotic-like compounds,3 and metabotropic glutamate receptor subtype 5 (mGluR5) antagonists.4
1,3-Thiazole rings are usually introduced into target molecules by use of a monohalo thiazole in an organometal-catalyzed coupling procedure (e.g., a Sonogashira, Heck, or Suzuki reaction). Traditionally, the required monohalo thiazoles have been synthesized from 1,3-thiazole or 2-bromo-1,3-thiazole in two or three steps via stannyl or silyl intermediates.5 2,4-Dibromo-1,3-thiazole has also been produced under harsher conditions with phosphorus oxybromide.6 Although these procedures give access to a variety of halo-1,3-thiazoles, only a limited number are commercially available. Moreover, these methods have seldom shown applicability to the syntheses of more complex thiazole-containing molecules bearing further functional groups. In the course of our research to produce new improved radioligands for the molecular imaging of brain mGluR5 in vivo with positron emission tomography (PET),7 we aimed to synthesize new 2-halo-1,3-thiazole derivatives with various appendages in position 4. One such compound, the 2-bromo-1,3-thiazole derivative (2), had previously been obtained in high yield from compound 1, copper(II) bromide, and tert-butyl nitrite under Sandmeyer conditions (Scheme 1).8
SCHEME 1.

Bromination of 4-((3-Fluorophenyl)ethynyl)-thiazol-2-amine (1) under Sandmeyer Conditions
Surprisingly, in our hands, this reaction led predominantly to the 2,5-dibromo adduct 3 with the targeted monobromo compound 2 as minor product (Scheme 1). Classically, the Sandmeyer reaction involves diazotization of an arylamine followed by reaction of the formed diazonium salt with copper(II) halide (CuX2, X = Cl, Br, or I). Although good yields of haloarenes are generally obtained, this procedure is usually complicated by numerous competing side reactions. The scope and limitations of the Sandmeyer reaction have been widely investigated with various arenes,9 but little was previously known about this reaction for the halogenation of 2-amino-1,3-thiazoles.
To investigate the reasons for the formation of the dibromo compound 3 from 1, we synthesized the des-fluoro analogue 4 and used it as a model in bromination reactions conducted with CuBr2 and n-butyl nitrite in acetonitrile (Scheme 2).
SCHEME 2.

Reactions of Copper Halides with 4-(Phenylethynyl)thiazol-2-amine (4)
The effects of order and method of addition of reagents, the temperature, and the nitrite to copper bromide mole ratio on the yield of the dibromo compound 6b were investigated. Temperature played a critical role in product outcome. Above room temperature, we found that 6b was almost always the major product (>70%), independent of the stoichiometry of reagents and other factors. Use of amyl, tert-butyl or n-butyl nitrite gave the same results; therefore the latter was used in the rest of this work. When the reaction was performed at 85 °C, 6b was formed rapidly, reaching 99% yield after 15 min (Table 1).
TABLE 1.
Yields of Products from the Bromination of 4 with Various Reagents under Sandmeyer Conditions
| brominating agenta | 5b (%)b | 6b (%)b | 7b (%)b |
|---|---|---|---|
| CuBr2c | 94 | ||
| CuBr2d | 99 | ||
| CuBrd | 98 | ||
| CuBr | 67 | ||
| KBr | 9 | 4 | |
| KBr-CuBr | 77 | ||
| Br2 | 58 | ||
| Alumina-KCuBr2 | 90 |
All reactions were performed overnight with n-butyl nitrite at room temperature, unless otherwise indicated.
Yields are conversion of 4 determined by HPLC.
Performed in the absence of n-butyl nitrite.
Performed at 85 °C for 15 min.
The fact that the dibromo compound 6b was produced suggested that the reaction occurred in two steps via a monobromo intermediate. To verify this hypothesis, 4 was submitted to a temperature study in which reaction progress was monitored by HPLC with the temperature rising by 10 °C at intervals of 30 min (±5 min) (Figure 1). Initially, after 10 min of reaction at – 40 °C, 5-bromo-4-(phenyl)ethynyl-1,3-thiazol-2-amine (7b) appeared in solution, showing bromination to occur first at position 5. The ratio of 7b to 4 increased with temperature. At – 10 °C, 7b was the only product. Above –10 °C, 7b converted into the dibromo compound, 6b. This conversion was slow below 0 °C but became rapid and complete at 60 °C. Thus, reaction of 4 with copper(II) bromide in the presence of n-butyl nitrite rapidly gives 7b and is followed by a second slower step, the diazotization of the amino group, and bromine insertion. The latter reaction can be avoided by omitting n-butyl nitrite, resulting in a high yield (94%) of 7b (Table 1).
FIGURE 1.

Yield of products with temperature for the reaction of 4 with copper(II) bromide and n-butyl nitrite (mole ratio, 1:1.2:1.2).
The desired 2-bromo compound 5b was not observed in the preceding temperature study. However, it reached almost 50% yield when the reaction was run at a low temperature (between –10 and 0 °C) with the molar ratio of copper(II) bromide to nitrite decreased to 0.5. At room temperature, under these conditions, yields of 5b never exceeded 30%.
To obtain the initially targeted 2-bromothiazole 5b in acceptable yield, we tested the reactivity of 4 with a variety of brominating agents under Sandmeyer conditions (Table 1). CuBr, KBr, KBr-CuBr, Br2, or alumina-supported-KCuBr2 was added to a solution of 4 and n-butyl nitrite. Remarkably, near quantitative yields of 5b were obtained with CuBr. KBr gave an inextricable mixture of products in which 5b was found in only low yield. A moderately high yield of 5b was obtained with KBr-CuBr. Molecular bromine selectively gave the dibromo-derivative 6b in moderate yield.
The supported copper(I) complex, alumina-KCuBr2, is an efficient reagent for halogen exchange in haloarenes.10 This reagent gave 5b selectively and in very high yield at room temperature after 24 h (Table 1). By contrast, reactions run with CuBr without alumina at this temperature gave lower yields. Technically, the use of a supported reagent is very attractive as the protocol for product extraction is simple filtration. Also this room temperature procedure might potentially allow the halogenation of more complex molecules that would either decompose or racemize in solution at higher temperatures.
We also prepared other alumina-supported copper(I) halides (alumina-KCuX2, X = Cl or I). Reaction of these reagents or of the available CuXn salts (X = Cl or Br for n = 1 or 2, or for I, n = 1) with 4 or the 3-cyanophenyl analogue 9 led to several new halo-1,3-thiazoles (Table 2). Iodination of 4 with alumina-supported-KCuI2 appeared to reach an equilibrium, since a second addition of reagent significantly improved the yield of 5c. Having demonstrated the regioselectivity of these reactions, we speculated that it would be possible to introduce different halogens selectively in two steps at two different positions. The synthesis of 2-iodo-5-chlorothiazole derivative 8 and 2-chloro-5-bromothiazole derivative 9 demonstrated that this was indeed possible with these versatile procedures. The methods are only limited by the availabilities of the halogenation reagents; therefore no 5-iodo analogue could be produced as unstable CuI2 is not commercially available.
TABLE 2.
Yields of Monohalo and Dihalo Derivatives of 1,3-Thiazol-4-arylacetylenes
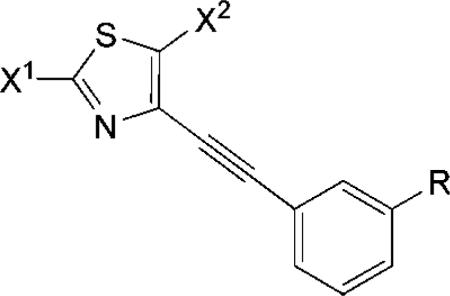 | |||||
|---|---|---|---|---|---|
| product | methoda | R | X1 | X2 | yieldb (%) |
| 5a | I | H | Cl | H | 33 |
| 5b | I | H | Br | H | 46 |
| 5c | I | H | I | H | 50 |
| 6a | II | H | Cl | Cl | 35 |
| 6b | II | H | Br | Br | 79 |
| 7a | III | H | NH2 | Cl | 51 |
| 7b | III | H | NH2 | Br | 94 |
| 8 | III then I | H | I | Cl | 42 |
| 9 | III then I | H | Cl | Br | 13 |
| 11 | I | CN | Br | H | 32 |
See Scheme 2 and the Experimental Section for details
Yields are for isolated pure materials.
It has been established that the Sandmeyer reaction proceeds via formation of radical species.11 Nevertheless, in the case of the 2-amino-1,3-thiazole derivative 4, addition of the radical trap TEMPO or of the radical initiator AIBN failed to modify the yields of reaction or the ratios of the products 5b, 6b, and 7b, suggesting that the reactions proceeded according to ionic mechanisms.
In conclusion, we have shown that halo-1,3-thiazole derivatives can be prepared regioselectively and efficiently with a variety of simple reagents. Regioselective halogenation in position 5 of 2-amino-1,3-thiazoles was achieved at room temperature by reaction with CuX2 (X = Cl or Br) in acetonitrile. Dibromination was realized in high yield with n-butyl nitrite and CuX2 at temperatures above 65 °C and halogenation in position 2 was achieved selectively by using an alumina-supported copper(I) material or CuX (X = Cl, Br, or I). The methods we described here for selective halogenations of 2-amino-1,3-thiazole derivatives were mild and were successfully used on variously substituted compounds and led to several new iodo, bromo, and chloro derivatives. These are especially interesting as potential intermediates for new mGluR5 ligands, and could lead to the development of new PET radioligands by radiofluorination12 or new SPECT radioligands by radioiodination.13
Experimental Section
Method I: Synthesis of 2-Halo-1,3-thiazolarylacetylenes 5a–c
The 2-aminothiazole 4 (182 mg, 0.91 mmol) and CuX (1.39 mmol, X = Cl, Br, or I) were dissolved in acetonitrile (8 mL) at room temperature. n-Butyl nitrite (162 μL, 1.39 mmol) was added with stirring, and the solution was heated to 60 °C. The reaction was complete after 15 min, as monitored by TLC. The reaction mixture was then evaporated to dryness in vacuo. The residue was dissolved in ethyl acetate (20 mL) and washed with ammonia solution (0.1 M; 2 × 50 mL). The organic layer was dried over MgSO4 and evaporated to dryness in vacuo. The residue was purified by chromatography on silica gel (hexane–ethyl acetate; 97: 3, v/v). This procedure gave 5a, 5b, and 5c in 33%, 46%, and 50% yield, respectively.
Method II: Synthesis of Dihalo 1,3-Thiazoles 6a and 6b
The 2-aminothiazole 4 (40 mg, 0.20 mmol) and CuX2 (X = Cl or Br; 0.30 mmol) were dissolved in acetonitrile (2 mL). This reaction mixture was stirred at room temperature for 15 to 120 min (gentle heating at 40 °C is necessary when using CuCl2 as the reaction proceeds at a slower rate). Reaction was monitored by TLC. After all starting material had been consumed, n-butyl nitrite (35 μL, 0.30 mmol) was added and the reaction was stirred for an additional 15 min at 65 °C. The reaction mixture was then cooled and the acetonitrile evaporated off in vacuo. The residue was dissolved in ethyl acetate (20 mL) and washed with ammonia solution (0.1 M; 2 × 50 mL). The organic layer was collected, dried over MgSO4, and evaporated to dryness in vacuo. The residue was purified by chromatography on silica gel (hexane–ethyl acetate; 97: 3, v/v). This procedure gave 6a and 6b in 35% and 79% yield, respectively.
Method III: Synthesis of 2-Amino-5-halo-1,3-thiazolarylacetylenes 7a and 7b
2-Aminothiazole derivative 4 (52 mg, 0.26 μmol) and CuX2 (X = Cl or Br; 0.26 μmol) were dissolved in acetonitrile (2.5 mL). This reaction mixture was stirred at rt for 10 h. The reaction mixture was then evaporated to dryness in vacuo, and the residue was dissolved in ethyl acetate (20 mL) and washed with aqueous ammonia (0.1 M; 2 × 50 mL). The organic layer was dried over MgSO4 then evaporated to dryness in vacuo. The residue was purified by chromatography on silica gel (hexane–ethyl acetate; 70: 30, v/v). This procedure gave 7a and 7b in 51% and 94% yield, respectively.
General Procedure for the Halogenation of 4 on Supported Copper(I) Salts
Potassium halide (1 mmol) and copper(I) halide (1 mmol) were mixed in a flask containing neutral aluminum oxide (3 g). A solution of water (0.1% v/v) in acetonitrile (5 mL) was added and the mixture was stirred in an open vessel until solvent had completely evaporated off. The residue was placed in an oven at 100 °C for 24 h and stored at room temperature before use. 2-Aminothiazole 4 (25 μmol) was dissolved in acetonitrile (3.5 mL) with n-butyl nitrite (30 μmol) at room temperature. The reaction mixture was stirred for 15 min and then a supported copper(I) halide (0.5 g) was added. The reaction mixture was stirred overnight at room temperature. Then acetonitrile was filtered off and the residue was rinsed with ethyl acetate. The organic solutions were combined and solvent evaporated off. The residue was purified by chromatography on silica gel. This procedure produced compounds 5a, 5b, and 5c in 56%, 83%, and 62% yield, respectively.
In the case of the Al2O3–KCuI2, the reaction was incomplete after 24 h at room temperature. Addition of more supported reagent (0.5 g) led to reaction completion after a further 12 h. The mixture was filtered and the filtrate was washed with sodium metabisulfate and dried over magnesium sulfate. Finally, solvents were evaporated off and product purified with chromatography.
Supplementary Material
Acknowledgement
This work was supported by the Intramural Research Program of the National Institutes of Health (NIMH).
Footnotes
Supporting Information Available: Other experimental procedures, characterization data for compounds 4–11, and copies of 1H and 13C spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nicolaou KC, King NP, Finlay MRV, He Y, Roschangar F, Vourloumis D, Vallberg H, Sarabia F, Ninkovic S, Hepworth D. Bioorg. Med. Chem. 1999;7:665. doi: 10.1016/s0968-0896(98)00153-9. [DOI] [PubMed] [Google Scholar]
- 2.Fyfe FMCT, Gardner LS, Nawano M, Procter JM, Rasamison CM, Shofield KL, Shah VK, Yasuda K. PCT Int. Appl. WO 2004/072031, 26 August 2004. Chem. Abstr. 2004;141:225496. [Google Scholar]
- 3.Kelly TR, Lang F. Tetrahedron Lett. 1995;36:5319. [Google Scholar]
- 4.a Cosford NDP, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson JJ, Bristow L, Brodkin J, Jiang XH, McDonald I, Rao S, Washburn M, Varney MA. J. Med. Chem. 2003;46:204. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]; b Hamill TG, Krause S, Ryan C, Bonnefous C, Govek S, Seiders TJ, Cosford NDP, Roppe J, Kamenecka T, Patel S, Gibson RE, Sanabria S, Riffel K, Eng W, King C, Yang X, Green MD, O'Malley SS, Hargreaves R, Burns HD. Synapse. 2005;56:205. doi: 10.1002/syn.20147. [DOI] [PubMed] [Google Scholar]
- 5.a Dondoni A, Mastellari AR, Medici A, Negrini E, Pedrini P. Synthesis. 1986;9:757. [Google Scholar]; b Dondoni A, Fantin G, Fogagnolo M, Medici A, Pedrini P. J. Org. Chem. 1988;53:1748. [Google Scholar]
- 6.a Stanetty P, Schnuerch M, Mihovilovic MD. J. Org. Chem. 2006;71:3754. doi: 10.1021/jo0601009. [DOI] [PubMed] [Google Scholar]; b Le Flohic A, Meyer C, Cossy J. Tetrahedron. 2006;62:9017. [Google Scholar]
- 7.a Siméon FG, Brown AK, Zoghbi SS, Patterson VM, Innis RB, Pike VW. J. Med. Chem. 2007;50:3256. doi: 10.1021/jm0701268. [DOI] [PubMed] [Google Scholar]; b Brown AK, Kimura Y, Zoghbi SS, Siméon FG, Liow J-S, Kreisl WC, Taku A, Fujita M, Pike VW, Innis RB. J. Nucl. Med. 2008;49:2042. doi: 10.2967/jnumed.108.056291. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shetty HU, Zoghbi SS, Siméon FG, Liow JS, Brown AK, Kannan P, Innis RB, Pike VW. J. Pharmacol. Exp. Ther. 2008;327:727. doi: 10.1124/jpet.108.143347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iso Y, Grajkowska E, Wroblewski JT, Davis J, Goeders NE, Johnson KM, Sanker S, Roth BL, Tueckmantel W, Kozikowski AP. J. Med. Chem. 2006;49:1080. doi: 10.1021/jm050570f. [DOI] [PubMed] [Google Scholar]
- 9.a Doyle MP, Siegfried B, Dellaria JF. J. Org. Chem. 1977;42:2426. [Google Scholar]; b Kochi JK. J. Am. Chem. Soc. 1957;79:2942. [Google Scholar]
- 10.Clark JH, Jones CW, Duke CVA, Miller JM. J. Chem. Res. (S) 1989:238. [Google Scholar]
- 11.Friedman L, Chlebowski JF. J. Org. Chem. 1968;33:1633. [Google Scholar]
- 12.a Siméon FG, Pike VW. J. Labelled Compd. Radiopharm. 2005;48(1):S158. [Google Scholar]; b Cai L, Lu SY, Pike VW. Eur. J. Org. Chem. 2008;17:285. [Google Scholar]
- 13.See: Kabalka GW, Gooch EE, Sastry KAR. J. Nucl. Med. 1981;22:908.Dewanjee MK. Radioiodination: Theory, Practice, and Biomedical Application, Developments in Nuclear Medicine. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1992.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.